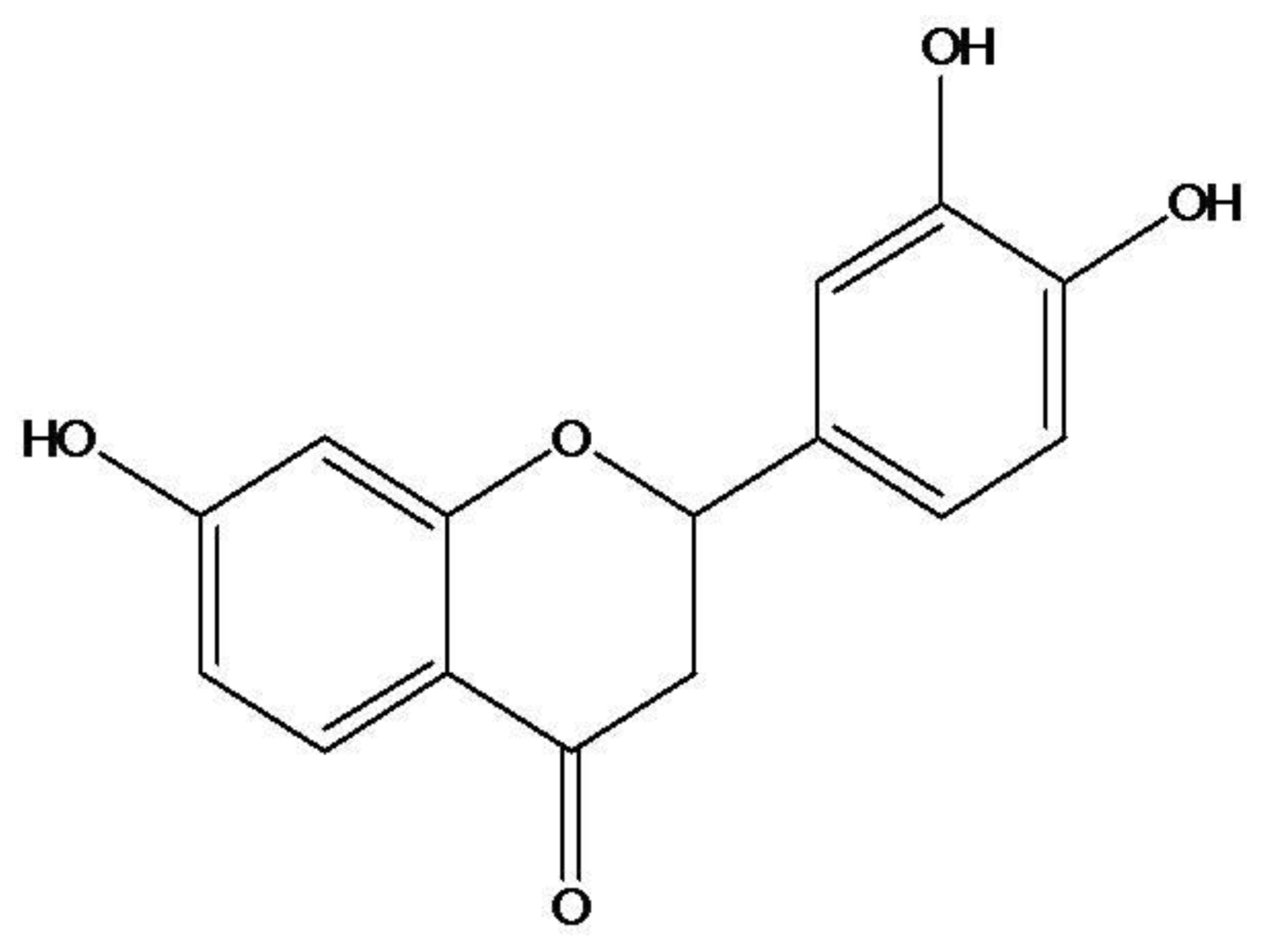
Butin (7,3′,4′-Trihydroxydihydroflavone) Reduces Oxidative Stress-Induced Cell Death via Inhibition of the Mitochondria-Dependent Apoptotic Pathway

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
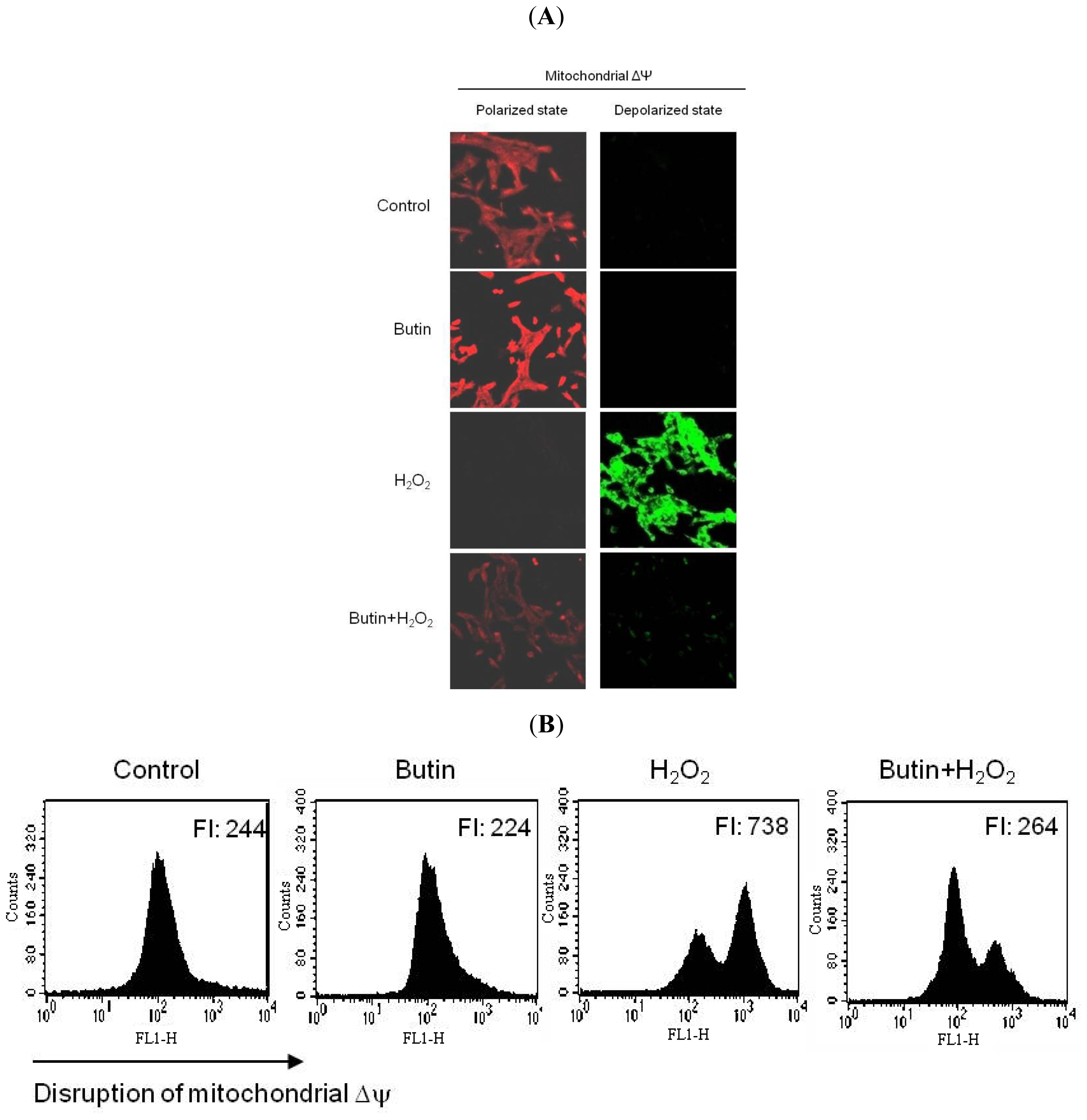
2.1. Effect of Butin on H2O2-Induced Δψm Depolarization
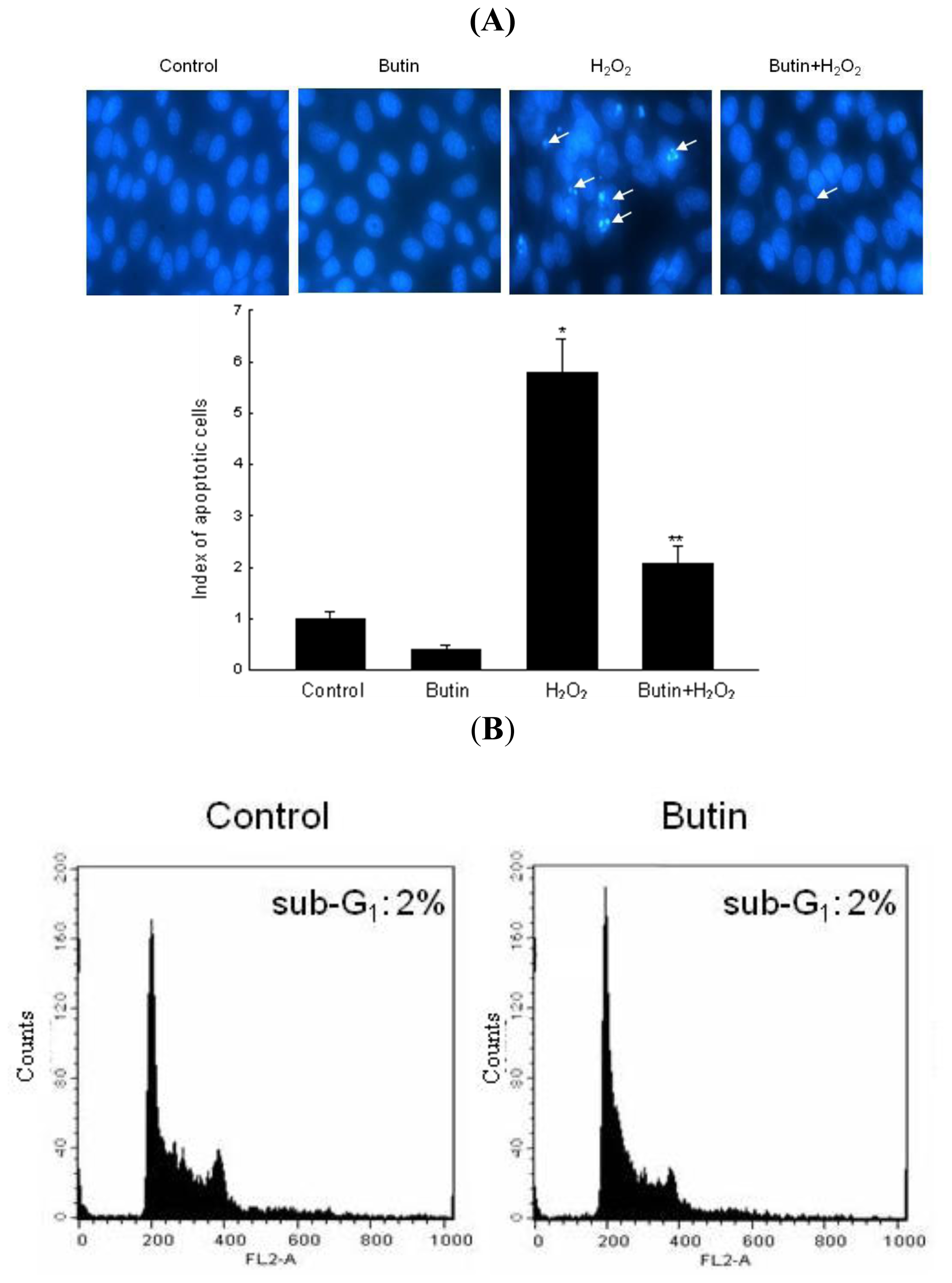
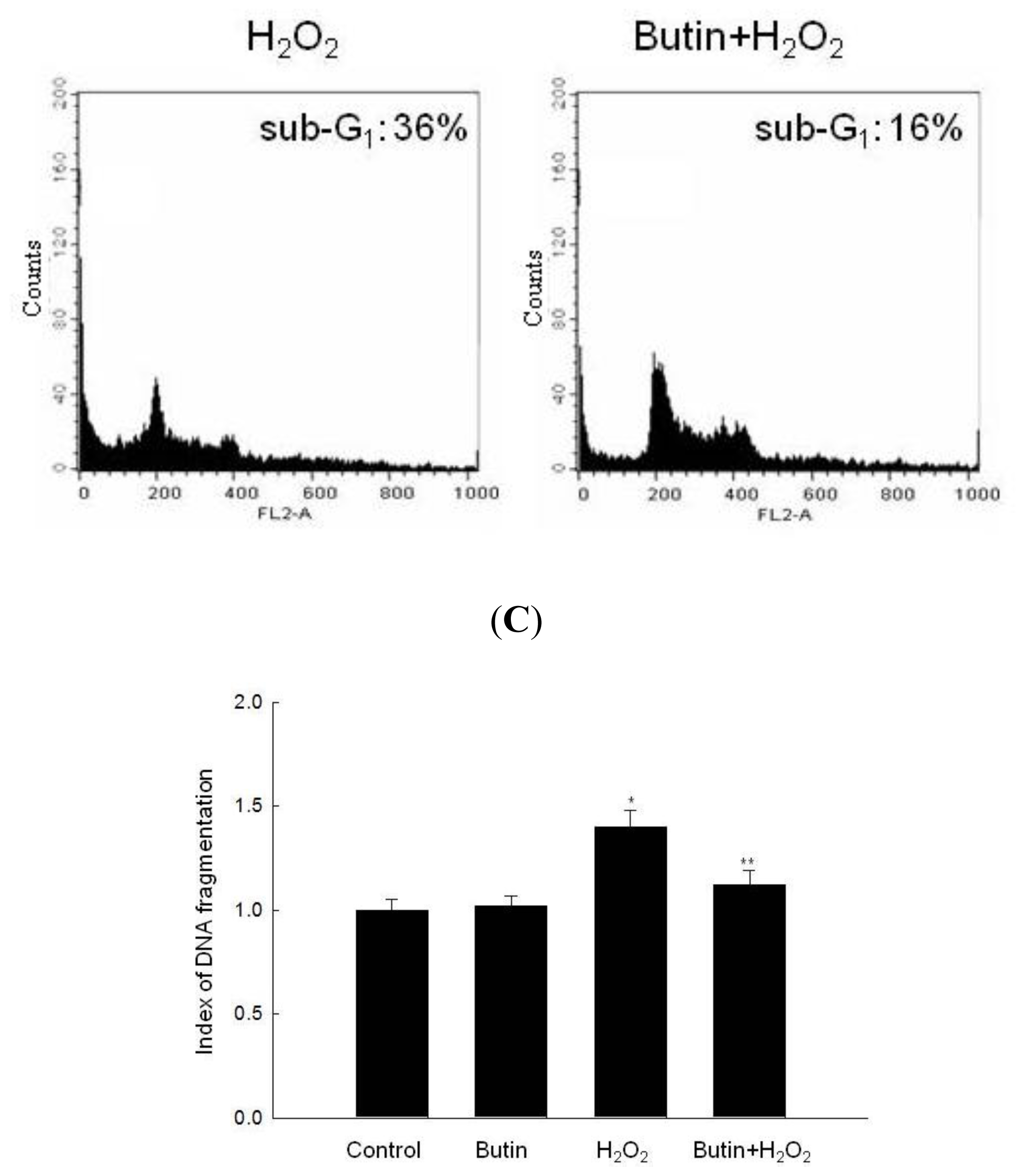
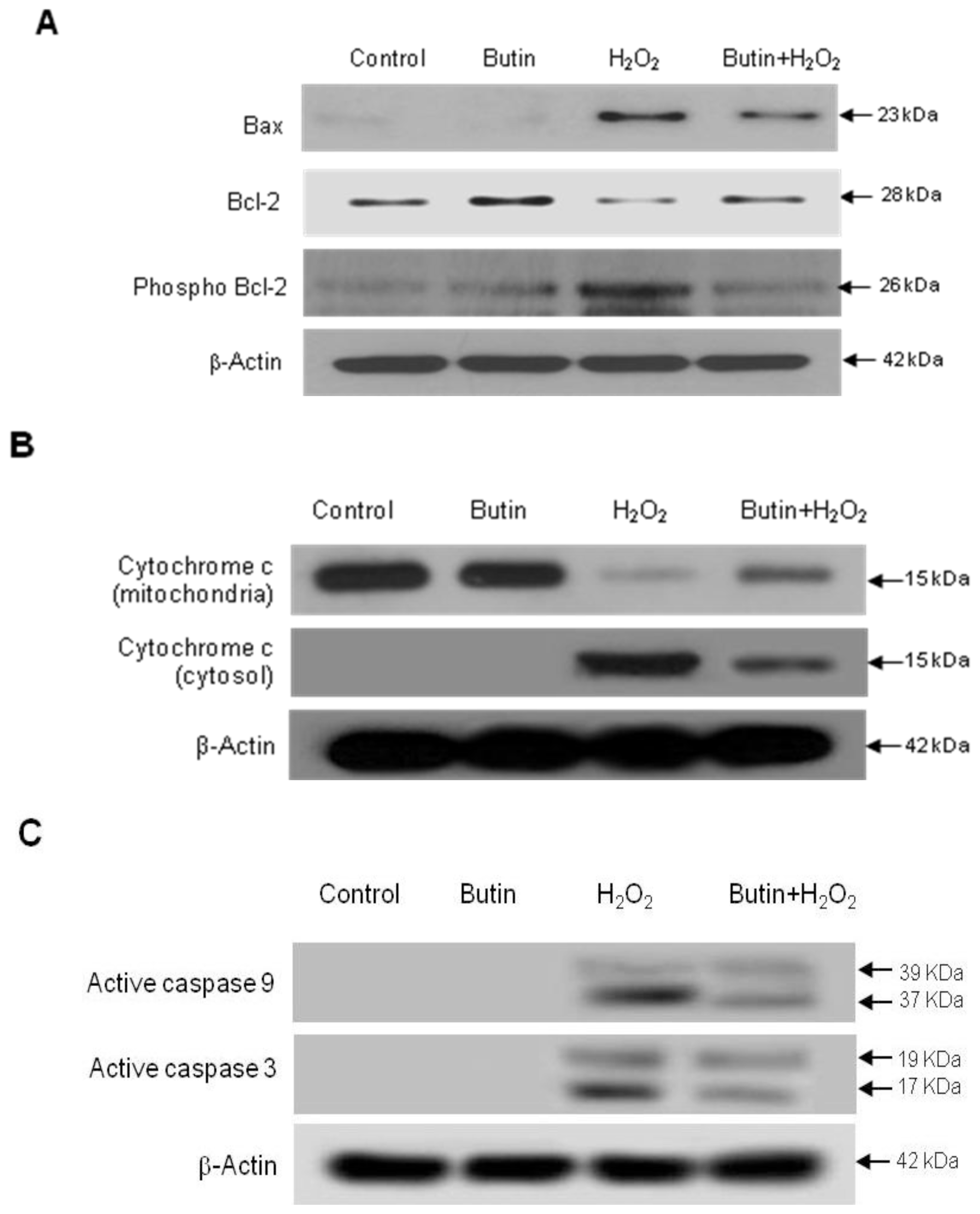
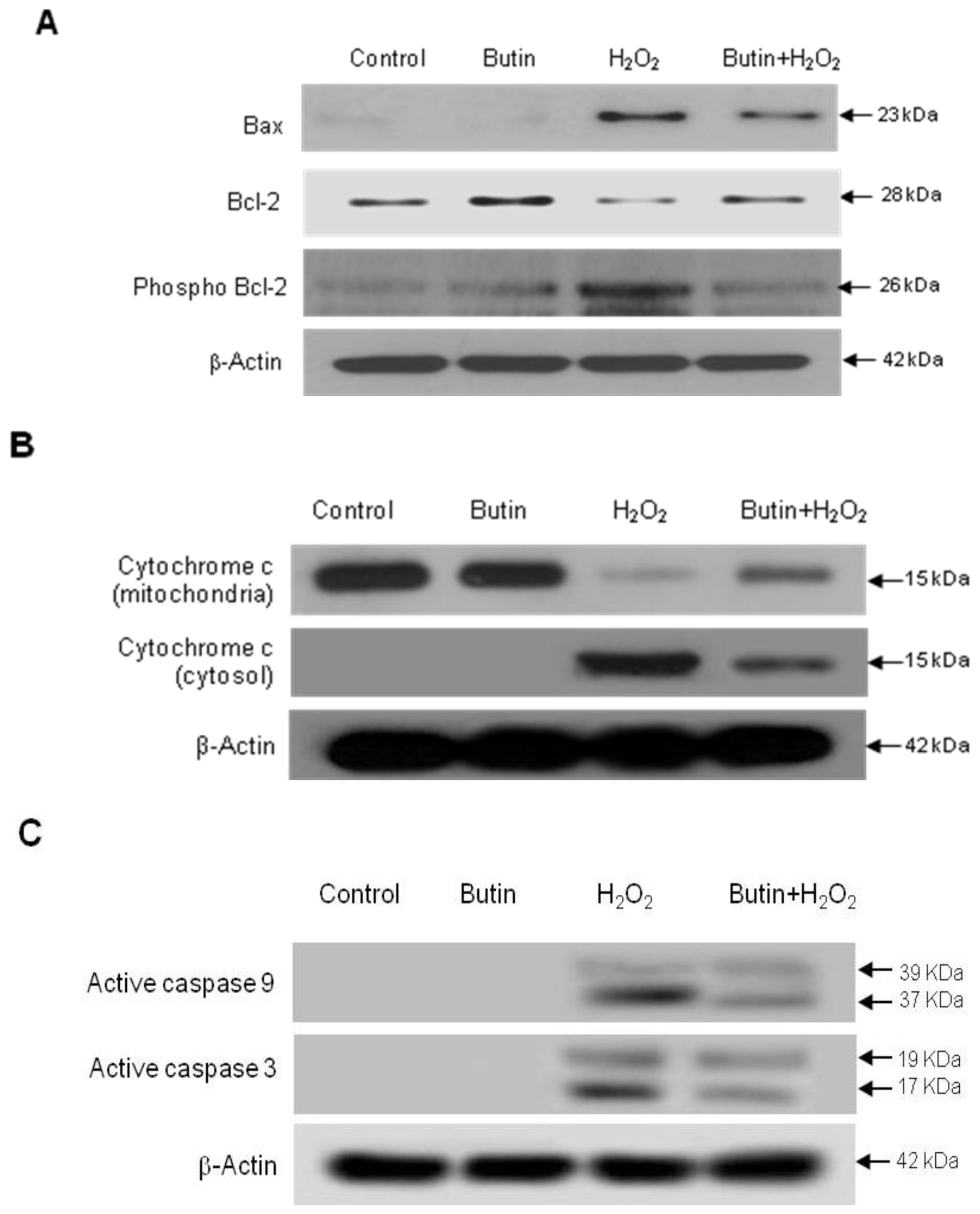
2.2. Effect of Butin against H2O2-Induced Apoptosis
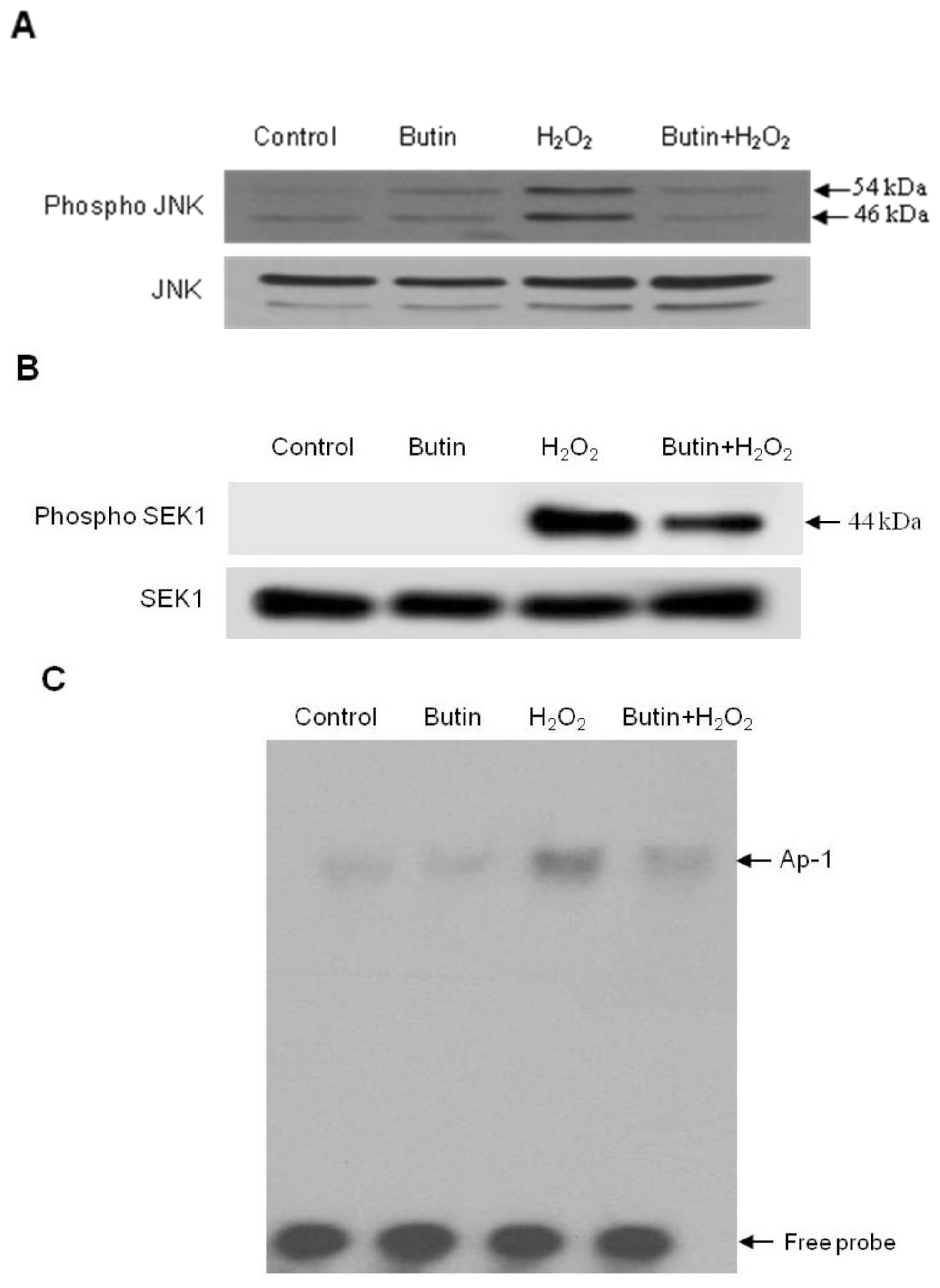
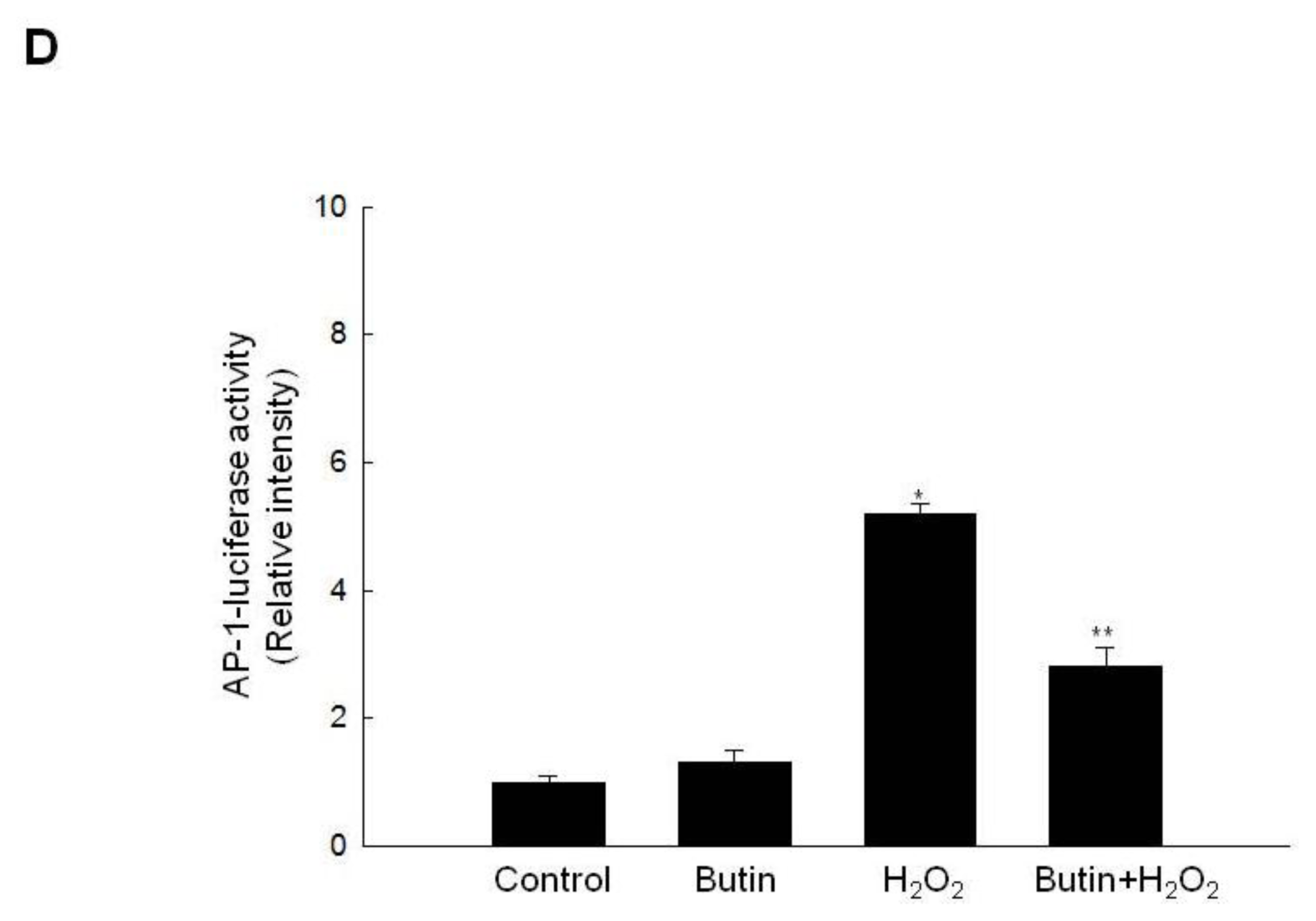
2.3. Effect of Butin on the SEK1-JNK-AP-1 Signaling Pathway
3. Experimental Section
3.1. Reagents
3.2. Cell Culture
3.3. Mitochondrial Membrane Potential (Δψm) Analysis
3.4. Western Blot Analysis
3.5. Nuclear Staining with Hoechst 33342
3.6. Detection of Apoptotic Sub-G1 Hypodiploid Cells
3.7. DNA Fragmentation
3.8. Preparation of the Nuclear Extract and Electrophoretic Mobility Shift Assay
3.9. Transient Transfection and AP-1 Luciferase Assay
3.10. Statistical Analysis
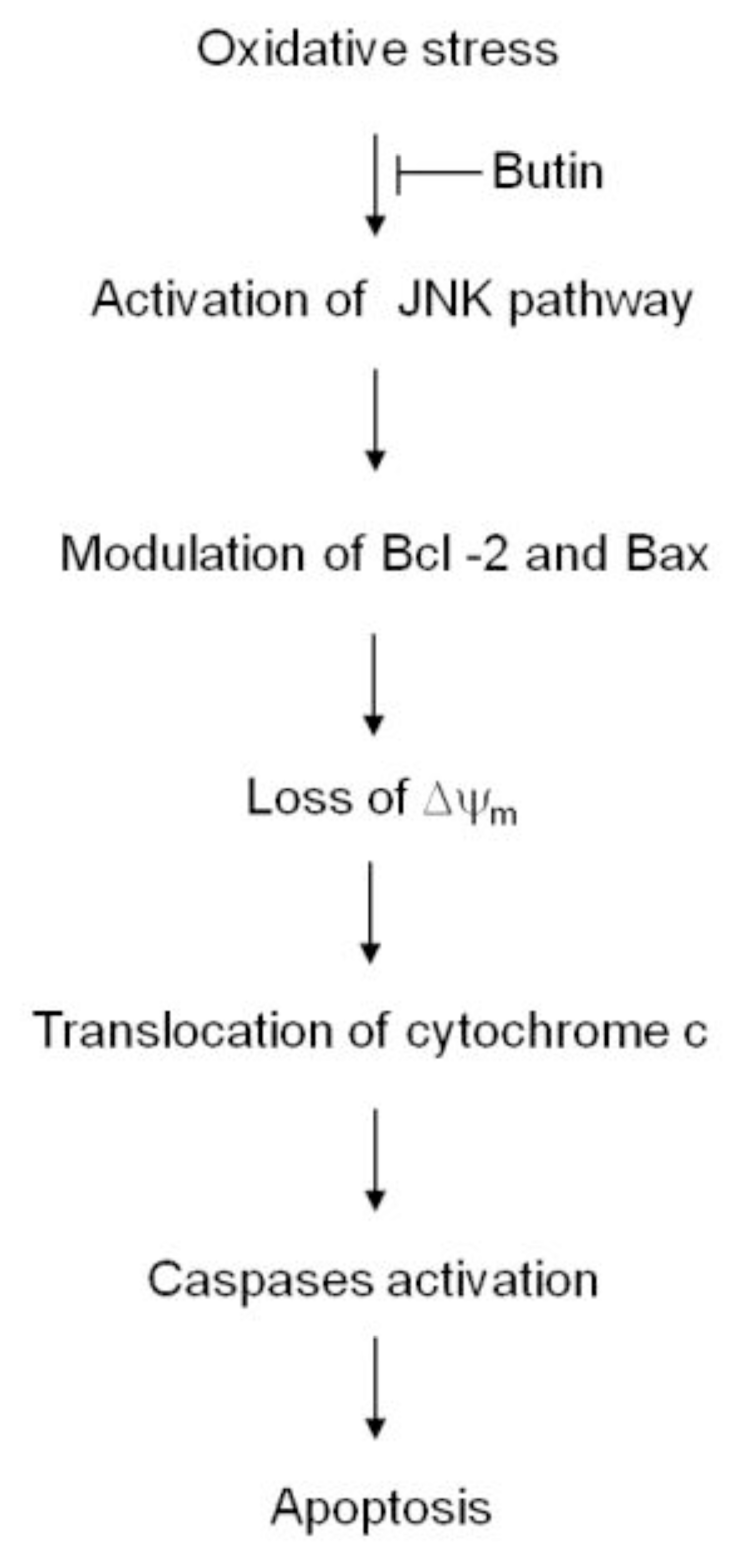
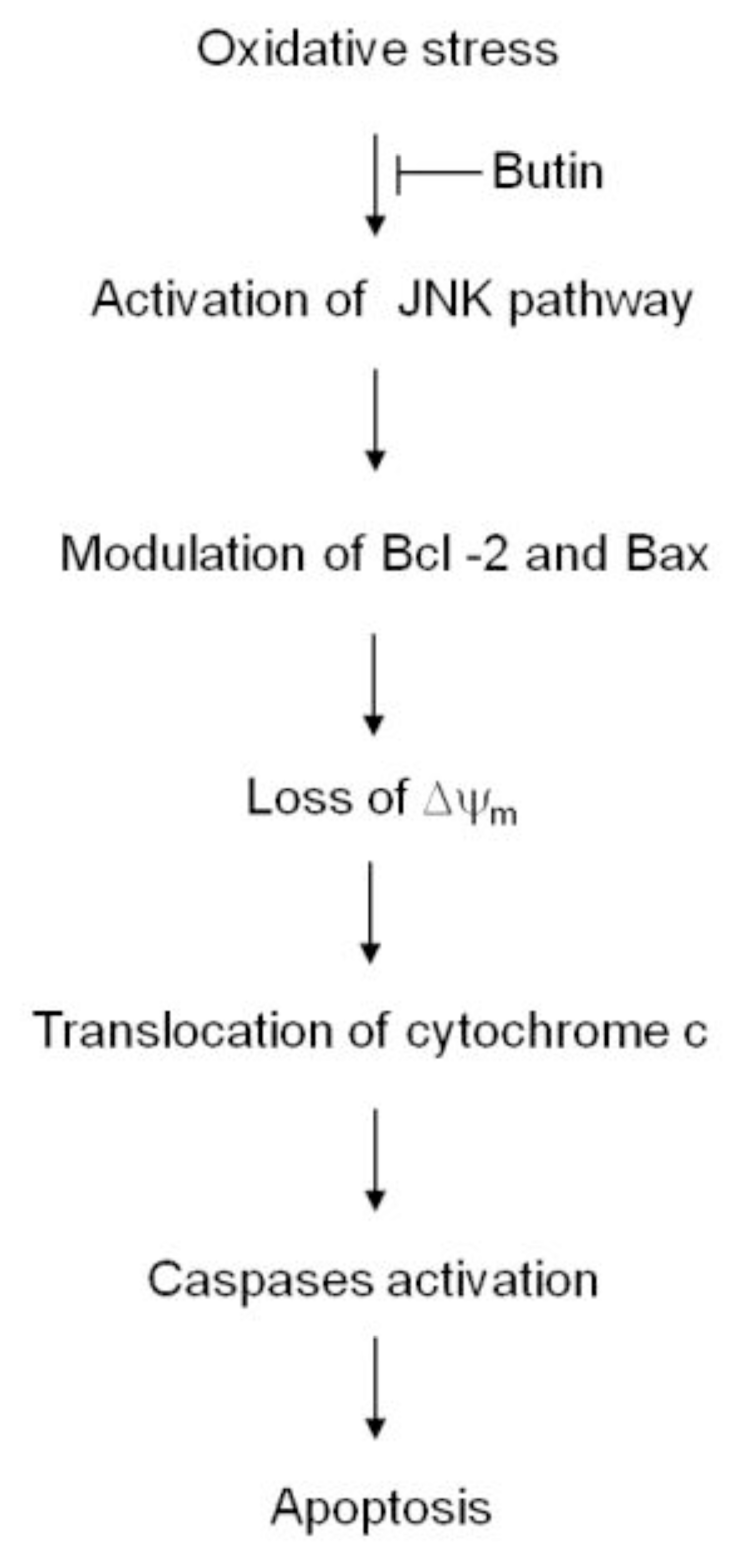
4. Conclusions
Acknowledgements
References
- Koblyakov, VA. Free radicals and inflammation (progress in inflammation research series, 1999, 260 p.). Biochemistry (Moscow) 2001, 66, 937–938. [Google Scholar]
- Loeb, LA; Wallace, DC; Martin, GM. The mitochondrial theory of aging and its relationship to reactive oxygen species damage and somatic mtDNA mutations. Proc. Natl. Acad. Sci. USA 2005, 102, 18769–18770. [Google Scholar]
- Schumacker, PT. Reactive oxygen species in cancer cells: Live by the sword, die by the sword. Cancer Cell 2006, 10, 175–176. [Google Scholar]
- Kannan, K; Jain, SK. Oxidative stress and apoptosis. Pathophysiology 2000, 7, 153–163. [Google Scholar]
- Jezek, P; Hlavata, L. Mitochondria in homeostasis of reactive oxygen species in cell, tissues, and organism. Int. J. Biochem. Cell Biol 2005, 37, 2478–2503. [Google Scholar]
- Green, DR; Reed, JC. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar]
- Li, J; Huang, CY; Zheng, RL; Cui, KR; Li, JF. Hydrogen peroxide induces apoptosis in human hepatoma cells and alters cell redox status. Cell Biol. Int 2000, 24, 9–23. [Google Scholar]
- Park, C; So, HS; Shin, CH; Baek, SH; Moon, BS; Shin, SH; Lee, HS; Lee, DW; Park, R. Quercetin protects the hydrogen peroxide-induced apoptosis via inhibition of mitochondrial dysfunction in H9c2 cardiomyoblast cells. Biochem. Pharmacol 2003, 66, 1287–1295. [Google Scholar]
- Adams, JM; Cory, S. The Bcl-2 protein family: arbiters of cell survival. Science 1998, 281, 1322–1326. [Google Scholar]
- Choi, WY; Choi, BT; Lee, WH; Choi, YH. Sulforaphane generates reactive oxygen species leading to mitochondrial perturbation for apoptosis in human leukemia U937 cells. Biomed. Pharmacother 2008, 62, 637–644. [Google Scholar]
- Brusselmans, K; Vrolix, R; Verhoeven, G; Swinnen, JV. Induction of cancer cell apoptosis by flavonoids is associated with their ability to inhibit fatty acid synthase activity. J. Biol. Chem 2005, 280, 5636–5645. [Google Scholar]
- Patil, CS; Singh, VP; Satyanarayan, PS; Jain, NK; Singh, A; Kulkarni, SK. Protective effect of flavonoids against aging- and lipopolysaccharide-induced cognitive impairment in mice. Pharmacology 2003, 69, 59–67. [Google Scholar]
- Kuzu, N; Bahcecioglu, IH; Dagli, AF; Ozercan, IH; Ustundag, B; Sahin, K. Epigallocatechin gallate attenuates experimental non-alcoholic steatohepatitis induced by high fat diet. J. Gastroenterol. Hepatol 2008, 23, e465–e470. [Google Scholar]
- Shu, XS; Lv, JH; Tao, J; Li, GM; Li, HD; Ma, N. Antihyperglycemic effects of total flavonoids from polygonatum odoratum in STZ and alloxan-induced diabetic rats. J. Ethnopharmacol 2009, 124, 539–543. [Google Scholar]
- Zhang, R; Chae, S; Kang, KA; Piao, MJ; Ko, DO; Wang, ZH; Park, DB; Park, JW; You, HJ; Hyun, JW. Protective effect of butin against hydrogen peroxide-induced apoptosis by scavenging reactive oxygen species and activating antioxidant enzymes. Mol. Cell. Biochem 2008, 318, 33–42. [Google Scholar]
- Kang, KA; Lee, JH; Chae, S; Zhang, R; Piao, MJ; Kim, HS; You, HJ; Hyun, JW. Butin decreases oxidative stress-induced 8-hydroxy-2′-deoxyguanosine levels via activation of oxoguanine glycosylase 1. Chem. Biol. Interact 2009, 181, 338–342. [Google Scholar]
- Zhang, R; Kang, KA; Piao, MJ; Chang, WY; Maeng, YH; Chae, S; Lee, IK; Kim, BJ; Hyun, JW. Butin reduces oxidative stress-induced mitochondrial dysfunction via scavenging of reactive oxygen species. Food Chem. Toxicol 2010, 48, 922–927. [Google Scholar]
- Wallace, DC. Mitochondrial defects in cardiomyopathy and neuromuscular disease. Am. Heart J 2000, 139, S70–S85. [Google Scholar]
- Lesnefsky, EJ; Moghaddas, S; Tandler, B; Kerner, J; Hoppel, CL. Mitochondrial dysfunction in cardiac disease: ischemia-reperfusion, aging, and heart failure. J. Mol. Cell. Cardiol 2001, 33, 1065–1089. [Google Scholar]
- Orrenius, S; Gogvadze, V; Zhivotovsky, B. Mitochondrial oxidative stress: Implications for cell death. Annu. Rev. Pharmacol. Toxicol 2007, 47, 143–183. [Google Scholar]
- Zamzami, N; Marchetti, P; Castedo, M; Zanin, C; Vayssiere, JL; Petit, PX; Kroemer, G. Reduction in mitochondrial potential constitutes an early irreversible step of programmed lymphocyte death in vivo. J. Exp. Med 1995, 181, 1661–1672. [Google Scholar]
- Cai, J; Yang, J; Jones, DP. Mitochondrial control of apoptosis: the role of cytochrome c. Biochim. Biophys. Acta 1998, 1366, 139–149. [Google Scholar]
- Perkins, CL; Fang, G; Kim, CN; Bhalla, KN. The role of Apaf-1, caspase-9, and Bid proteins in etoposide- or paclitaxel-induced mitochondrial events during apoptosis. Cancer Res 2000, 60, 1645–1653. [Google Scholar]
- Inanami, O; Takahashi, K; Yoshito, A; Kuwabara, M. Hydrogen peroxide-induced activation of SAPK/JNK regulated by phosphatidylinositol 3-kinase in Chinese hamster V79 cells. Antioxid. Redox Signal 1999, 1, 113–121. [Google Scholar]
- Fan, M; Goodwin, M; Vu, T; Brantley-Finley, C; Gaarde, WA; Chambers, TC. Vinblastine-induced phosphorylation of Bcl-2 and Bcl-XL is mediated by JNK and occurs in parallel with inactivation of the Raf-1/MEK/ERK cascade. J. Biol. Chem 2000, 275, 29980–29985. [Google Scholar]
- Malhi, H; Bronk, SF; Werneburg, NW; Gores, GJ. Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. J. Biol. Chem 2006, 281, 12093–12101. [Google Scholar]
- Mann, KK; Davison, K; Colombo, M; Colosimo, AL; Diaz, Z; Padovani, AM; Guo, Q; Scrivens, PJ; Gao, W; Mader, S; et al. Antimony trioxide-induced apoptosis is dependent on SEK1/JNK signaling. Toxicol. Lett 2006, 160, 158–170. [Google Scholar]
- Whitmarsh, AJ; Davis, RJ. Transcription factor AP-1 regulation by mitogen-activated protein kinase signal transduction pathways. J. Mol. Med 1996, 74, 589–607. [Google Scholar]
- Cossarizza, A; Baccarani-Contri, M; Kalashnikova, G; Franceschi, C. A new method for the cytofluorimetric analysis of mitochondrial membrane potential using the J-aggregate forming lipophilic cation 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolcarbocyanine iodide (JC-1). Biochem. Biophys. Res. Commun 1993, 197, 40–45. [Google Scholar]
- Troiano, L; Ferraresi, R; Lugli, E; Nemes, E; Roat, E; Nasi, M; Pinti, M; Cossarizza, A. Multiparametric analysis of cells with different mitochondrial membrane potential during apoptosis by polychromatic flow cytometry. Nat. Protoc 2007, 2, 2719–2727. [Google Scholar]
- Nicoletti, I; Migliorati, G; Pagliacci, MC; Grignani, F; Riccardi, C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J. Immunol. Methods 1991, 139, 271–279. [Google Scholar]
- Zu, K; Hawthorn, L; Ip, C. Up-regulation of c-jun-NH2-kinase pathway contributes to the induction of mitochondria-mediated apoptosis by alpha-tocopheryl succinate in human prostate cancer cells. Mol. Cancer Ther 2005, 4, 43–50. [Google Scholar]
- Rice-Evans, CA; Miller, NJ; George, P. Structure-antioxidant activity relationships of flavonoids and phenolic acids. Free Radic. Biol. Med 1996, 20, 933–956. [Google Scholar]
- Arora, A; Nair, MG; Strasburg, GM. Structure-activity relationships for antioxidant activities of a series of flavonoids in a liposomal system. Free Radic. Biol. Med 1998, 24, 1355–1363. [Google Scholar]
- Williams, RJ; Spencer, JP; Rice-Evans, C. Flavonoids: Antioxidants or signalling molecules? Free Radic. Biol. Med 2004, 36, 838–849. [Google Scholar]
- Maruta, A; Enaka, K; Umeda, M. Mutagenicity of quercetin and kaempferol on cultured mammalian cells. Gann 1979, 70, 273–276. [Google Scholar]
- Zhang, R; Kang, KA; Piao, MJ; Maeng, YH; Lee, KH; Chang, WY; You, HJ; Kim, JS; Kang, SS; Hyun, JW. Cellular protection of morin against the oxidative stress induced by hydrogen peroxide. Chem. Biol. Interact 2009, 177, 21–27. [Google Scholar]
- Zhang, R; Kang, KA; Kang, SS; Park, JW; Hyun, JW. Morin (2′,3,4′5,7- pentahydroxyflavone) protected cells against γ-radiation-induced oxidative stress. Basic Clin. Pharmacol. Toxicol 2011, 108, 63–72. [Google Scholar]
- Kang, KA; Wang, ZH; Zhang, R; Piao, MJ; Kim, KC; Kang, SS; Kim, YW; Lee, J; Park, D; Hyun, JW. Myricetin protects cells against oxidative stress-induced apoptosis via regulation of PI3K/Akt and MAPK signaling pathways. Int. J. Mol. Sci 2010, 11, 4348–4360. [Google Scholar]
- Huang, KL; Chen, CS; Hsu, CW; Li, MH; Chang, H; Tsai, SH; Chu, SJ. Therapeutic effects of baicalin on lipopolysaccharide-induced acute lung injury in rats. Am. J. Chin. Med 2008, 36, 301–311. [Google Scholar]
- Harborne, JB; Williams, CA. Advances in flavonoid research since 1992. Phytochemistry 2000, 55, 481–504. [Google Scholar]
- Yao, LH; Jiang, YM; Shi, J; Tomas-Barberan, FA; Datta, N; Singanusong, R; Chen, SS. Flavonoids in food and their health benefits. Plant Foods Hum. Nutr 2004, 59, 113–122. [Google Scholar]
- Kang, YJ; Min, HY; Hong, JY; Kim, YS; Kang, SS; Sang, KL. Ochnaflavone, a natural biflavonoid, induces cell cycle arrest and apoptosis in HCT-15 human colon cancer cells. Biomol. Ther 2009, 17, 282–287. [Google Scholar]






© 2011 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhang, R.; Lee, I.K.; Piao, M.J.; Kim, K.C.; Kim, A.D.; Kim, H.S.; Chae, S.; Kim, H.S.; Hyun, J.W. Butin (7,3′,4′-Trihydroxydihydroflavone) Reduces Oxidative Stress-Induced Cell Death via Inhibition of the Mitochondria-Dependent Apoptotic Pathway. Int. J. Mol. Sci. 2011, 12, 3871-3887. https://doi.org/10.3390/ijms12063871
Zhang R, Lee IK, Piao MJ, Kim KC, Kim AD, Kim HS, Chae S, Kim HS, Hyun JW. Butin (7,3′,4′-Trihydroxydihydroflavone) Reduces Oxidative Stress-Induced Cell Death via Inhibition of the Mitochondria-Dependent Apoptotic Pathway. International Journal of Molecular Sciences. 2011; 12(6):3871-3887. https://doi.org/10.3390/ijms12063871
Chicago/Turabian StyleZhang, Rui, In Kyung Lee, Mei Jing Piao, Ki Cheon Kim, Areum Daseul Kim, Hye Sun Kim, Sungwook Chae, Hee Sun Kim, and Jin Won Hyun. 2011. "Butin (7,3′,4′-Trihydroxydihydroflavone) Reduces Oxidative Stress-Induced Cell Death via Inhibition of the Mitochondria-Dependent Apoptotic Pathway" International Journal of Molecular Sciences 12, no. 6: 3871-3887. https://doi.org/10.3390/ijms12063871