Self-Assembly of Protein Monolayers Engineered for Improved Monoclonal Immunoglobulin G Binding

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Cloning
2.2. Purification of Inclusion Bodies and Refolding
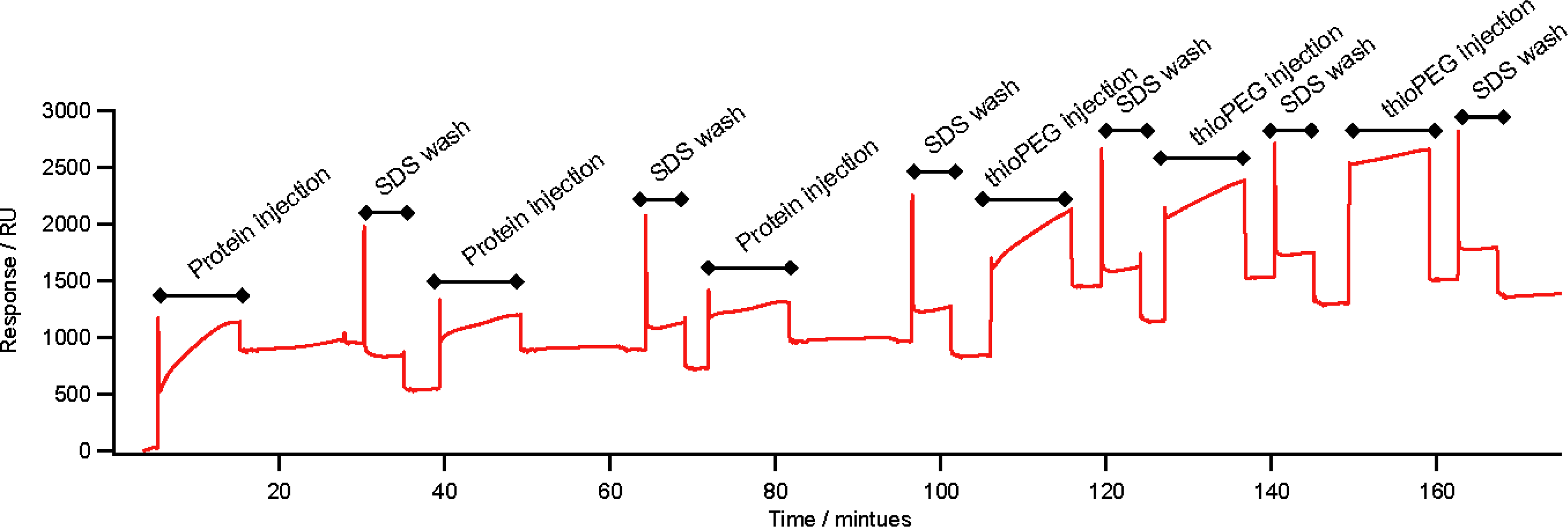
2.3. Self-Assembly on Gold
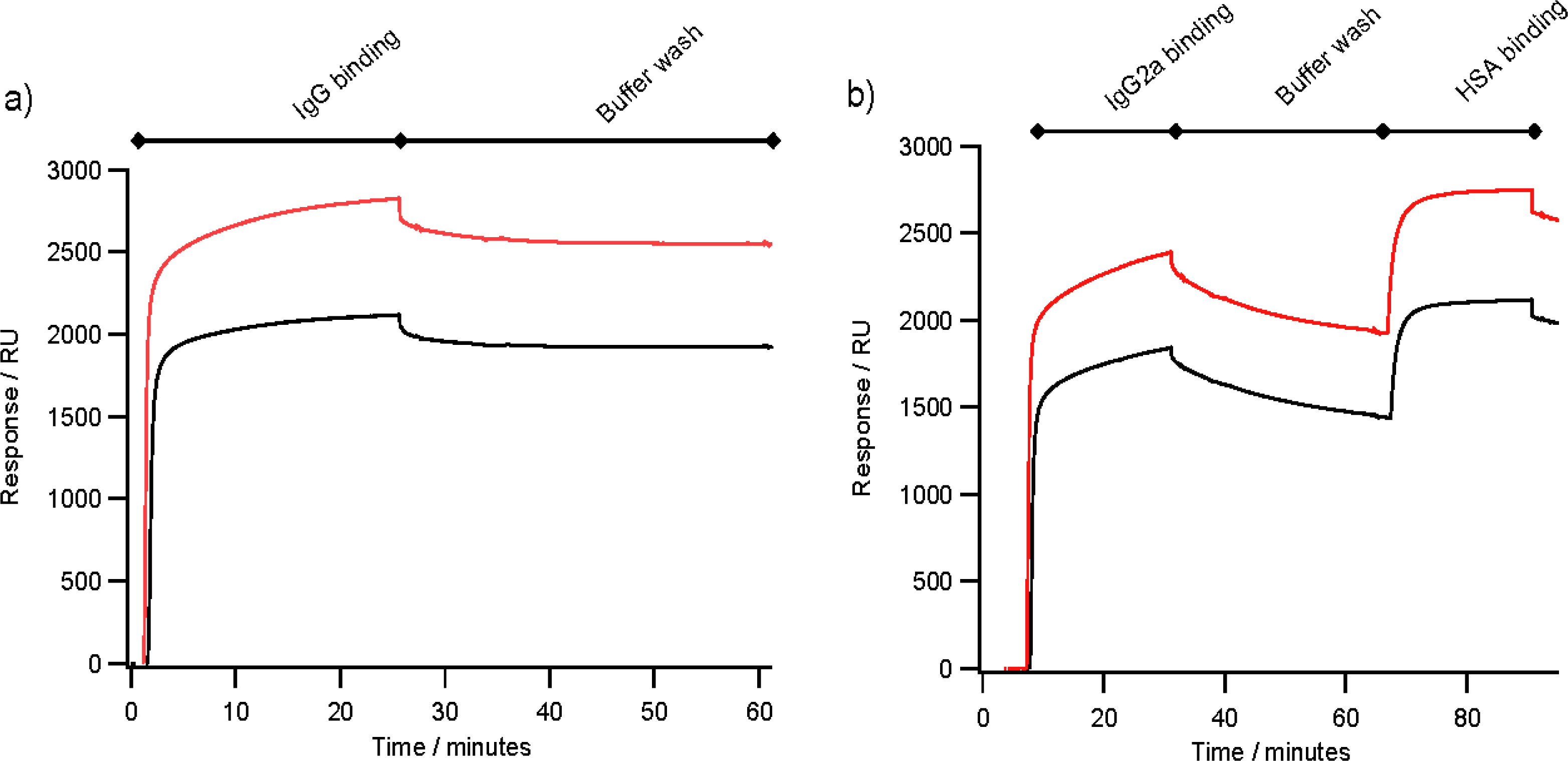
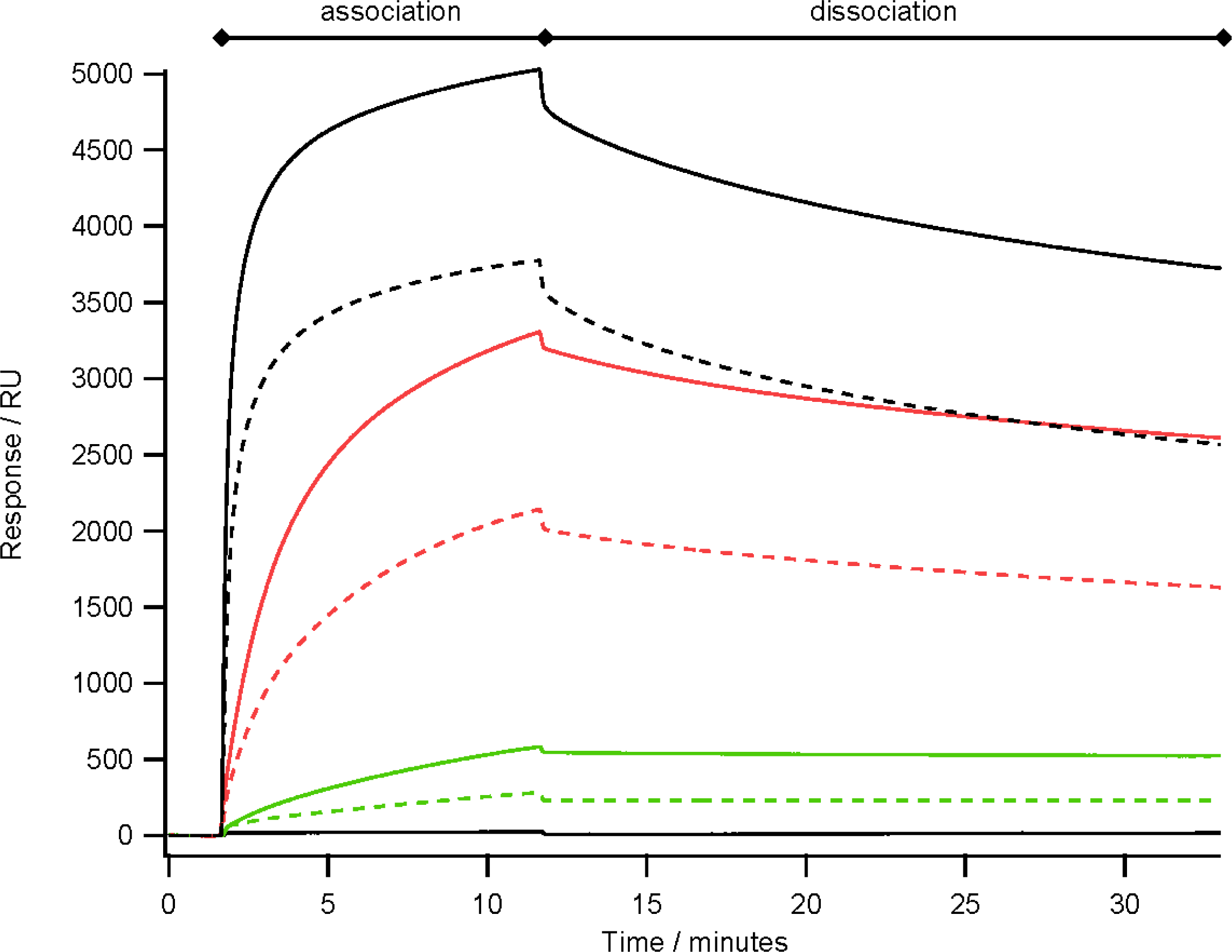
2.4. Antibody Binding
3. Experimental Details
3.1. Materials
3.2. Molecular Biology
3.3. Protein Expression, Purification and Refolding
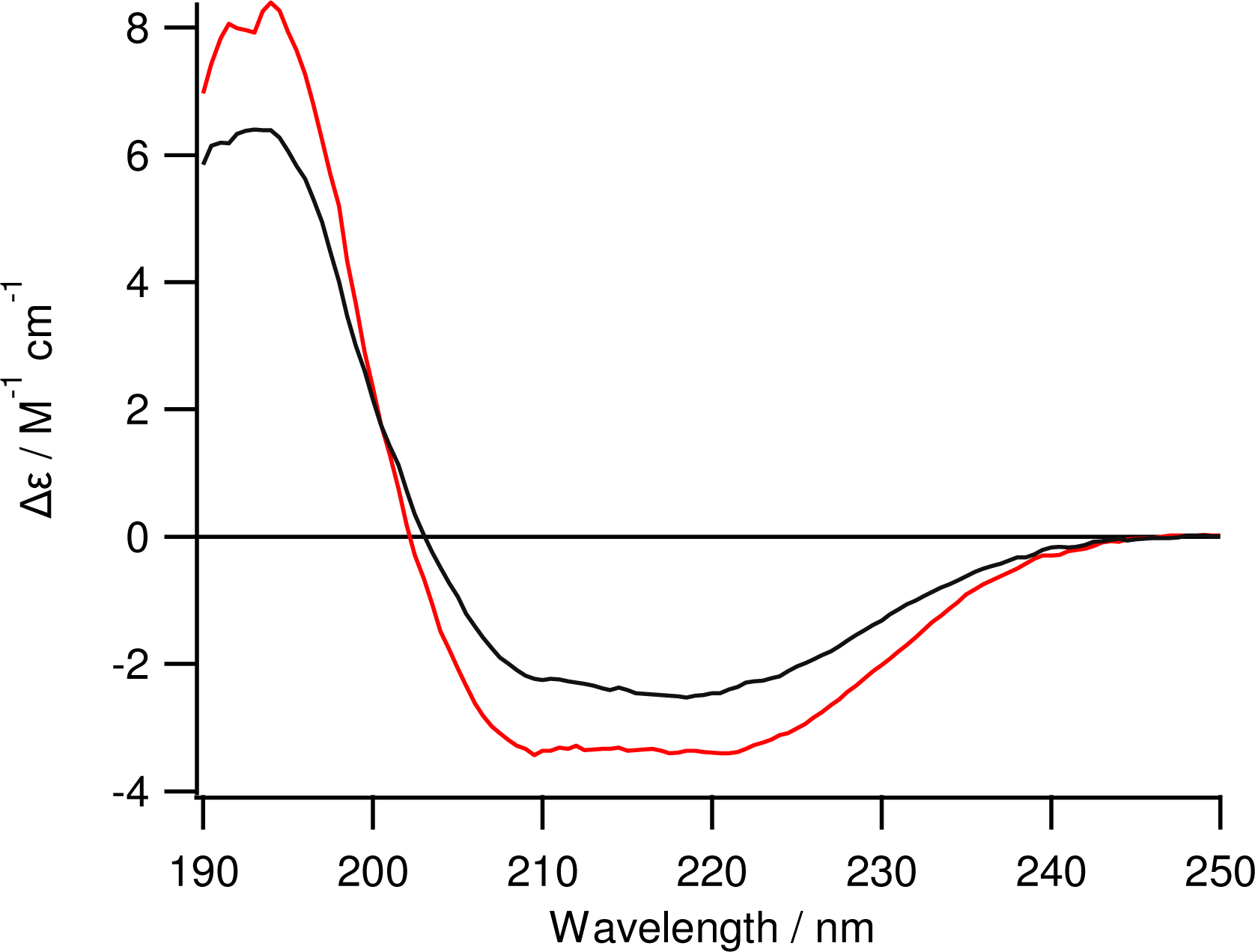
3.4. Circular Dichroism
3.5. Surface Plasmon Resonance
4. Conclusions
Acknowledgments
References
- Mrksich, M; Whitesides, GM. Using self-assembled monolayers to understand the interactions of man-made surfaces with proteins and cells. Annu. Rev. Biophys. Biomol. Struct 1996, 25, 55–78. [Google Scholar]
- Dubois, LH; Nuzzo, RG. Synthesis, structure, and properties of model organic-surfaces. Annu. Rev. Phys. Chem 1992, 43, 437–463. [Google Scholar]
- Chaffey, BT; Mitchell, E; Birch, MA; Lakey, JH. A generic expression system to produce proteins that co-assemble with alkane thiol SAM. Int. J. Nanomed 2008, 3, 287–293. [Google Scholar]
- Mitchell, EA; Chaffey, BT; McCaskie, AW; Lakey, JH; Birch, MA. Controlled spatial and conformational display of immobilised bone morphogenetic protein-2 and osteopontin signalling motifs regulates osteoblast adhesion and differentiation in vitro. BMC Biol 2010, 8. [Google Scholar] [CrossRef]
- Terrettaz, S; Ulrich, WP; Vogel, H; Hong, Q; Dover, LG; Lakey, JH. Stable self-assembly of a protein engineering scaffold on gold surfaces. Protein Sci 2002, 11, 1917–1925. [Google Scholar]
- Shah, DS; Thomas, MB; Phillips, S; Cisneros, DA; Le Brun, AP; Holt, SA; Lakey, JH. Self-assembling layers created by membrane proteins on gold. Biochem. Soc. Trans 2007, 35, 522–526. [Google Scholar]
- Pautsch, A; Schulz, GE. Structure of the outer membrane protein A transmembrane domain. Nat. Struct. Biol 1998, 5, 1013–1017. [Google Scholar]
- Pautsch, A; Schulz, GE. High-resolution structure of the OmpA membrane domain. J. Mol. Biol 2000, 298, 273–282. [Google Scholar]
- Le Brun, AP; Holt, SA; Shah, DS; Majkrzak, CF; Lakey, JH. Monitoring the assembly of antibody-binding membrane protein arrays using polarised neutron reflection. Eur. Biophys. J. Biophys. Lett 2008, 37, 639–645. [Google Scholar]
- Le Brun, AP; Holt, SA; Shah, DSH; Majkrzak, CF; Lakey, JH. The structural orientation of antibody layers bound to engineered biosensor surfaces. Biomaterials 2011, 32, 3303–3311. [Google Scholar]
- Argos, P. An investigation of the oligopeptides linking domains in protein tertiary structures and possible candidates for general gene fusion. J. Mol. Biol 1990, 211, 943–958. [Google Scholar]
- Huston, JS; Levinson, D; Mudgetthunter, M; Tai, MS; Novotny, J; Margolies, MN; Ridge, RJ; Bruccoleri, RE; Haber, E; Crea, R; et al. Protein engineering of antibody-binding sites — recovery of specific activity in an anti-digoxin single-chain Fv analog produced in Escherichia coli. Proc. Natl. Acad. Sci. USA 1988, 85, 5879–5883. [Google Scholar]
- Heikoop, JC; vanBeuningendeVaan, M; vandenBoogaart, P; Grootenhuis, PDJ. Evaluation of subunit truncation and the nature of the spacer for single chain human gonadotropins. Eur. J. Biochem 1997, 245, 656–662. [Google Scholar]
- Yu, Y; Lutz, S. Circular permutation: A different way to engineer enzyme structure and function. Trends Biotechnol 2011, 29, 18–25. [Google Scholar]
- Lowenadler, B; Jansson, B; Paleus, S; Holmgren, E; Nilsson, B; Moks, T; Palm, G; Josephson, S; Philipson, L; Uhlen, M. A gene fusion system for generating antibodies against short peptides. Gene 1987, 58, 87–97. [Google Scholar]
- George, RA; Heringa, J. An analysis of protein domain linkers: Their classification and role in protein folding. Protein Eng 2002, 15, 871–879. [Google Scholar]
- Stenberg, E; Persson, B; Roos, H; Urbaniczky, C. Quantitative determination of surface concentration of protein with surface plasmon resonance using radiolabeled proteins. J. Colloid Interface Sci 1991, 143, 513–526. [Google Scholar]
- Prime, KL; Whitesides, GM. Adsorption of proteins onto surfaces containing end-attached oligo(ethylene oxide): A model system using self-assembled monolayers. J. Am. Chem. Soc 1993, 115, 10714–10721. [Google Scholar]
- Holt, SA; Le Brun, AP; Majkrzak, CF; McGillivray, DJ; Heinrich, F; Losche, M; Lakey, JH. An ion-channel-containing model membrane: Structural determination by magnetic contrast neutron reflectometry. Soft Matter 2009, 5, 2576–2586. [Google Scholar]
- Arai, R; Wriggers, W; Nishikawa, Y; Nagamune, T; Fujisawa, T. Conformations of variably linked chimeric proteins evaluated by synchrotron X-ray small-angle scattering. Proteins Struct. Funct. Bioinf 2004, 57, 829–838. [Google Scholar]
- Guex, N; Peitsch, MC. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar]
- Tashiro, M; Tejero, R; Zimmerman, DE; Celda, B; Nilsson, B; Montelione, GT. High-resolution solution NMR structure of the Z domain of staphylococcal protein A. J. Mol. Biol 1997, 272, 573–590. [Google Scholar]





© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Le Brun, A.P.; Shah, D.S.H.; Athey, D.; Holt, S.A.; Lakey, J.H. Self-Assembly of Protein Monolayers Engineered for Improved Monoclonal Immunoglobulin G Binding. Int. J. Mol. Sci. 2011, 12, 5157-5167. https://doi.org/10.3390/ijms12085157
Le Brun AP, Shah DSH, Athey D, Holt SA, Lakey JH. Self-Assembly of Protein Monolayers Engineered for Improved Monoclonal Immunoglobulin G Binding. International Journal of Molecular Sciences. 2011; 12(8):5157-5167. https://doi.org/10.3390/ijms12085157
Chicago/Turabian StyleLe Brun, Anton P., Deepan S. H. Shah, Dale Athey, Stephen A. Holt, and Jeremy H. Lakey. 2011. "Self-Assembly of Protein Monolayers Engineered for Improved Monoclonal Immunoglobulin G Binding" International Journal of Molecular Sciences 12, no. 8: 5157-5167. https://doi.org/10.3390/ijms12085157