Multilocus Phylogenetics Show High Intraspecific Variability within Fusarium avenaceum


Abstract
:1. Introduction
2. Results
2.1. Sequence Characterization
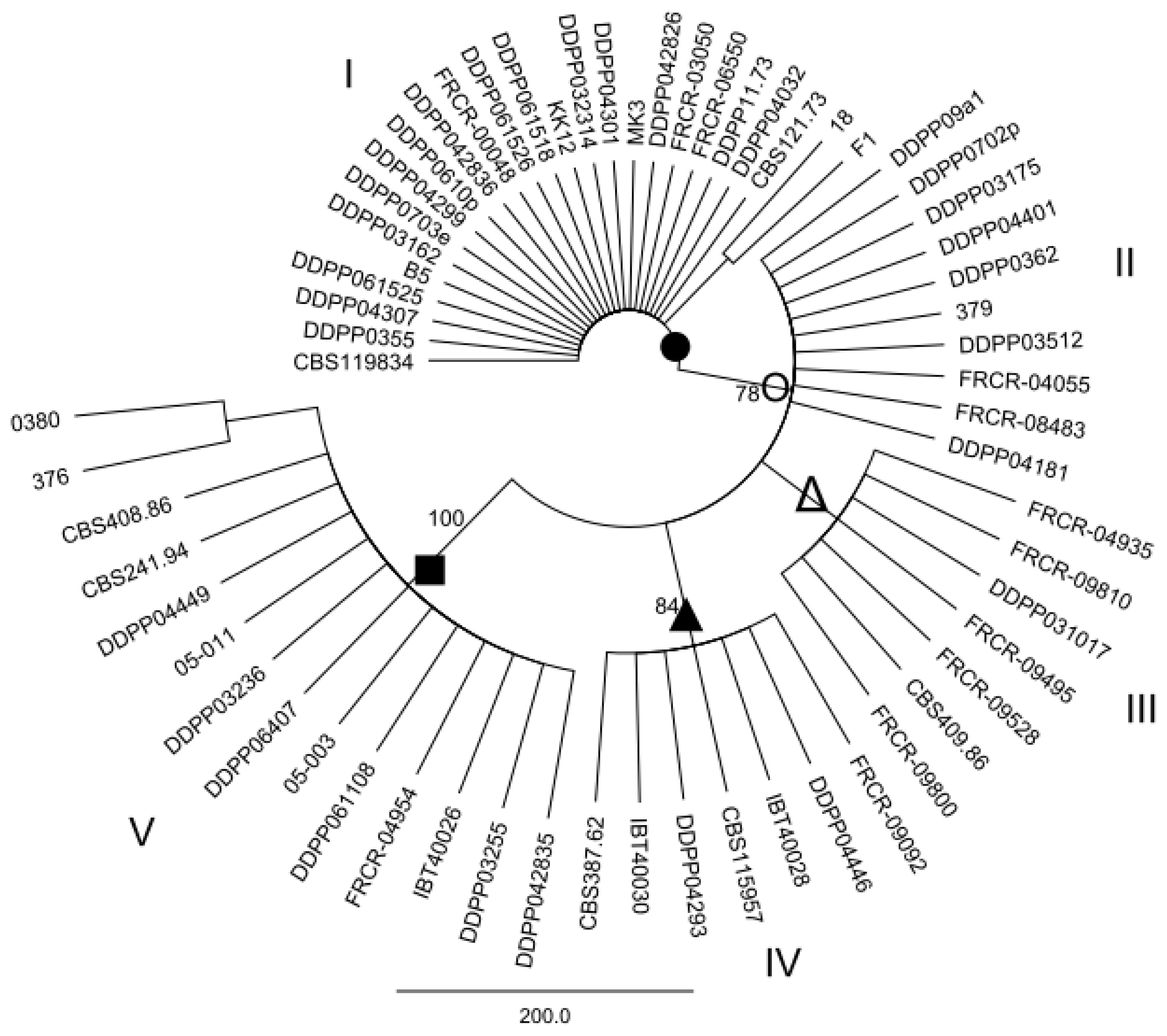
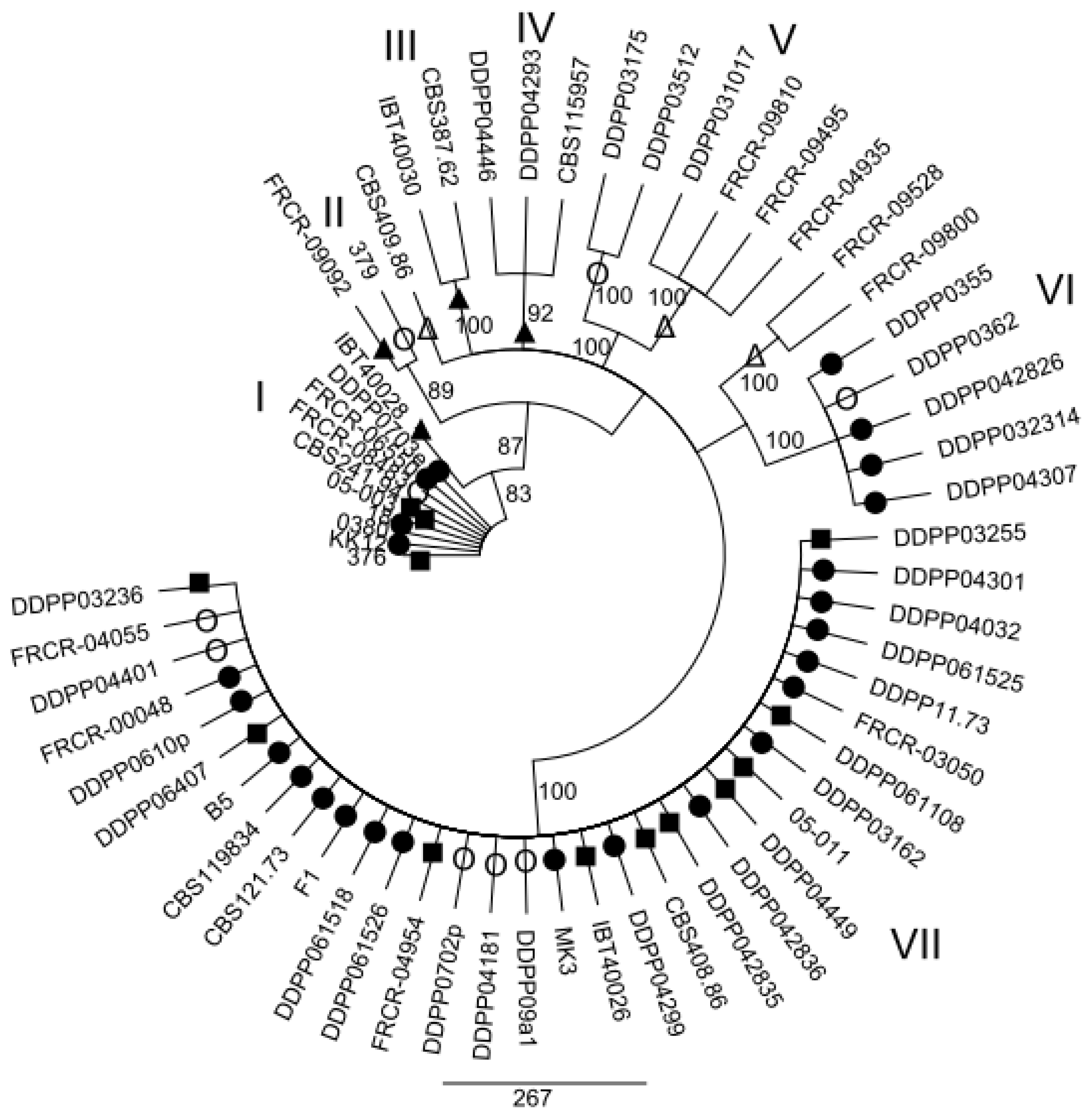
2.2. Maximum parsimony (MP) Analysis of Individual and Combined Gene Sequences
2.3. Partition Homogeneity Test
3. Discussion
4. Materials and Methods
4.1. Collection of F. avenaceum Isolates
4.2. DNA Extraction, PCR and DNA Sequencing
4.3. Phylogenetic Analyses
4.4. Partition Homogeneity Test
5. Conclusions
Acknowledgements
References
- Leslie, JF; Summerell, BA. The Fusarium Laboratory Manual; Blackwell Publishing: Ames, IA, USA, 2006. [Google Scholar]
- Spanic, V; Lemmens, M; Drezner, G. Morphological and molecular identification of Fusarium species associated with head blight on wheat in East Croatia. Eur J Plant Pathol 2010, 128, 511–516. [Google Scholar]
- Tan, DC; Flematti, GR; Ghisalberti, EL; Sivasithamparam, K; Barbetti, MJ. Toxigenicity of enniatins from Western Australian Fusarium species to brine shrimp (Artemia franciscana). Toxicon 2011, 57, 817–825. [Google Scholar]
- Tunali, B; Nicol, JM; Hodson, D; Uçkun, Z; Büyük, O; Erdurmuþ, D; Hekimhan, H; Aktaþ, H; Akbudak, MA; Bađcý, SA. Root and crown rot fungi associated with spring, facultative, and winter wheat in Turkey. Plant Dis 2008, 92, 1299–1306. [Google Scholar]
- Vogelgsang, S; Sulyok, M; Hecker, A; Jenny, E; Krska, R; Schuhmacher, R; Forrer, HR. Toxigenicity and pathogenicity of Fusarium poae and Fusarium avenaceum on wheat. Eur J Plant Pathol 2008, 122, 265–276. [Google Scholar]
- Jestoi, M; Rokka, M; Yli-Mattila, T; Parikka, P; Pizz, A; Peltonen, K. Presence and concentrations of the Fusarium-related mycotoxins beauvericin, enniatins and moniliformin in Finnish grain samples. Food Addit Contam 2004, 21, 794–802. [Google Scholar]
- Logrieco, A; Rizzo, A; Ferracane, R; Ritieni, A. Occurrence of beauvericin and enniatins in wheat affected by Fusarium avenaceum head blight. Appl Environ Microbiol 2002, 68, 82–85. [Google Scholar]
- Ivanova, L; Skjerv, E; Eriksen, GS; Uhlig, S. Cytotoxicity of enniatins A, A1, B, B1, B2 and B3 from Fusarium avenaceum. Toxicon 2006, 47, 868–876. [Google Scholar]
- Kamyar, M; Rawnduzi, P; Studenik, P; Kouri, CR; Lemmens-Gruber, R. Investigation of the electrophysiological properties of enniatins. Arch Biochem Biophys 2004, 429, 215–223. [Google Scholar]
- Macchia, L; Dipaola, R; Fornelli, F; Nenna, S; Moretti, A; Napolitano, R; Logrieco, A; Caiaffa, MF; Tursi, A; Bottalico, A. Cytotoxity of Beauvericin to Mammalian Cells. Proceedings of Book of Abstracts for International Seminar on Fusarium: Mycotoxins, Taxonomy And Pathogenicity Martina Franca, Bari, Italy, 9–13 May 1995.
- Yli-Mattila, T; Paavanen-Huhtala, S; Bulat, SA; Alekhina, IA; Nirenberg, HI. Molecular, morphological and phylogenetic analysis of the Fusarium avenaceum/F. arthrosporioides/F. tricinctum species complex—a polyphasic approach. Mycol Res 2002, 106, 655–669. [Google Scholar]
- Satyaprasad, K; Bateman, GL; Ward, E. Comparisons of isolates of Fusarium avenaceum from white lupin and other crops by pathogenicity tests, DNA analyses, and vegetative compatibility tests. J Phytopathol 2000, 148, 211–219. [Google Scholar]
- Nalim, FA; Elmer, WH; McGovern, RJ; Geiser, DM. Multilocus phylogenetic diversity of Fusarium avenaceum pathogenic on Lisianthus. Phytopathology 2009, 99, 462–468. [Google Scholar]
- Kerényi, Z; Hornok, L. Structure and function of mating type genes in Fusarium species. Acta Microbiol Immunol Hung 2002, 49, 313–314. [Google Scholar]
- Kerényi, Z; Moretti, A; Waalwijk, C; Oláh, B; Hornok, L. Mating type sequences in asexually reproducing Fusarium species. Appl Environ Microbiol 2004, 70, 4419–4423. [Google Scholar]
- Glenn, AE; Bacon, CW. FDB2 encodes a member of the arylamine N-acetyltransferase family and is necessary for biotransformation of benzoxazolinones by Fusarium verticillioides. J Appl Microbiol 2009, 107, 657–671. [Google Scholar]
- Obanor, F; Erginbas-Orakci, G; Tunali, B; Nicol, JM; Chakraborty, S. Fusarium culmorum is a single phylogenetic species based on multilocus sequence analysis. Fungal Biol 2010, 114, 753–765. [Google Scholar]
- Kulik, T; Pszczółkowska, A. Multilocus sequence analysis of Fusarium poae. J Plant Pathol 2011, 93, 119–126. [Google Scholar]
- Stenglein, SA; Rodriguero, MS; Chandler, E; Jennings, P; Salerno, GL; Nicholson, P. Phylogenetic relationships of Fusarium poae based on EF-1a and mtSSU sequences. Fungal Biol 2009, 114, 96–106. [Google Scholar]
- Scott, JB; Chakraborty, S. Multilocus sequence analysis of Fusarium pseudograminearum reveals a single phylogenetic species. Mycol Res 2006, 110, 1413–1425. [Google Scholar]
- Geiser, DM; Pitt, JI; Taylor, JW. Cryptic speciation and recombination in the aflatoxin producing fungus Aspergillus flavus. Proc Natl Acad Sci USA 1998, 95, 388–393. [Google Scholar]
- Khaldi, N; Collemare, J; Lebrun, M-H; Wolf, KH. Evidence for horizontal transfer of a secondary metabolite gene cluster between fungi. Genome Biol 2008, 9, R18. [Google Scholar]
- Ward, TJ; Bielawski, JP; Kistler, HC; Sullivan, E; O’Donnell, K. Ancestral polymorphism and adaptive evolution in the trichothecene mycotoxin gene cluster of phytopathogenic Fusarium. Proc Natl Acad Sci USA 2002, 99, 9278–9283. [Google Scholar]
- Booth, C. The Genus. In Fusarium; CABI Publishing: Oxford, UK, 1971. [Google Scholar]
- Altschul, SF; Gish, W; Miller, W; Myers, EW; Lipman, DJ. Basic local alignment search tool. J Mol Biol 1990, 215, 403–410. [Google Scholar]
- Kulik, T; Pszczółkowska, A; Fordoński, G; Olszewski, J. PCR approach based on the esyn1 gene for the detection of potential enniatin-producing Fusarium species. Int J Food Microbiol 2007, 116, 319–324. [Google Scholar]
- Thompson, JD; Higgins, DG; Gibson, TJ. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position specific gap penalties and weight matrix choice. Nucleic Acids Res 1994, 22, 4673–4680. [Google Scholar]
- Drummond, AJ; Ashton, B; Cheung, M; Heled, J; Kearse, M; Moir, R; Stones-Havas, S; Thierer, T; Wilson, A. Geneious. 4.7. Available online: http://www.geneious.com/ (accessed on 4 February 2011).
- Librado, P; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar]
- Kelly, JK. A test of neutrality based on interlocus associations. Genetics 1997, 146, 1197–1206. [Google Scholar]
- Swofford, DL. PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods), Version 4th ed; Sinauer Associates: Sunderland, MA, USA, 2001. [Google Scholar]
- Posada, D; Crandall, KA. MODELTEST: Testing the model of DNA substitution. Bioinformatics 1998, 14, 817–818. [Google Scholar]
- Akaike, H. Information Theory and an Extension of the Maximum Likelihood Principle. In Second International Symposium on Information Theory; Petrov, PN, Csaki, F, Eds.; Akad Kiado: Budapest, Hungary, 1973; pp. 267–281. [Google Scholar]
- Farris, JS; Kallersjo, M; Kluge, AG; Bult, C. Testing significance of incongruence. Cladistics 1995, 10, 315–319. [Google Scholar]
- Cunningham, CW. Can three incongruence tests predict when data should be combined? Mol Biol Evol 1997, 14, 733–740. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Sequence Range | % GC | No. Variable Sites (%) | No. Parsimony Informative Sites (%) | Nucleotide Diversity | No. of Haplotypes | Haplotype Diversity |
|---|---|---|---|---|---|---|---|
| EF1α | 510–514 | 53.1 | 22 (4.3%) | 15 (2.9%) | 0.00931 | 11 | 0.814 |
| ESYN1 | 508–510 | 56 | 72 (14.1%) | 65 (12.7%) | 0.03303 | 13 | 0.710 |
| IGS rDNA | 482–516 | 49.1 | 108 (20.9%) | 86 (16.7%) | 0.04662 | 29 | 0.929 |
| NAT2 | 513 | 50.9 | 9 (1.8%) | 7 (1.4%) | 0.00354 | 6 | 0.598 |
| RPB2 | 518 | 51.3 | 1 (0.2%) | 0 | 0.00006 | 2 | 0.032 |
| MLS combined | 2023–2572 | 51.8 | 138 (5.4%) | 106 (4.1%) | 0.01395 | 51 | 0.991 |
| Isolate Number 1 | Geographical Origin, Host/Habitat of Origin | F. avenaceum Haplotypes 2 | |||||
|---|---|---|---|---|---|---|---|
| EF1α | ESYN1 | IGS rDNA | NAT2 | RPB2 | MLS | ||
| KK12 | Hungary, wheat kerne | 1 | 1 | 24 | 1 | 1 | 1 |
| MK3 | 1 | 13 | 5 | 1 | 1 | 2 | |
| FRC R-03050 | Australia, soil | 1 | 13 | 13 | 5 | 1 | 3 |
| FRC R-00048 | Pennsylvania, turf | 1 | 13 | 19 | 1 | 1 | 4 |
| DDPP 061526 a* | Poland, wheat kernel | 1 | 13 | 27 | 1 | 1 | 5 |
| DDPP 0615251 a* | 1 | 13 | 27 | 1 | 1 | 5 | |
| DDPP 03162 * | 1 | 13 | 27 | 1 | 1 | 5 | |
| DDPP 0703e | England, wheat kernel | 1 | 1 | 15 | 5 | 1 | 6 |
| CBS 121.73 * | United Kingdom, Dianthus caryophyllus | 1 | 13 | 26 | 1 | 1 | 7 |
| DDPP 11.73 * | unknown | 1 | 13 | 26 | 1 | 1 | 7 |
| DDPP 04301 b | Poland, wheat kernel | 1 | 13 | 6 | 6 | 1 | 8 |
| DDPP 04307 b | 1 | 12 | 6 | 6 | 1 | 8 | |
| DDPP 0610p | Poland, currant | 1 | 13 | 28 | 6 | 1 | 9 |
| DDPP 061518 a | Poland, wheat kernel | 1 | 13 | 27 | 2 | 1 | 10 |
| DDPP 04299 d | 1 | 13 | 18 | 1 | 1 | 11 | |
| DDPP 042836 c | 1 | 13 | 27 | 6 | 1 | 12 | |
| DDPP 042826 c | 1 | 12 | 15 | 1 | 1 | 13 | |
| DDPP 04032 | 1 | 13 | 14 | 1 | 1 | 14 | |
| DDPP 0355 * | 1 | 12 | 11 | 6 | 1 | 15 | |
| DDPP 032314 e* | 1 | 12 | 11 | 6 | 1 | 15 | |
| CBS 119834 | unknown | 1 | 13 | 15 | 1 | 1 | 16 |
| B5 | Hungary, wheat kernel | 1 | 13 | 23 | 1 | 1 | 17 |
| 18 | 3 | 1 | 18 | 1 | 1 | 18 | |
| F1 | 3 | 13 | 2 | 1 | 1 | 19 | |
| FRC R-06550 | California, carnation | 2 | 1 | 25 | 1 | 1 | 20 |
| FRC R-08483 | Sweden, Salix viminalis | 5 | 1 | 22 | 6 | 1 | 21 |
| FRC R-04055 | South Africa, carnation | 5 | 13 | 16 | 6 | 1 | 22 |
| DDPP 0702p | Poland, currant | 5 | 13 | 13 | 1 | 1 | 23 |
| DDPP 04401 | Poland, wheat kernel | 5 | 13 | 15 | 1 | 1 | 24 |
| 379 | Switzerland, wheat kernel | 6 | 3 | 15 | 1 | 1 | 24 |
| DDPP 04181 | Poland, wheat kernel | 5 | 13 | 27 | 5 | 1 | 25 |
| CBS 115957 | Italy, Fagus sylvatica | 8 | 8 | 24 | 3 | 1 | 26 |
| CBS 387.62 | Turkey, Camellia sinensis | 8 | 6 | 23 | 3 | 1 | 27 |
| DDPP 04293 d | Poland, wheat kernel | 8 | 7 | 20 | 4 | 1 | 28 |
| DDPP 04446 f | 8 | 8 | 9 | 3 | 1 | 29 | |
| IBT 40028 | Denmark, wheat kernel | 8 | 2 | 21 | 1 | 1 | 30 |
| IBT 40030 | Denmark rye kernel | 8 | 5 | 10 | 3 | 1 | 31 |
| FRC R-09092 | Sweden, barley | 9 | 3 | 17 | 5 | 1 | 32 |
| DDPP 0362 | Poland, wheat kernel | 5 | 12 | 27 | 1 | 1 | 33 |
| CBS 409.86 | USA, barley kernel | 7 | 4 | 7 | 1 | 1 | 34 |
| FRC R-09495 | California, lisianthus | 7 | 10 | 7 | 1 | 1 | 34 |
| FRC R-09800 | Connecticut, lisianthus | 7 | 11 | 7 | 1 | 1 | 34 |
| DDPP 031017 | Poland, wheat kernel | 7 | 10 | 27 | 1 | 1 | 35 |
| FRC R-09528 | North Dakota, barley | 7 | 11 | 4 | 1 | 1 | 36 |
| FRC R-09810 | Florida, lisianthus | 7 | 10 | 1 | 1 | 1 | 37 |
| FRC R-04935 | Brazil, wheat | 7 | 10 | 3 | 1 | 1 | 38 |
| DDPP 03512 | Poland, wheat kernel | 5 | 9 | 19 | 6 | 1 | 39 |
| DDPP 03175 | 5 | 9 | 27 | 1 | 2 | 40 | |
| DDPP 09a1 | 4 | 13 | 15 | 1 | 1 | 41 | |
| 0380 | Switzerland, wheat kernel | 11 | 1 | 8 | 1 | 1 | 42 |
| 376 | 11 | 1 | 8 | 5 | 1 | 43 | |
| 05-003 | 10 | 1 | 27 | 1 | 1 | 44 | |
| DDPP 03236 e* | Poland, wheat kernel | 10 | 13 | 27 | 1 | 1 | 44 |
| DDPP 042835 c* | 10 | 13 | 27 | 1 | 1 | 44 | |
| DDPP 06407 * | 10 | 13 | 27 | 1 | 1 | 44 | |
| 05-011 | Switzerland, wheat kernel | 10 | 13 | 27 | 5 | 1 | 45 |
| CBS 241.94 | The Netherlands, Dianthus caryophyllus | 10 | 1 | 12 | 1 | 1 | 46 |
| CBS 408.86 | Denmark, barley kernel | 10 | 13 | 18 | 5 | 1 | 47 |
| DDPP 03255 | Poland, wheat kernel | 10 | 13 | 19 | 6 | 1 | 48 |
| IBT 40026 * | Denmark, wheat kernel | 10 | 13 | 19 | 1 | 1 | 49 |
| DDPP 04449 f* | Poland, wheat kernel | 10 | 13 | 19 | 1 | 1 | 49 |
| DDPP 061108 | 10 | 13 | 2 | 5 | 1 | 50 | |
| FRC R-04954 | Germany, barley | 10 | 13 | 27 | 3 | 1 | 51 |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kulik, T.; Pszczółkowska, A.; Łojko, M. Multilocus Phylogenetics Show High Intraspecific Variability within Fusarium avenaceum. Int. J. Mol. Sci. 2011, 12, 5626-5640. https://doi.org/10.3390/ijms12095626
Kulik T, Pszczółkowska A, Łojko M. Multilocus Phylogenetics Show High Intraspecific Variability within Fusarium avenaceum. International Journal of Molecular Sciences. 2011; 12(9):5626-5640. https://doi.org/10.3390/ijms12095626
Chicago/Turabian StyleKulik, Tomasz, Agnieszka Pszczółkowska, and Maciej Łojko. 2011. "Multilocus Phylogenetics Show High Intraspecific Variability within Fusarium avenaceum" International Journal of Molecular Sciences 12, no. 9: 5626-5640. https://doi.org/10.3390/ijms12095626