Reviewing Ligand-Based Rational Drug Design: The Search for an ATP Synthase Inhibitor


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Pharmacophore
2.1. Construction of a Pharmacophore Model
2.2. Applications of Pharmacophore Models
3. QSAR
3.1. Building a QSAR Model
3.2. Applications of QSAR
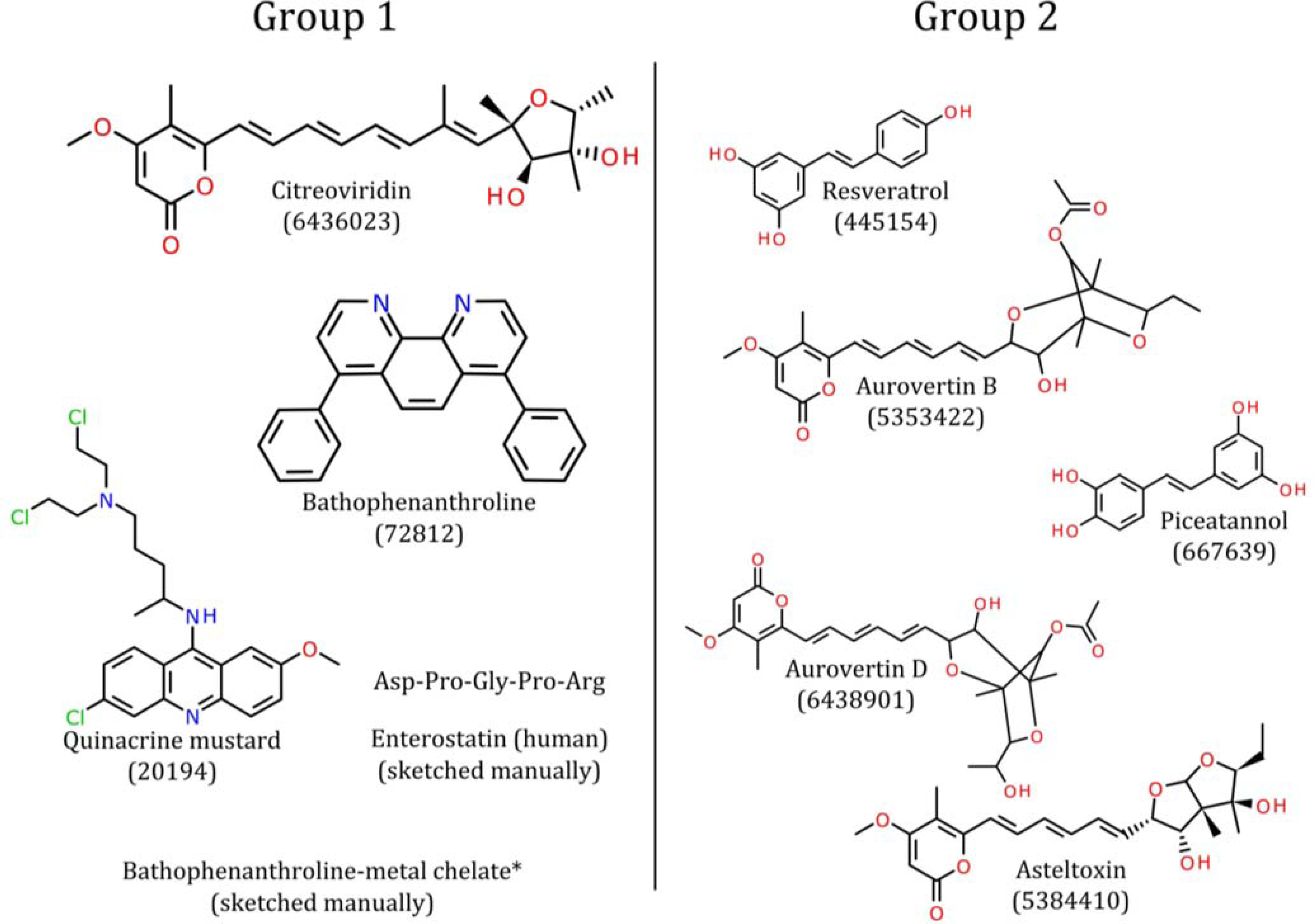
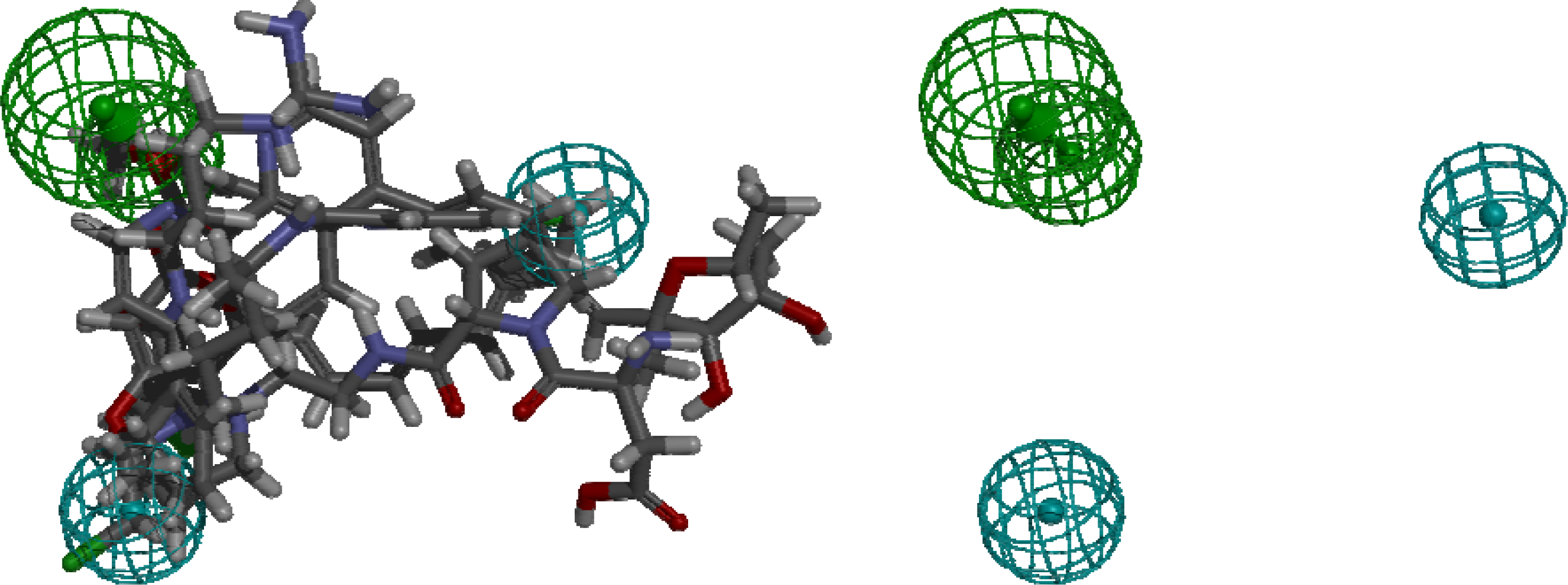
4. Pharmacophore Models of ATP Synthase Beta Subunit-Binding Ligands
5. Conclusions
Acknowledgments
References
- Sneader, W. Drug Discovery: A History; John Wiley & Sons, Ltd: Chichester, UK, 2005. [Google Scholar]
- Drews, J. Drug discovery: a historical perspective. Science 2000, 287, 1960–1964. [Google Scholar]
- Butcher, EC; Berg, EL; Kunkel, EJ. Systems biology in drug discovery. Nat. Biotechnol 2004, 22, 1253–1259. [Google Scholar]
- Chen, YP; Chen, F. Identifying targets for drug discovery using bioinformatics. Expert Opin. Ther. Targets 2008, 12, 383–389. [Google Scholar]
- Engel, T. Basic overview of chemoinformatics. J. Chem. Inf. Model 2006, 46, 2267–2277. [Google Scholar]
- Moon, JB; Howe, WJ. Computer design of bioactive molecules: a method for receptor-based de novo ligand design. Proteins 1991, 11, 314–328. [Google Scholar]
- Sleno, L; Emili, A. Proteomic methods for drug target discovery. Curr. Opin. Chem. Biol 2008, 12, 46–54. [Google Scholar]
- Cavasotto, CN; Phatak, SS. Homology modeling in drug discovery: current trends and applications. Drug Discov. Today 2009, 14, 676–683. [Google Scholar]
- Ivanov, AS; Veselovsky, AV; Dubanov, AV; Skvortsov, VS. Bioinformatics platform development: from gene to lead compound. Methods Mol. Biol 2006, 316, 389–431. [Google Scholar]
- Chen, Z; Li, HL; Zhang, QJ; Bao, XG; Yu, KQ; Luo, XM; Zhu, WL; Jiang, HL. Pharmacophore-based virtual screening versus docking-based virtual screening: a benchmark comparison against eight targets. Acta. Pharmacol. Sin 2009, 30, 1694–1708. [Google Scholar]
- Wang, Y; Shaikh, SA; Tajkhorshid, E. Exploring transmembrane diffusion pathways with molecular dynamics. Physiology 2010, 25, 142–154. [Google Scholar]
- Bemporad, D; Luttmann, C; Essex, JW. Behaviour of small solutes and large drugs in a lipid bilayer from computer simulations. Biochim. Biophys. Acta 2005, 1718, 1–21. [Google Scholar]
- Tintori, C; Manetti, F; Botta, M. Pharmacophoric models and 3D QSAR studies of the adenosine receptor ligands. Curr. Top. Med. Chem 2010, 10, 1019–1035. [Google Scholar]
- Vogt, M; Bajorath, J. Predicting the performance of fingerprint similarity searching. Methods Mol. Biol 2011, 672, 159–173. [Google Scholar]
- Langer, T; Hoffmann, RD. Virtual screening: an effective tool for lead structure discovery? Curr. Pharm. Des 2001, 7, 509–527. [Google Scholar]
- Scior, T; Medina-Franco, JL; Do, QT; Martínez-Mayorga, K; Yunes Rojas, JA; Bernard, P. How to recognize and workaround pitfalls in QSAR studies: a critical review. Curr. Med. Chem 2009, 16, 4297–4313. [Google Scholar]
- Ehrlich, P. Ueber den jetzigen Stand der Chemotherapie. Ber. Dtsch. Chem. Ges 1909, 42, 17–47. [Google Scholar]
- van Drie, JH. Pharmacophore discovery-lessons learned. Curr. Pharm. Des 2003, 9, 1649–1664. [Google Scholar]
- Wolber, G; Seidel, T; Bendix, F; Langer, T. Molecule-pharmacophore superpositioning and pattern matching in computational drug design. Drug Discov. Today 2008, 13, 23–19. [Google Scholar]
- Yang, SY. Pharmacophore modeling and applications in drug discovery: challenges and recent advances. Drug Discov. Today 2010, 15, 444–450. [Google Scholar]
- Kubinyi, H. Success Stories of Computer-Aided Design. In Computer Applications in Pharmaceutical Research and Development; Ekins, S, Wang, B, Eds.; Wiley-Interscience: Hoboken, NJ, USA, 2006; pp. 377–424. [Google Scholar]
- McNaught, AD; Wilkinson, A. Compendium of Chemical Terminology-IUPAC Recommendations, 2nd ed; Blackwell Science: Hoboken, NJ, USA, 1997. [Google Scholar]
- Rankovic, Z; Morphy, R. Lead Generation Approaches in Drug Discovery; John Wiley & Sons, Inc: Hoboken, NJ, USA, 2011; pp. 86–90. [Google Scholar]
- Chen, IJ; Foloppe, N. Is conformational sampling of drug-like molecules a solved problem? Drug Dev. Res 2011, 72, 85–94. [Google Scholar]
- Güner, OF. History and evolution of the pharmacophore concept in computer-aided drug design. Curr. Top. Med. Chem 2002, 2, 1321–1332. [Google Scholar]
- Mustata, G; Follis, AV; Hammoudeh, DI; Metallo, SJ; Wang, H; Prochownik, EV; Lazo, JS; Bahar, I. Discovery of novel Myc-Max heterodimer disruptors with a three-dimensional pharmacophore model. J. Med. Chem 2009, 52, 1247–1250. [Google Scholar]
- Petersen, RK; Christensen, KB; Assimopoulou, AN; Fretté, X; Papageorgiou, VP; Kristiansen, K; Kouskoumvekaki, I. Pharmacophore-driven identification of PPARγ agonists from natural sources. J. Comput. Aided Mol. Des 2011, 25, 107–116. [Google Scholar]
- Barroso, I; Gurnell, M; Crowley, VE; Agostini, M; Schwabe, JW; Soos, MA; Maslen, GL; Williams, TD; Lewis, H; Schafer, AJ. Dominant negative mutations in human PPARγ associated with severe insulin resistance, diabetes mellitus and hypertension. Nature 1999, 402, 880–883. [Google Scholar]
- Lyne, PD; Kenny, PW; Cosgrove, DA; Deng, C; Zabludoff, S; Wendoloski, JJ; Ashwell, S. Identification of compounds with nanomolar binding affinity for checkpoint kinase-1 using knowledge-based virtual screening. J. Med. Chem 2004, 47, 1962–1968. [Google Scholar]
- Peach, ML; Nicklaus, MC. Combining docking with pharmacophore filtering for improved virtual screening. J Cheminform 2009, 1, 6:1–6:15. [Google Scholar]
- Esposito, EX; Hopfinger, AJ; Madura, JD. Methods for applying the quantitative structure-activity relationship paradigm. Methods. Mol. Biol 2004, 275, 131–214. [Google Scholar]
- Perkins, R; Fang, H; Tong, W; Welsh, WJ. Quantitative structure-activity relationship methods: perspectives on drug discovery and toxicology. Environ. Toxicol. Chem 2003, 22, 1666–1679. [Google Scholar]
- Du, QS; Huang, RB; Chou, KC. Recent advances in QSAR and their applications in predicting the activities of chemical molecules, peptides and proteins for drug design. Curr. Protein Pept. Sci 2008, 9, 248–260. [Google Scholar]
- Bradbury, SP. Quantitative structure-activity relationships and ecological risk assessment: an overview of predictive aquatic toxicology research. Toxicol. Lett 1995, 79, 229–237. [Google Scholar]
- Martinez-Mayorga, K; Medina-Franco, JL. Chemoinformatics-applications in food chemistry. Adv. Food. Nutr. Res 2009, 58, 33–56. [Google Scholar]
- Myint, KZ; Xie, XQ. Recent advances in fragment-based QSAR and multi-dimensional QSAR methods. Int. J. Mol. Sci 2010, 11, 3846–3866. [Google Scholar]
- De Melo, EB. Multivariate SAR/QSAR of 3-aryl-4-hydroxyquinolin-2(1H)-one derivatives as type I fatty acid synthase (FAS) inhibitors. Eur. J. Med. Chem 2010, 45, 5817–5826. [Google Scholar]
- Zhou, T; Shi, Q; Chen, CH; Zhu, H; Huang, L; Ho, P; Lee, KH. Anti-AIDS agents 79. Design, synthesis, molecular modeling and structure-activity relationships of novel dicamphanoyl-2′,2′-dimethyldihydropyranochromone (DCP) analogs as potent anti-HIV agents. Bioorg. Med. Chem 2010, 18, 6678–6689. [Google Scholar]
- Karolidis, DA; Agatonovic-Kustrin, S; Morton, DW. Artificial neural network (ANN) based modelling for D1 like and D2 like dopamine receptor affinity and selectivity. Med. Chem 2010, 6, 259–270. [Google Scholar]
- Chen, C; Yang, J. MI-QSAR models for prediction of corneal permeability of organic compounds. Acta Pharmacol. Sin 2006, 27, 193–204. [Google Scholar]
- Tropsha, A; Zheng, W. Identification of the descriptor pharmacophores using variable selection QSAR: applications to database mining. Curr. Pharm. Des 2001, 7, 599–612. [Google Scholar]
- Cramer, RD, III; Patterson, DE; D Bunce, JD. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc 1988, 110, 5959–5967. [Google Scholar]
- Klebe, G; Abraham, U; Mietzner, T. Molecular similarity indices in a comparative analysis (CoMSIA) of drug molecules to correlate and predict their biological activity. J. Med. Chem 1994, 37, 4130–4146. [Google Scholar]
- Turner, DB; Willett, P. The EVA spectral descriptor. Eur. J. Med. Chem 2000, 35, 367–375. [Google Scholar]
- Katritzky, AR; Gordeeva, EV. Traditional topological indices vs. electronic, geometrical, and combined molecular descriptors in QSAR/QSPR research. J. Chem. Inf. Comput. Sci 1993, 33, 835–857. [Google Scholar]
- Whitley, DC; Ford, MG; Livingstone, DJ. Unsupervised forward selection: a method for eliminating redundant variables. J. Chem. Inf. Comput. Sci 2000, 40, 1160–1168. [Google Scholar]
- Chen, H; Zhou, J; Xie, G. PARM: a genetic evolved algorithm to predict bioactivity. J. Chem. Inf. Comput. Sci 1998, 38, 243–250. [Google Scholar]
- Rogers, D; Hopfinger, AJ. Application of genetic function approximation to quantitative structure-activity relationships and quantitative structure-property relationships. J. Chem. Inf. Comput. Sci 1994, 34, 854–866. [Google Scholar]
- Xue, L; Godden, JW; Bajorath, J. Evaluation of descriptors and mini-fingerprints for the identification of molecules with similar activity. J. Chem. Inf. Comput. Sci 2000, 40, 1227–1234. [Google Scholar]
- Wikel, JH; Dow, ER. The use of neural networks for variable selection in QSAR. Bioorg. Med. Chem. Lett 1993, 3, 645–651. [Google Scholar]
- Itskowitz, P; Tropsha, A. k nearest neighbors QSAR modeling as a variational problem: theory and applications. J. Chem. Inf. Model 2005, 45, 777–785. [Google Scholar]
- Jain, AN; Koile, K; Chapman, D. Compass: Predicting biological activities from molecular surface properties. performance comparisons on a steroid benchmark. J. Med. Chem 1994, 37, 2315–2327. [Google Scholar]
- Shahlaei, M; Fassihi, A; Saghaie, L. Application of PC-ANN and PC-LS-SVM in QSAR of CCR1 antagonist compounds: a comparative study. Eur. J. Med. Chem 2010, 45, 1572–1582. [Google Scholar]
- Silverman, BD; Platt, DE. Comparative molecular moment analysis (CoMMA): 3D-QSAR without molecular superposition. J. Med. Chem 1996, 39, 2129–2140. [Google Scholar]
- Kurogi, Y; Güner, OF. Pharmacophore modeling and three-dimensional database searching for drug design using catalyst. Curr. Med. Chem 2001, 8, 1035–1055. [Google Scholar]
- Verma, J; Khedkar, VM; Coutinho, EC. 3D-QSAR in drug design-a review. Curr. Top. Med. Chem 2010, 10, 95–115. [Google Scholar]
- Cramer, RD, III; Bunce, JD; Patterson, DE; Frank, IE. Crossvalidation, bootstrapping, and partial least squares compared with multiple regression in conventional QSAR studies. Quant. Struct.-Act. Relat 1988, 7, 18–25. [Google Scholar]
- Tropsha, A; Gramatica, P; Gombar, VK. The importance of being earnest: validation is the absolute essential for successful application and interpretation of QSPR models. QSAR Comb. Sci 2003, 22, 69–77. [Google Scholar]
- Consonni, V; Ballabio, D; Todeschini, R. Comments on the definition of the Q2 parameter for QSAR validation. J. Chem. Inf. Model 2009, 49, 1669–1678. [Google Scholar]
- Saghaie, L; Shahlaei, M; Fassihi, A; Madadkar-Sobhani, A; Gholivand, MB; Pourhossein, A. QSAR analysis for some diaryl-substituted pyrazoles as CCR2 inhibitors by GA-stepwise MLR. Chem. Biol. Drug. Des 2011, 77, 75–85. [Google Scholar]
- Obiol-Pardo, C; Gomis-Tena, J; Sanz, F; Saiz, J; Pastor, M. A multiscale simulation system for the prediction of drug-induced cardiotoxicity. J. Chem. Inf. Model 2011, 51, 483–492. [Google Scholar]
- Huang, TC; Chang, HY; Hsu, CH; Kuo, WH; Chang, KJ; Juan, HF. Targeting therapy for breast carcinoma by ATP synthase inhibitor aurovertin B. J. Proteome Res 2008, 7, 1433–1444. [Google Scholar]
- Sayers, EW; Barrett, T; Benson, DA; Bolton, E; Bryant, SH; Canese, K; Chetvernin, V; Church, DM; Dicuccio, M; Federhen, S; et al. Database resources of the national center for biotechnology information. Nucleic Acids Res 2010, 38, D5–D16. [Google Scholar]
- Hong, S; Pedersen, PL. ATP synthase and the actions of inhibitors utilized to study its roles in human health, disease, and other scientific areas. Microbiol. Mol. Biol. Rev 2008, 72, 590–641. [Google Scholar]
- Barnum, D; Greene, J; Smellie, A; Sprague, P. Identification of common functional configurations among molecules. J. Chem. Inf. Comput. Sci 1996, 36, 563–571. [Google Scholar]
- Smellie, A; Teig, SL; Towbin, P. Poling: promoting conformational variation. J. Comp. Chem 1995, 16, 171–187. [Google Scholar]



© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lee, C.-H.; Huang, H.-C.; Juan, H.-F. Reviewing Ligand-Based Rational Drug Design: The Search for an ATP Synthase Inhibitor. Int. J. Mol. Sci. 2011, 12, 5304-5318. https://doi.org/10.3390/ijms12085304
Lee C-H, Huang H-C, Juan H-F. Reviewing Ligand-Based Rational Drug Design: The Search for an ATP Synthase Inhibitor. International Journal of Molecular Sciences. 2011; 12(8):5304-5318. https://doi.org/10.3390/ijms12085304
Chicago/Turabian StyleLee, Chia-Hsien, Hsuan-Cheng Huang, and Hsueh-Fen Juan. 2011. "Reviewing Ligand-Based Rational Drug Design: The Search for an ATP Synthase Inhibitor" International Journal of Molecular Sciences 12, no. 8: 5304-5318. https://doi.org/10.3390/ijms12085304