An Unstructured Phylogeographic Pattern with Extensive Gene Flow in an Endemic Bird of South China: Collared Finchbill (Spizixos semitorques)

Abstract
:1. Introduction
2. Results and Discussion
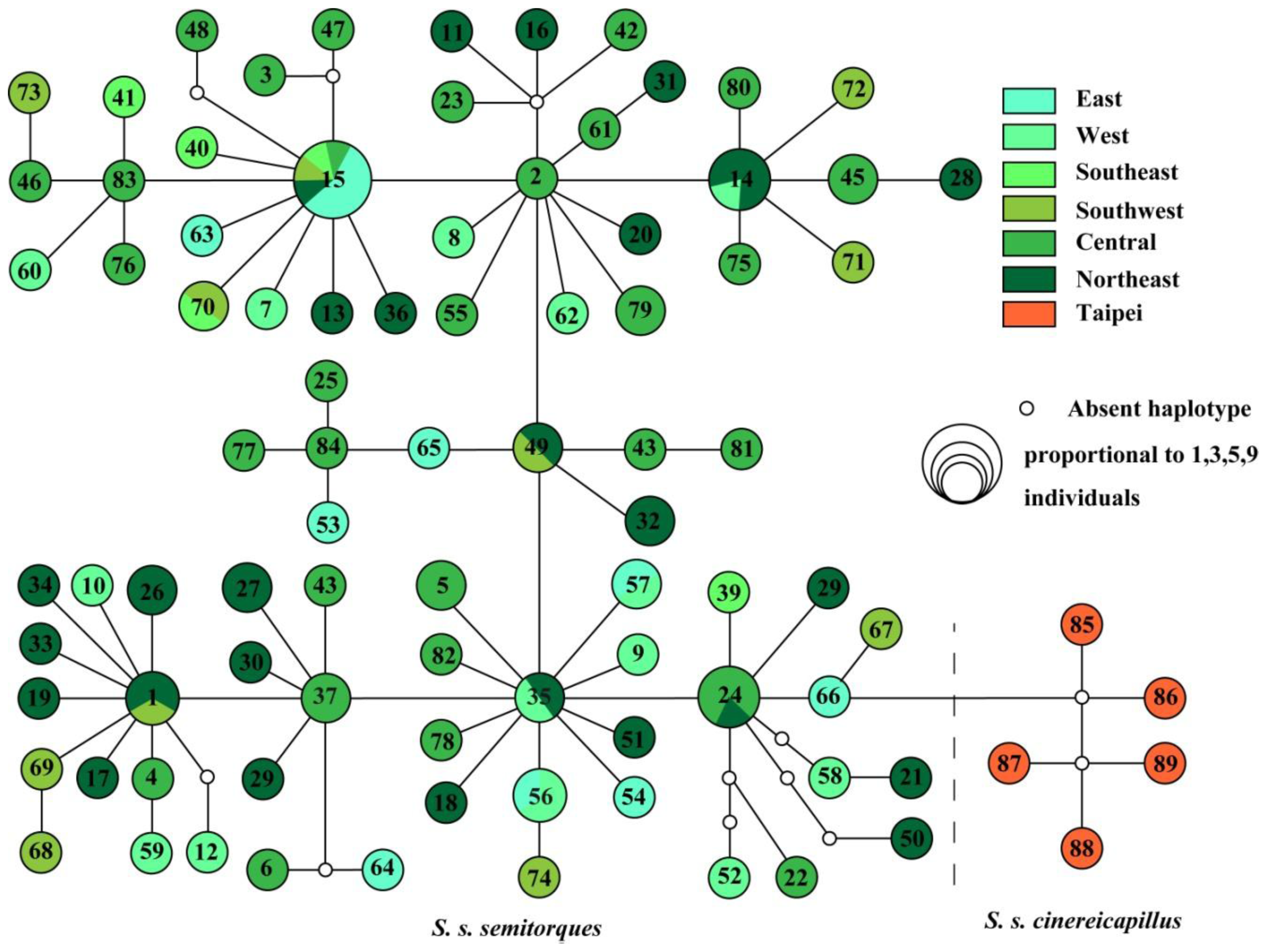
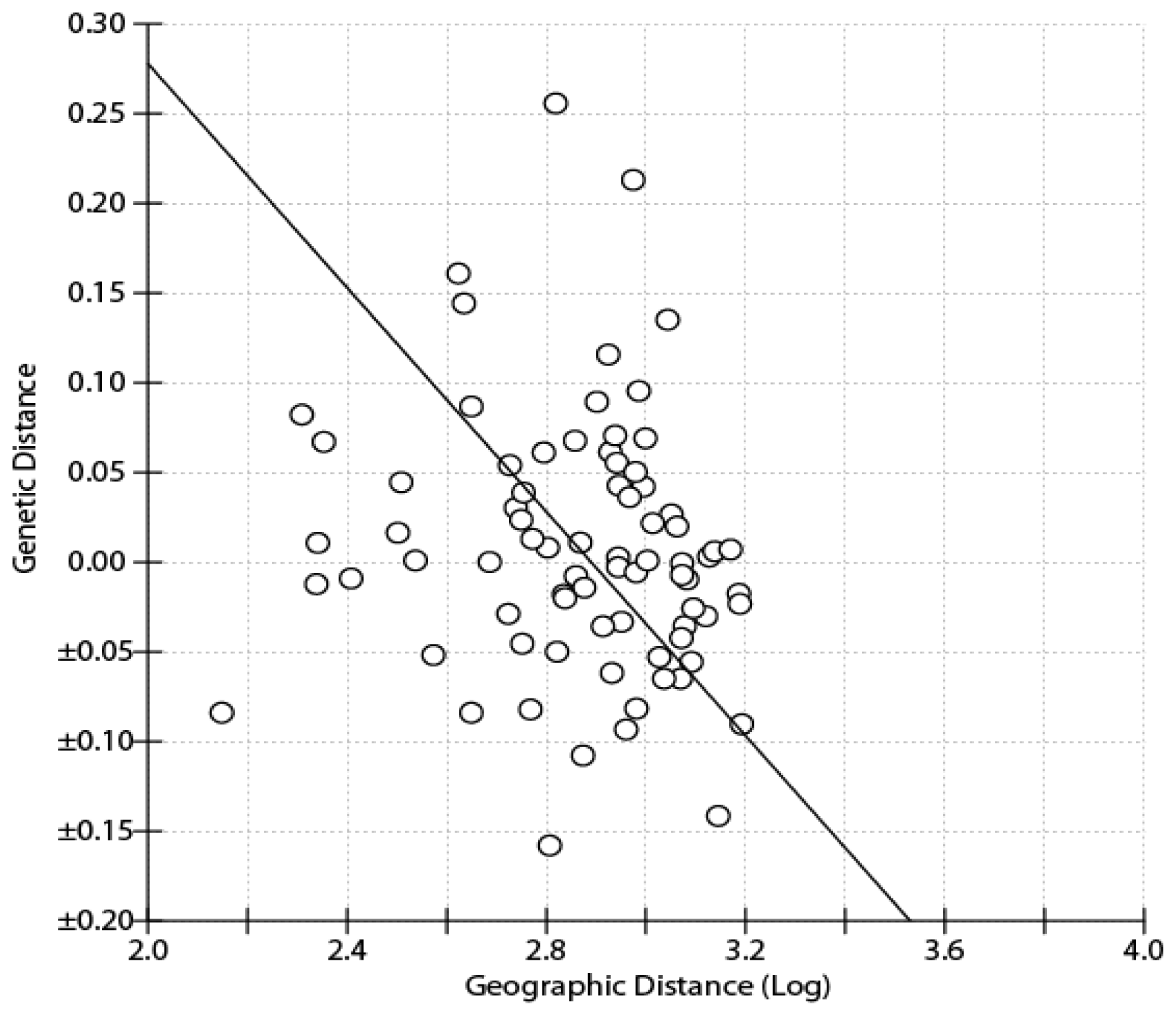
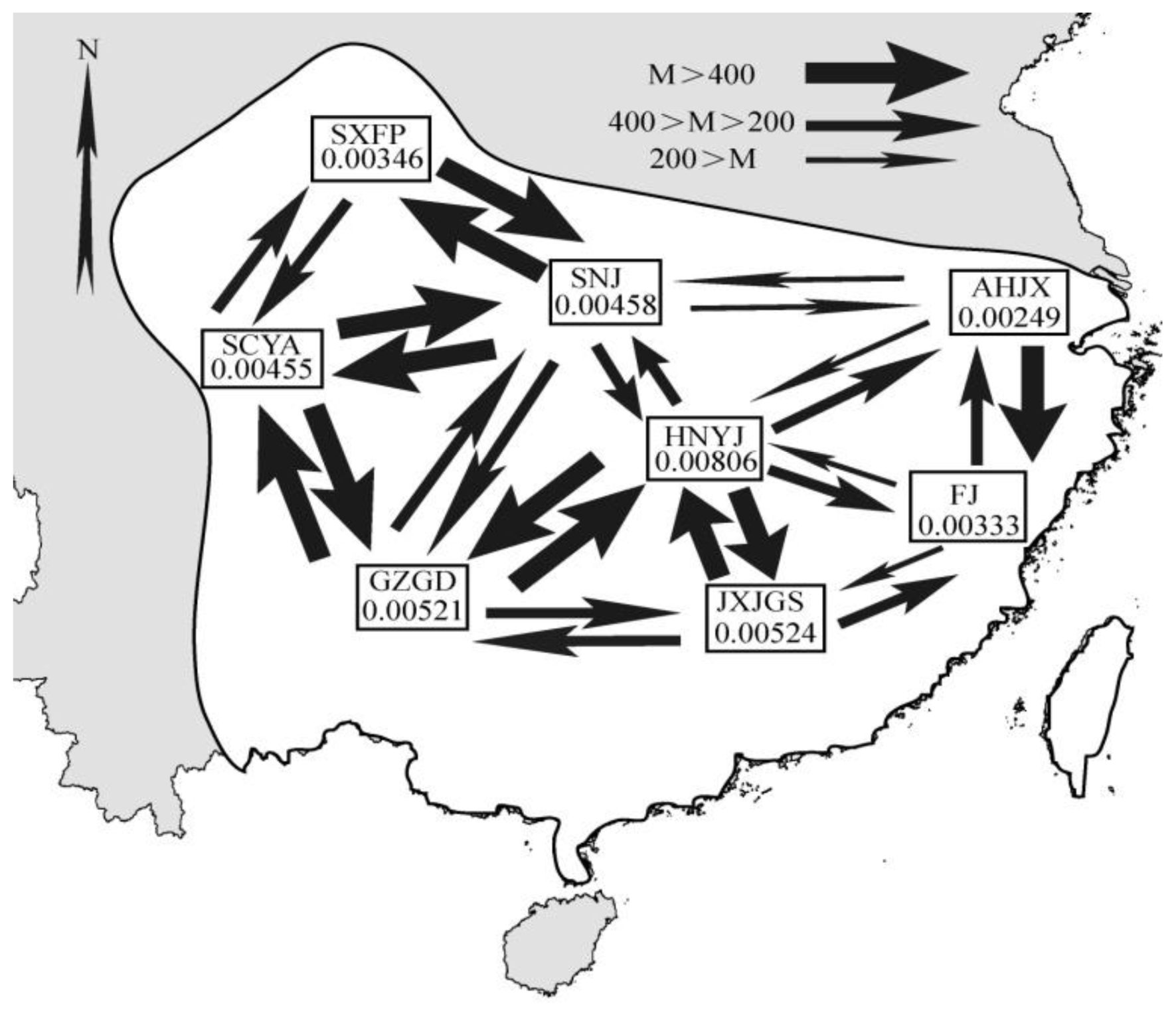
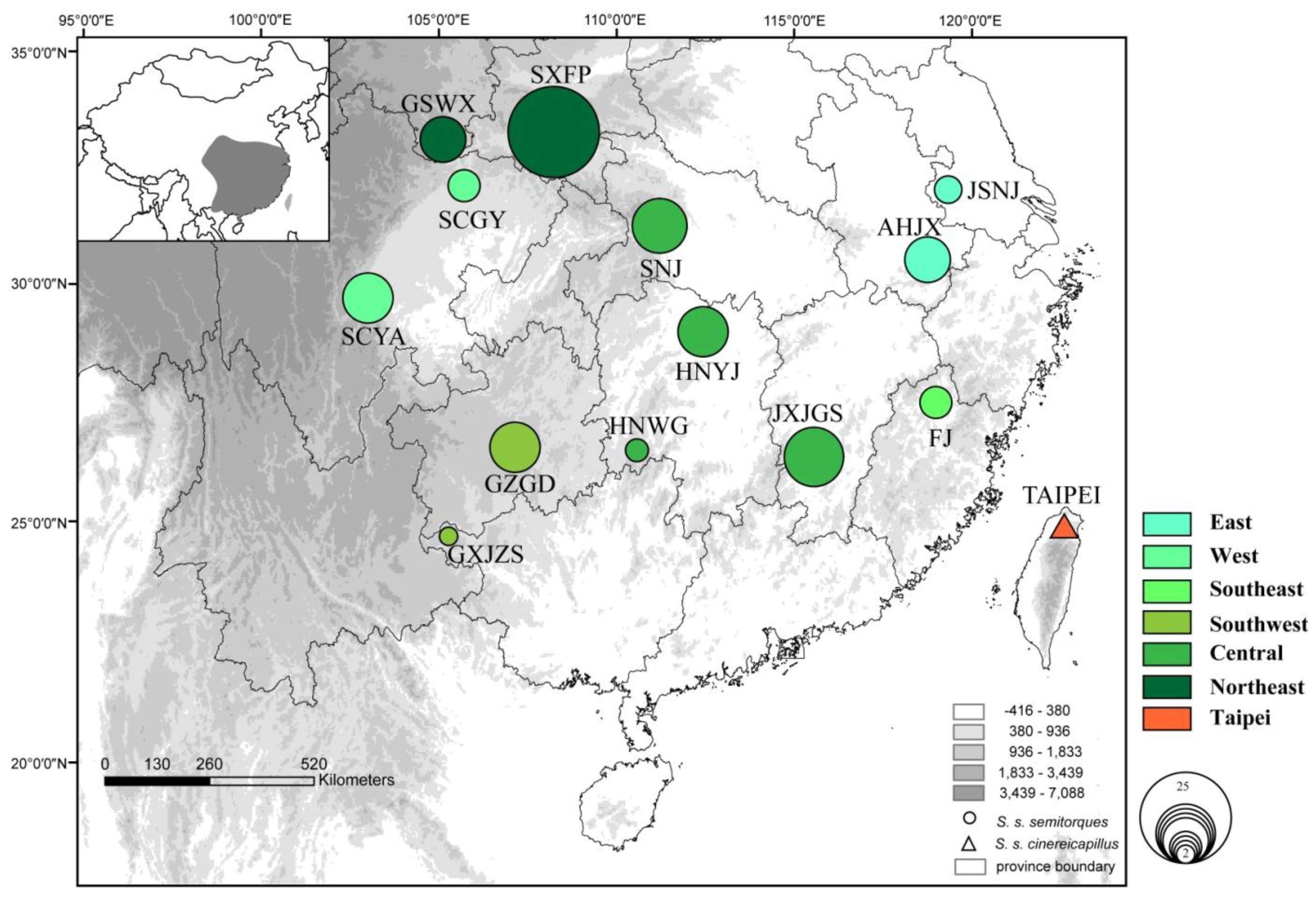
2.1. Results
2.2. Discussion
Demographic History
3. Materials and Methods
3.1. Sample Collection, DNA Extraction, Amplification and Sequencing
3.2. Sequence Analysis
4. Conclusions
Acknowledgements
References
- Avise, J. Phylogeography: The History and Formation of Species; Harvard University Press: London, UK, 2000; pp. 145–208. [Google Scholar]
- Hewitt, G. The genetic legacy of the Quaternary ice ages. Nature 2000, 405, 907–913. [Google Scholar]
- Hewitt, G. Genetic consequences of climatic oscillations in the Quaternary. Phil. Trans. Roy. Soc. London B 2004, 359, 183–195. [Google Scholar]
- Taberlet, P; Fumagalli, L; Wust-Saucy, AG; Cosson, JF. Comparative phylogeography and postglacial colonization routes in Europe. Mol. Ecol 1998, 7, 453–464. [Google Scholar]
- Lessa, EP; Cook, JA; Patton, JL. Genetic footprints of demographic expansion in North America, but not Amazonia, during the Late Quaternary. Proc. Nat. Acad. Sci. USA 2003, 100, 10331–10334. [Google Scholar]
- Zhang, Y; Ge, S. Molecular evolution study in China: Progress and future promise. Phil. Trans. Roy. Soc. London B 2007, 362, 973–986. [Google Scholar]
- Qiu, YX; Fu, CX; Comes, HP. Plant molecular phylogeography in China and adjacent regions: Tracing the genetic imprints of Quaternary climate and environmental change in the world’s most diverse temperate flora. Mol. Phylogenet. Evol 2011, 59, 225–244. [Google Scholar]
- Soltis, DE; Morris, AB; McLachlan, JS; Manos, PS; Soltis, PS. Comparative phylogeography of unglaciated eastern North America. Mol. Ecol 2006, 15, 4261–4293. [Google Scholar]
- Shi, Y. Quaternary glaciation in China. Quatern. Sci. Rev 1986, 5, 503–507. [Google Scholar]
- Liu, K. Quaternary history of the temperate forests of China. Quatern. Sci. Rev 1988, 7, 1–20. [Google Scholar]
- Weaver, A; Eby, M; Fanning, A; Wiebe, E. Simulated influence of carbon dioxide, orbital forcing and ice sheets on the climate of the last glacial maximum. Nature 1998, 394, 847–853. [Google Scholar]
- Pinot, S; Ramstein, G; Harrison, S; Prentice, I; Guiot, J; Stute, M; Joussaume, S. Tropical paleoclimates at the last glacial maximum: comparison of Paleoclimate Modeling Intercomparison Project (PMIP) simulations and paleodata. Clim. Dynam 1999, 15, 857–874. [Google Scholar]
- Ju, L; Wang, H; Jiang, D. Simulation of the last glacial maximum climate over East Asia with a regional climate model nested in a general circulation model. Palaeogeogr. Palaeoclimatol 2007, 248, 376–390. [Google Scholar]
- Li, S; Yeung, C. Sailing through the Late Pleistocene: unusual historical demography of an East Asian endemic, the Chinese Hwamei (Leucodioptron canorum canorum), during the last glacial period. Mol. Ecol 2009, 18, 622–633. [Google Scholar]
- Qian, H; Ricklefs, RE. Large-scale processes and the Asian bias in species diversity of temperate plants. Nature 2000, 407, 180–182. [Google Scholar]
- Song, G; Qu, Y; Yin, Z; Li, S; Liu, N; Lei, F. Phylogeography of the Alcippe morrisonia (Aves: Timaliidae): Long population history beyond late Pleistocene glaciations. BMC Evol. Biol 2009, 9, 143. [Google Scholar]
- Huang, Z; Liu, N; Liang, W; Zhang, Y; Liao, X; Ruan, L; Yang, Z. Phylogeography of Chinese bamboo partridge, Bambusicola thoracica thoracica (Aves: Galliformes) in south China: Inference from mitochondrial DNA control-region sequences. Mol. Phylogenet. Evol 2010, 56, 273–280. [Google Scholar]
- MacKinnon, J; Phillipps, K; He, F. A Field Guide to the Birds of China; Oxford University Press: New York, NY, USA, 2000; pp. 362–363. [Google Scholar]
- Grant, W; Bowen, B. Shallow population histories in deep evolutionary lineages of marine fishes: insights from sardines and anchovies and lessons for conservation. J. Hered 1998, 89, 415–427. [Google Scholar]
- Hewitt, G. Some genetic consequences of ice ages, and their role, in divergence and speciation. Biol. J. Linn. Soc 1996, 58, 247–276. [Google Scholar]
- Kryukov, A; Iwasa, M; Kakizawa, R; Suzuki, H; Pinsker, W; Haring, E. Synchronic east-west divergence in azure-winged magpies (Cyanopica cyanus) and magpies (Pica pica)*. J. Zool. Syst. Evol. Res 2004, 42, 342–351. [Google Scholar]
- Zink, RM; Drovetski, SV; Questiau, S; Fadeev, IV; Nesterov, EV; Westberg, MC; Rohwer, S. Recent evolutionary history of the bluethroat (Luscinia svecica) across Eurasia. Mol. Ecol 2003, 12, 3069–3075. [Google Scholar]
- Pavlova, A; Zink, RM; Drovetski, SV; Red’kin, Y; Rohwer, S. Phylogeographic patterns in Motacilla flava and Motacilla citreola: species limits and population history. Auk 2003, 120, 744–758. [Google Scholar]
- Zou, F; Chen, G. A study of understory bird communities in tropical mountain rain forest of Jianfengling, Hainan Island, China. Acta Ecol. Sinica 2004, 24, 510–516. [Google Scholar]
- Taiwanica, AZ. Diet analysis of the gray-cheeked Fulvetta (Alcippe morrisonia) at Fushan Experimental Forest in Taiwan. Acta Zool. Taiwan 1998, 9, 59–66. [Google Scholar]
- Yuan, D; Cheng, H; Edwards, R; Dykoski, C; Kelly, M; Zhang, M; Qing, J; Lin, Y; Wu, J. Timing, duration, and transitions of the last interglacial Asian monsoon. Science 2004, 304, 575–578. [Google Scholar]
- Kelly, M; Edwards, R; Cheng, H; Yuan, D; Cai, Y; Zhang, M; Lin, Y; An, Z. High resolution characterization the Asian Monsoon between 146,000 and 99,000 years BP from Dongge Cave, China and global correlation of events surrounding Termination II. Palaeogeogr. Palaeoclimatol 2006, 236, 20–38. [Google Scholar]
- Yu, G; Gui, F; Shi, Y; Zheng, Y. Late marine isotope stage 3 palaeoclimate for East Asia: A data-model comparison. Palaeogeogr. Palaeoclimatol 2007, 250, 167–183. [Google Scholar]
- Zhao, B; Wang, Z; Chen, J; Chen, Z. Marine sediment records and relative sea level change during late Pleistocene in the Changjiang delta area and adjacent continental shelf. Quatern. Int 2008, 186, 164–172. [Google Scholar]
- Hearty, PJ; Hollin, JT; Neumann, AC; O’Leary, MJ; McCulloch, M. Global sea-level fluctuations during the Last Interglaciation (MIS 5e). Quatern. Sci. Rev 2007, 26, 2090–2112. [Google Scholar]
- Qu, Y; Lei, F. Comparative phylogeography of two endemic birds of the Tibetan plateau, the white-rumped snow finch (Onychostruthus taczanowskii) and the Hume’s ground tit (Pseudopodoces humilis). Mol. Phylogenet. Evol 2009, 51, 312–326. [Google Scholar]
- Tarr, C. Primers for amplification and determination of mitochondrial control-region sequences in oscine passerines. Mol. Ecol 1995, 4, 527–530. [Google Scholar]
- Hall, T. BioEdit: A User-Friendly Biological Sequence Alignment Editor and Analysis Program for Windows 95/98/NT. Nucleic Acids Symp 1999, 41, 95–98. [Google Scholar]
- Librado, P; Rozas, J. DnaSP version 5.0: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar]
- Nei, M. Molecular Evolutionary Genetics; Columbia Universiy Press: New York, NY, USA, 1987. [Google Scholar]
- Nei, M; Tajima, F. DNA polymorphism detectable by restriction endonucleases. Genetics 1981, 97, 145–163. [Google Scholar]
- Excoffier, L; Laval, G; Schneider, S. Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol. Bioinform 2005, 1, 47–50. [Google Scholar]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar]
- Fu, Y; Li, W. Statistical tests of neutrality of mutations. Genetics 1993, 133, 693–709. [Google Scholar]
- Fu, YX. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 1997, 147, 915–925. [Google Scholar]
- Bandelt, H; Forster, P; Röhl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol 1999, 16, 37–48. [Google Scholar]
- Jensen, J; Bohonak, A; Kelley, S. Isolation by distance, web service. BMC Genet 2005, 6, 13. [Google Scholar]
- Nielsen, R; Wakeley, J. Distinguishing migration from isolation: A Markov chain Monte Carlo approach. Genetics 2001, 158, 885–896. [Google Scholar]
- Weir, J; Schluter, D. Calibrating the avian molecular clock. Mol. Ecol 2008, 17, 2321–2328. [Google Scholar]
- Weir, JT. Divergent timing and patterns of species accumulation in lowland and highland neotropical birds. Evolution 2006, 60, 842–855. [Google Scholar]
- Lovette, IJ. Mitochondrial dating and mixed support for the “2% rule” in birds. Auk 2004, 121, 1–6. [Google Scholar]
- Sæther, BE; Lande, R; Engen, S; Weimerskirch, H; Lillegard, M; Altwegg, R; Becker, PH; Bregnballe, T; Brommer, JE; McCleery, RH; Merilä, J; Nyholm, E; Rendell, W; Tryjanowski, P. Generation time and temporal scaling of bird population dynamics. Nature 2005, 436, 99–102. [Google Scholar]
- Kuhner, M. LAMARC 2.0: Maximum likelihood and Bayesian estimation of population parameters. Bioinformatics 2006, 22, 768–770. [Google Scholar]
- Posada, DC; Crandall, KA. MODELTEST: Testing the model of DNA substitution. Bioinformatics 1998, 14, 817–818. [Google Scholar]
- Rambaut, A; Drummond, AJ. Tracer v1.5. 2007. Available online: http://beast.bio.ed.ac.uk/Tracer accessed on 29 March 2011.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. s. semitorques | S. s. cinereicapillus | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Statistics | GSWX | SXFP | SCGY | SCYA | SNJ | JXJGS | HNYJ | GZGD | AHJX | FJ | TAIPEI |
| Tajima’s D | −0.933 | −1.633 | −0.109 | −0.7064 | −0.996 | −0.694 | −0.591 | −0.691 | −0.986 | −1.199 | −0.894 |
| P | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS |
| Fu’s Fs | −4.843 | −12.75 | −1.283 | −3.336 | −2.752 | −1.908 | −5.719 | −6.401 | −2.764 | −1.554 | −1.633 |
| P | <0.01 | 0.00 | NS | NS | NS | NS | NS | NS | NS | NS | NS |
| Fu and Li’s D | −1.313 | −2.803 | −0.109 | −0.640 | −1.270 | −0.798 | −0.765 | −1.360 | −1.370 | −1.199 | −0.894 |
| P | NS | < 0.05 | NS | NS | NS | NS | < 0.01 | < 0.01 | NS | NS | NS |
| Group | Among groups (AG) | Among populations within groups(AP) | Within populations (WP) | Percentage of variation |
|---|---|---|---|---|
| FCT | FSC | FST | ||
| 2 Groups S. s. semitorques (mainland sites): S. s. cinereicapillus (TAIPEI) | 0.5043 | 0.0098 | 0.5092 | Among groups:50.44 Among populations within group: 0.49 Within populations: 49.08 |
| 6 Groups East(AHJX, JSNJ): West(SCYA, SCGY): Southeast(FJ): Southwest(GZGD, GXJZS): Central (JXJGS, HNYJ, HNWG, SNJ): Northwest(GSWX, SXFP) | 0.0029 | 0.0067 | 0.0096 | Among groups: 0.29 Among populations within group: 0.67 Within populations: 99.04 |
| Subspecies | Localities | Latitude | Longitude | Sample size | Haplotype numbers | Nucleotide diversity (π) | Haplotype diversity (Hd) |
|---|---|---|---|---|---|---|---|
| S. s. semitorques | GSWX | 32°56′37.74″ | 104°41′0.16″ | 9 | 1/13/14/15/16/17/18/19/21 | 0.00236 | 1.000 |
| SXFP | 33°11′24.0″ | 108°12′0.0″ | 25 | 1/11/14/20/24/26/27/28/29/30/31/32/33/34/35/36/38/49/50/51 | 0.00231 | 0.980 | |
| SCGY | 32°25′44.51″ | 105°54′4.61″ | 5 | 7/8/9/10/12 | 0.00243 | 1.000 | |
| SCYA | 29°35′24.0″ | 102°35′24.0″ | 10 | 14/35/52/56/57/58/59/60/62 | 0.00252 | 0.978 | |
| SNJ | 31°24′35.6″ | 110°33′18.5″ | 11 | 2/3/4/5/6/37/45/55/61 | 0.00236 | 0.964 | |
| HNYJ | 28°55′53.0″ | 112°17′39.7″ | 10 | 22/24/25/42/43/44/46/47/48 | 0.00240 | 0.978 | |
| HNWG | 26°25′48.0″ | 110°22′12.0″ | 3 | 15/23/24 | 0.00187 | 1.000 | |
| GZGD | 26°35′4.96″ | 107°14′3.46″ | 10 | 1/15/49/68/69/70/71/72/73/ | 74 0.00245 | 1.000 | |
| GXJZS | 24°39′59.36″ | 104°52′17.8″ | 2 | 37/67 | 0.00187 | 1.000 | |
| JXJGS | 26°32′38.44″ | 114°8′50.45″ | 12 | 24/75/76/77/78/79/80/81/82/83/84 | 0.00195 | 0.985 | |
| JSNJ | 32°1′48.00″ | 118°27′36.0″ | 4 | 53/54/56/57 | 0.00210 | 1.000 | |
| AHJX | 30°4′15.12″ | 118°35′44.45″ | 9 | 15/63/64/65/66 | 0.00132 | 0.786 | |
| FJ | 27°19′54.86″ | 118°7′13.85″ | 5 | 15/39/40/41/70 | 0.00205 | 1.000 | |
| S. s. cinereicapillus | TAIPEI | 24°54′56.56″ | 121°40′26.17″ | 5 | 85/86/87/88/89 | 0.00196 | 1.000 |
© 2011 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gao, B.; Yu, L.; Qu, Y.; Song, G.; Dai, C.; Zhang, R.; Yin, Z.; Wang, K.; Gao, X.; Li, S.-H.; et al. An Unstructured Phylogeographic Pattern with Extensive Gene Flow in an Endemic Bird of South China: Collared Finchbill (Spizixos semitorques). Int. J. Mol. Sci. 2011, 12, 3635-3647. https://doi.org/10.3390/ijms12063635
Gao B, Yu L, Qu Y, Song G, Dai C, Zhang R, Yin Z, Wang K, Gao X, Li S-H, et al. An Unstructured Phylogeographic Pattern with Extensive Gene Flow in an Endemic Bird of South China: Collared Finchbill (Spizixos semitorques). International Journal of Molecular Sciences. 2011; 12(6):3635-3647. https://doi.org/10.3390/ijms12063635
Chicago/Turabian StyleGao, Bin, Lijiang Yu, Yanhua Qu, Gang Song, Chuanyin Dai, Ruiying Zhang, Zuohua Yin, Kaifeng Wang, Xuebin Gao, Shou-Hsien Li, and et al. 2011. "An Unstructured Phylogeographic Pattern with Extensive Gene Flow in an Endemic Bird of South China: Collared Finchbill (Spizixos semitorques)" International Journal of Molecular Sciences 12, no. 6: 3635-3647. https://doi.org/10.3390/ijms12063635