Targeting Protective Autophagy Exacerbates UV-Triggered Apoptotic Cell Death

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
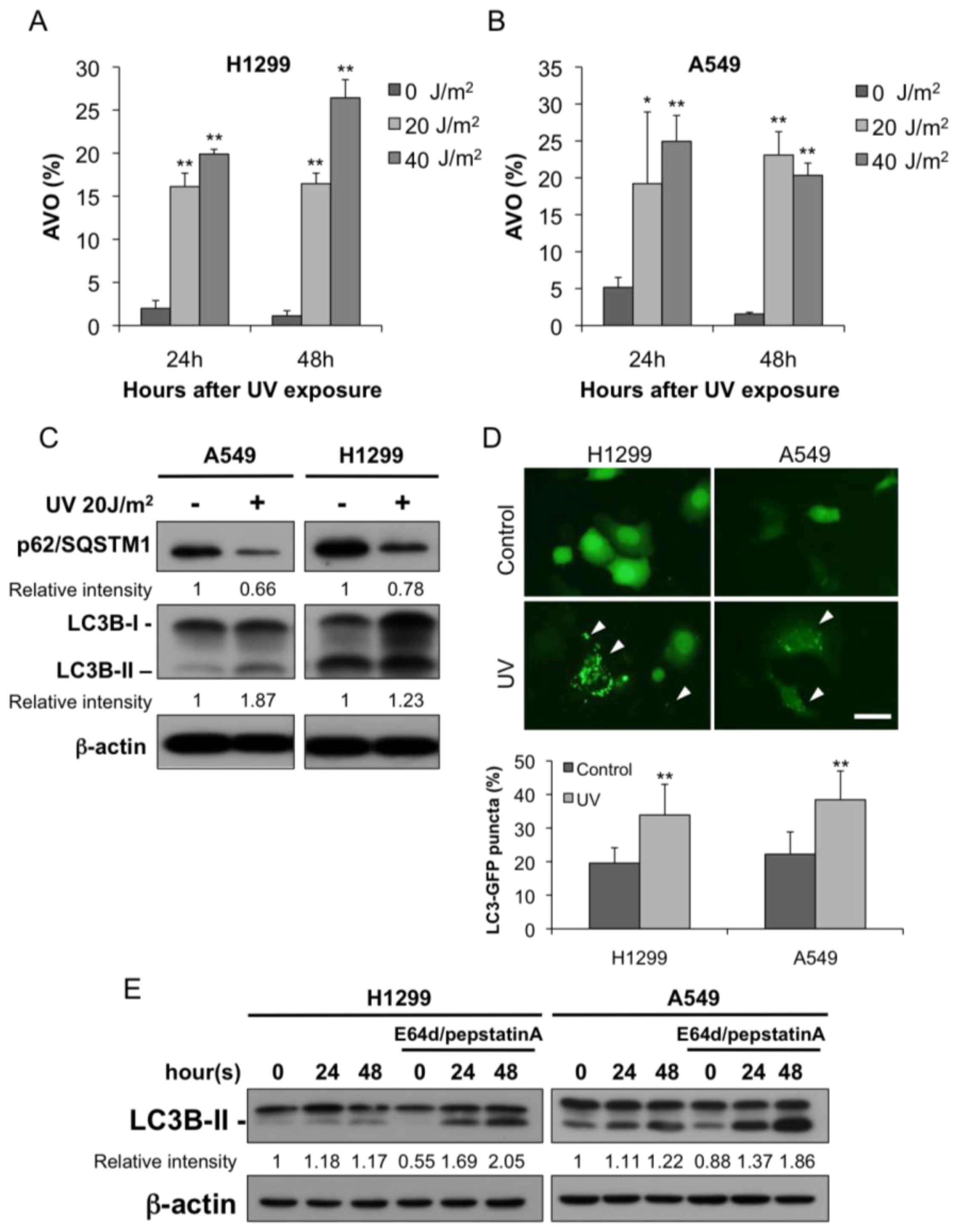
2.1. Autophagy Was Induced after UV Irradiation
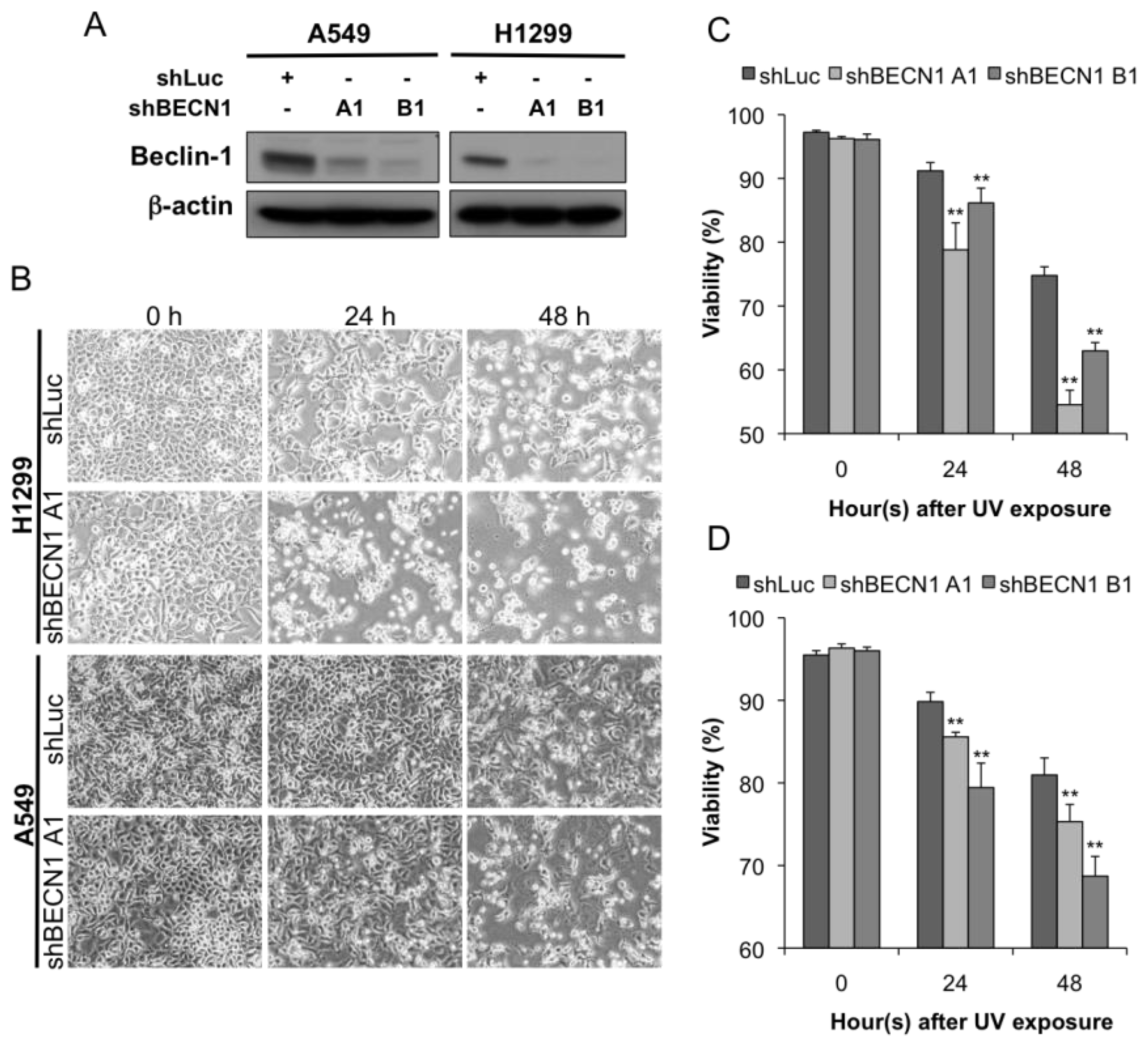
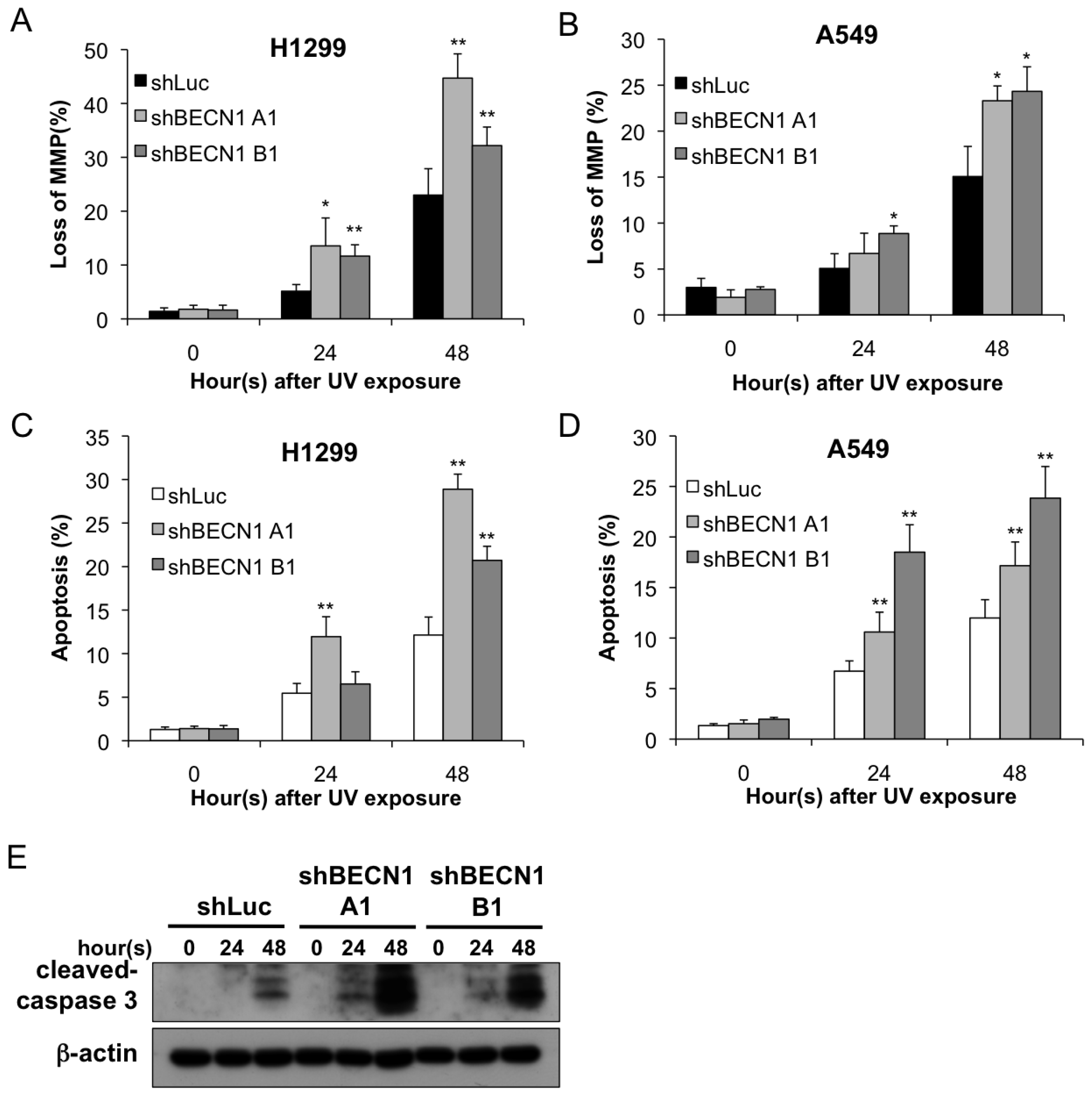
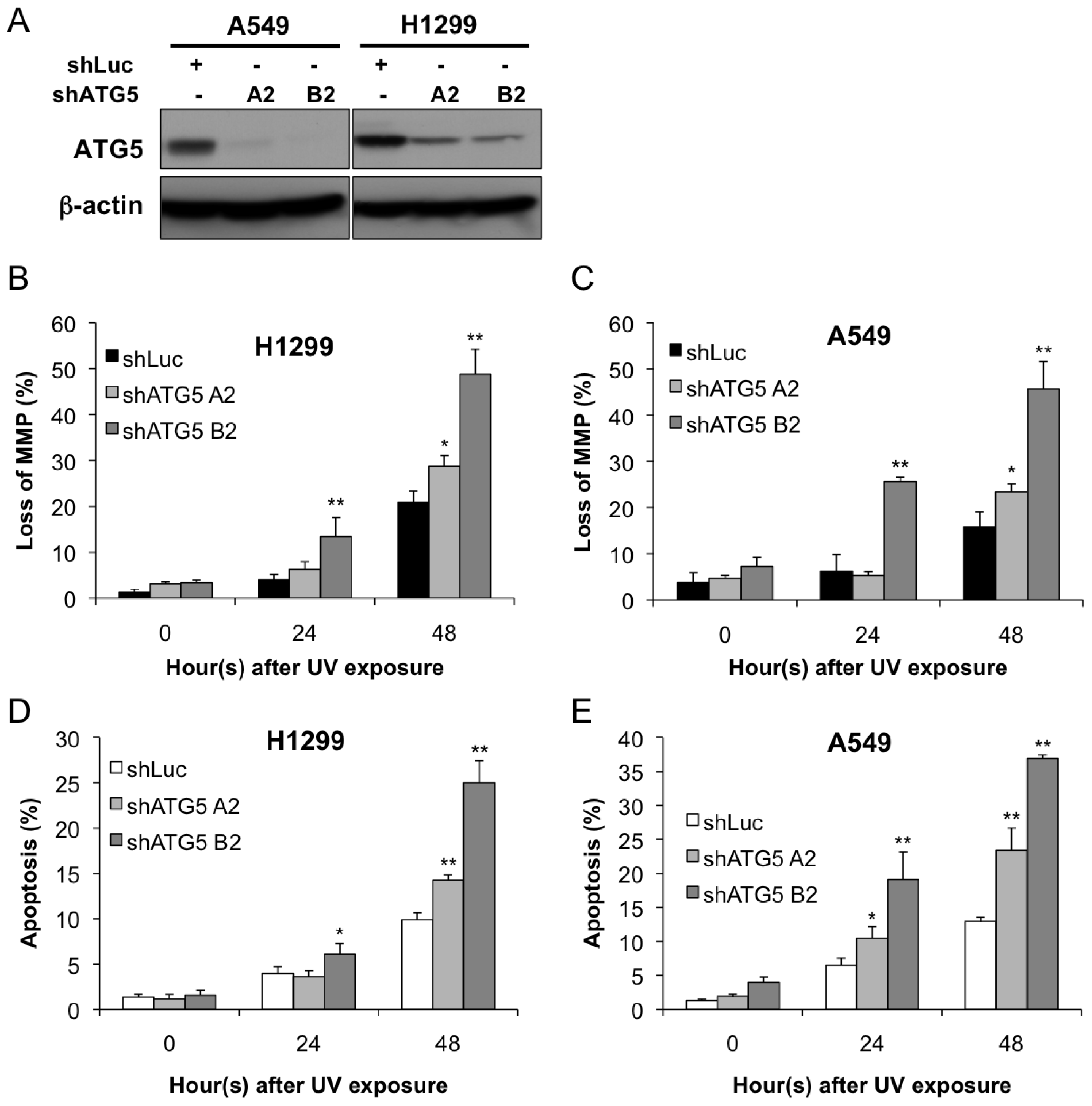
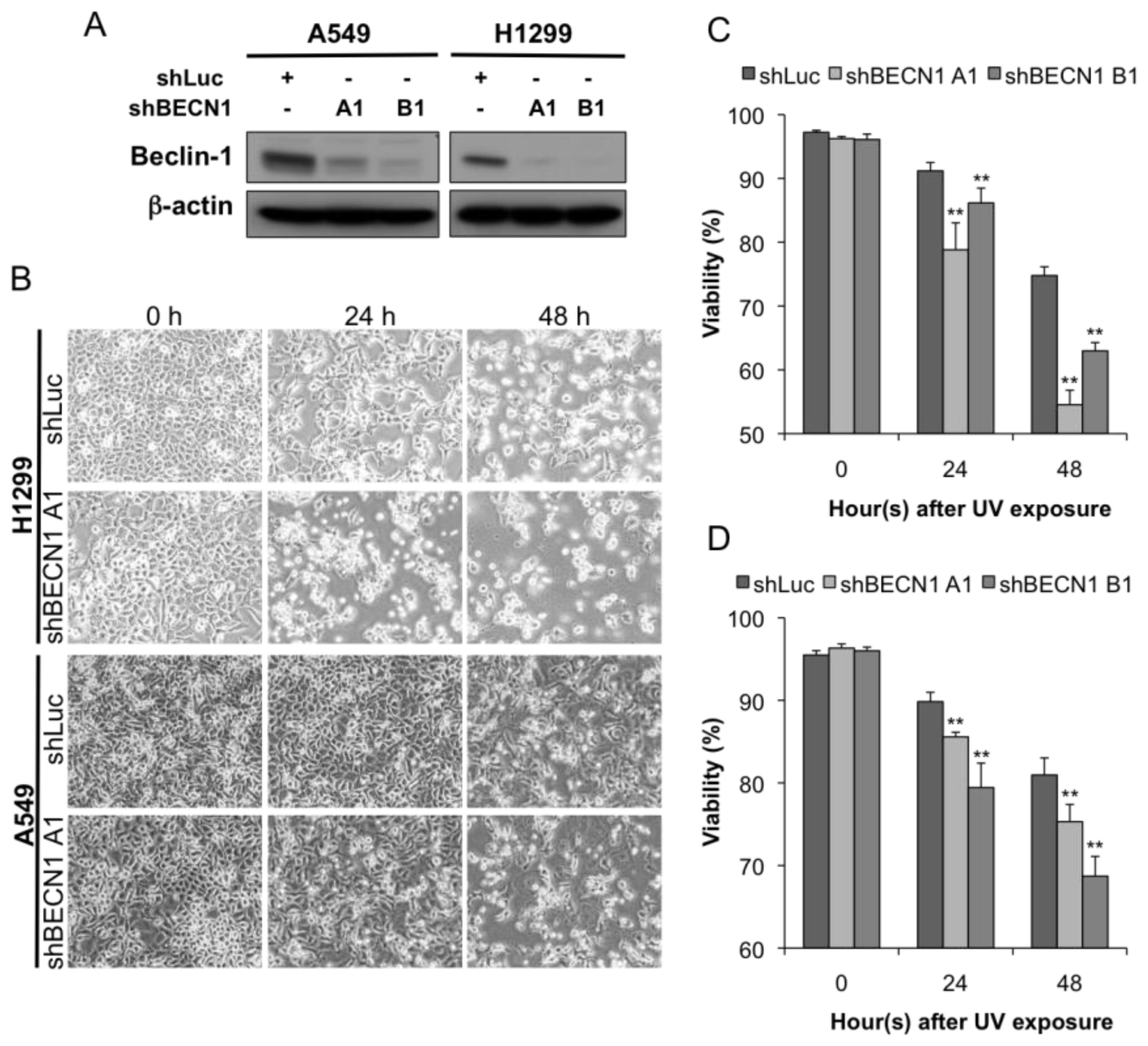
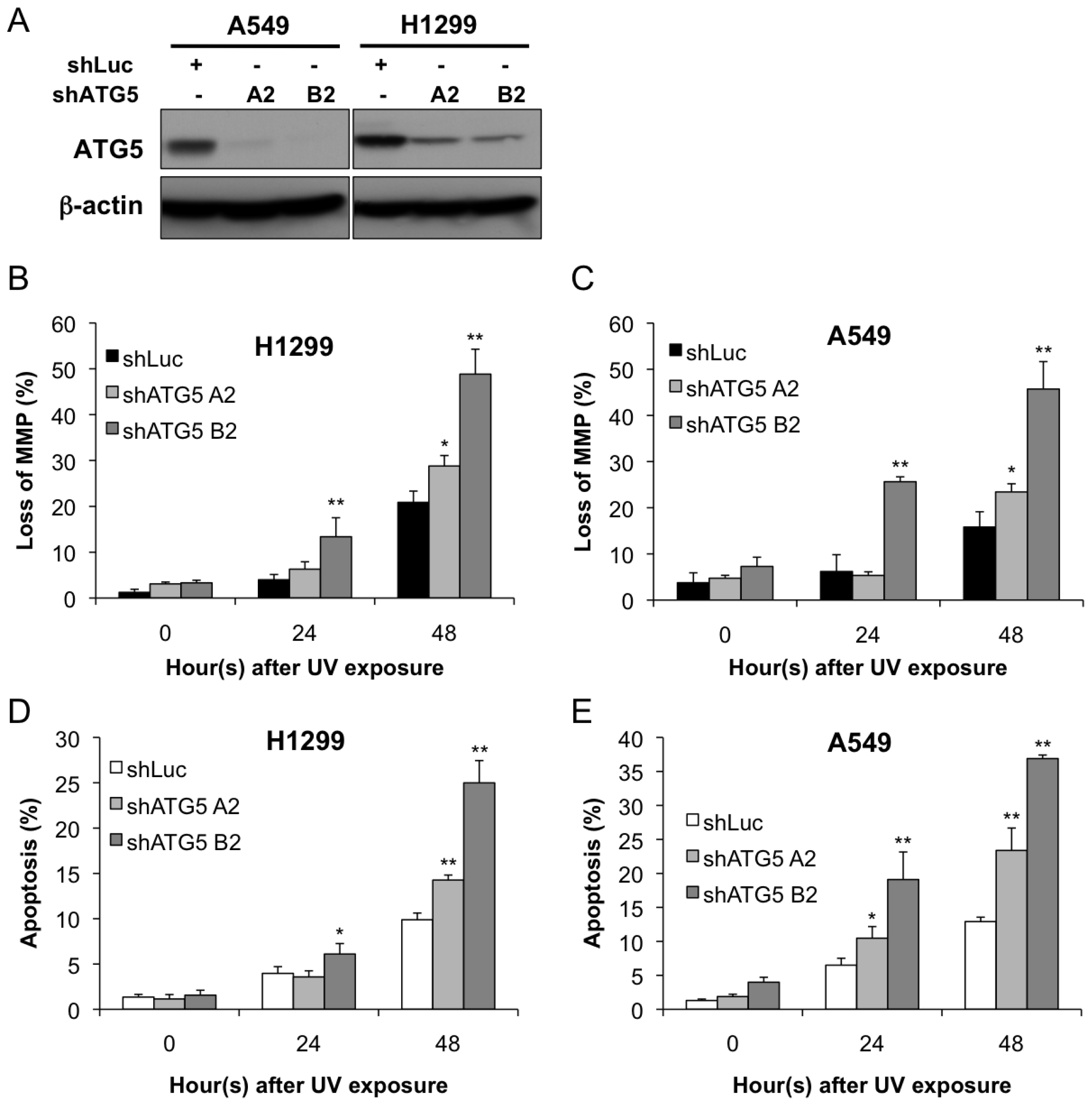
2.2. Autophagy Inhibition Enhances UV-Induced Cell Death
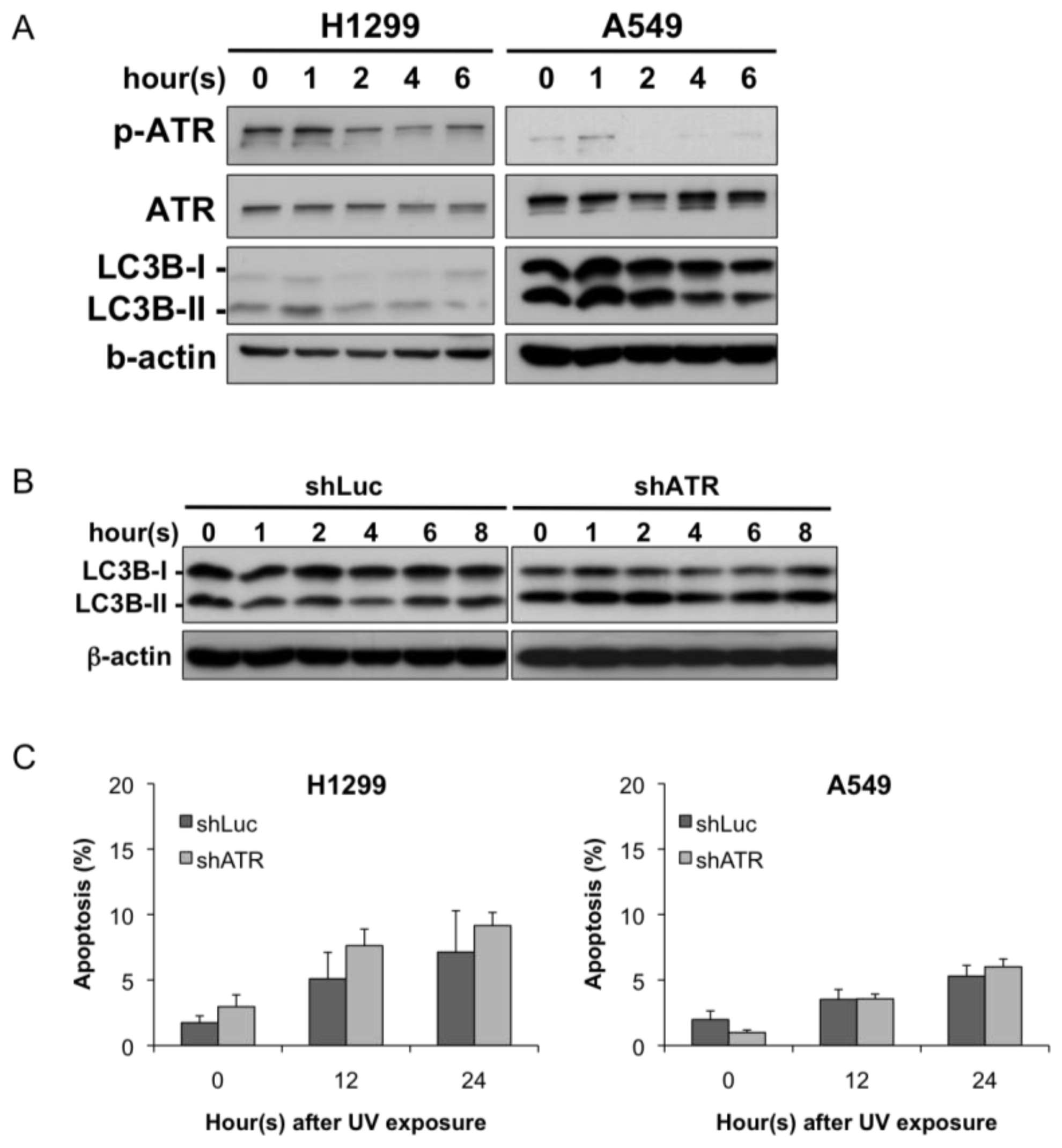
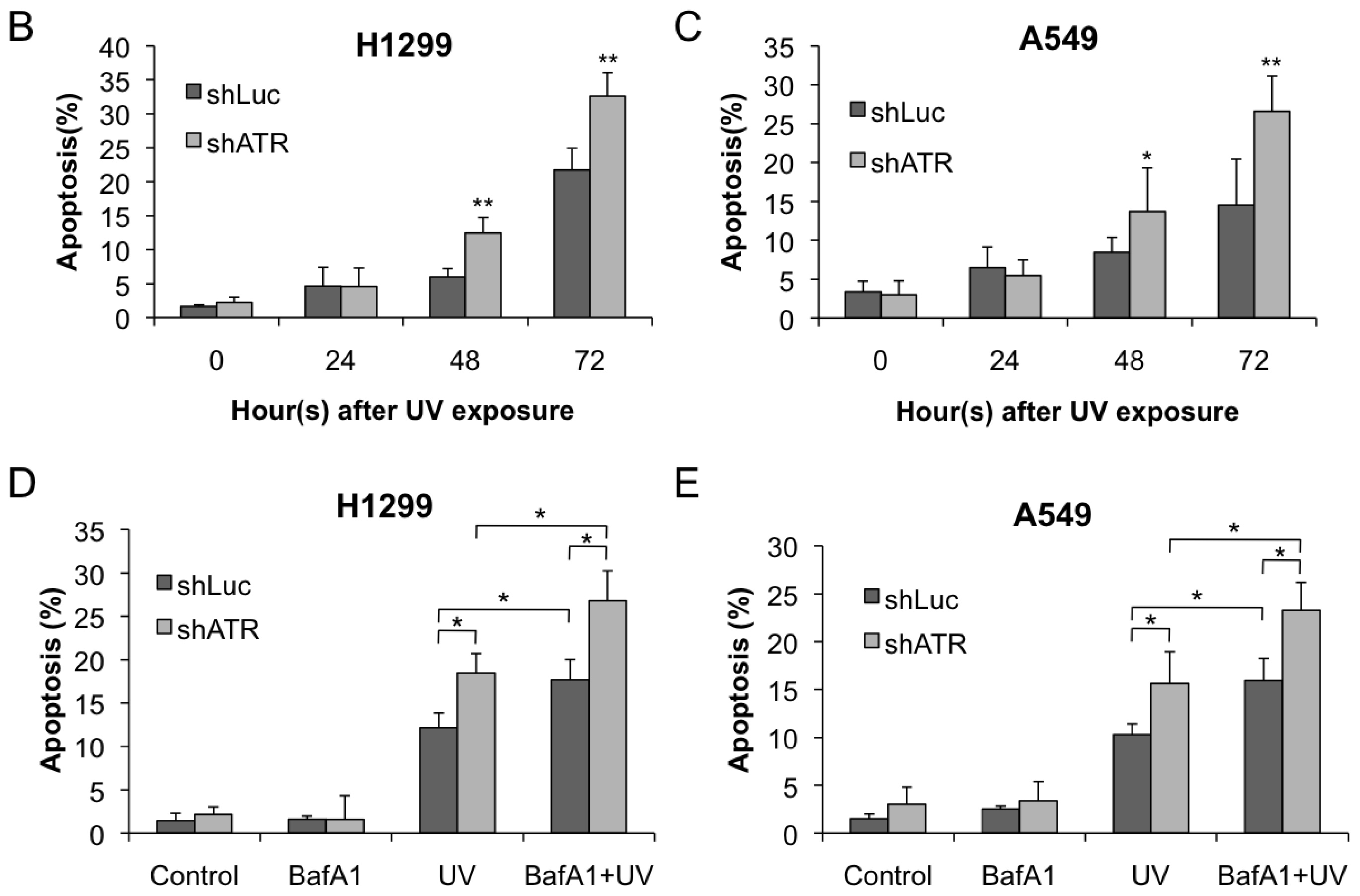
2.3. ATR Activation Parallels the Induction of Autophagy at Early Stages Following UV Treatment
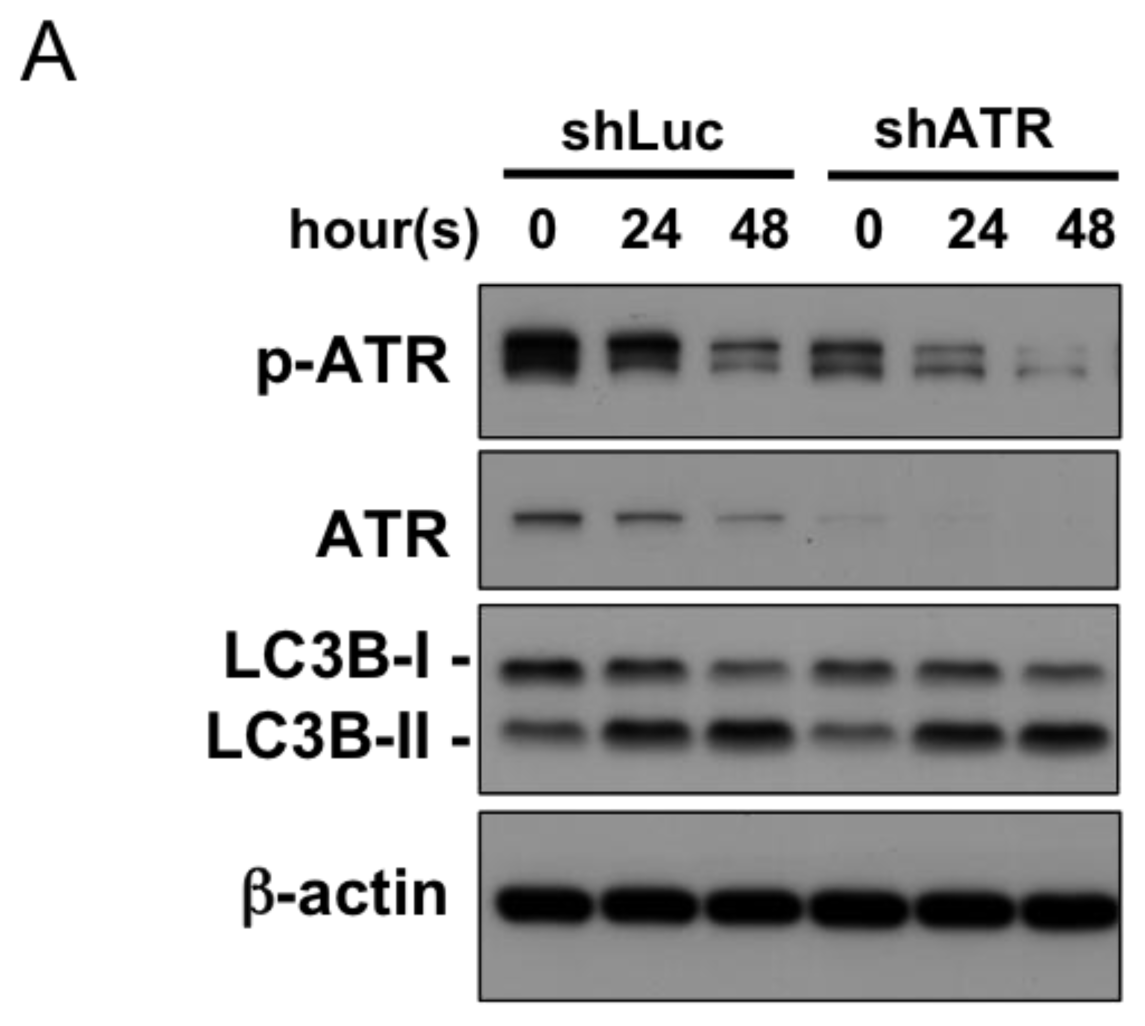
2.4. Autophagy Induction at Late Stages Is Independent of ATR
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture and UV Irradiation
4.3. Detection of Acidic Vesicular Organelles (AVOs) with Acridine Orange
4.4. Immunoblotting
4.5. Apoptosis Assay
4.6. Mitochondrial Membrane Potential Analysis
4.8. LC3-GFP Puncta
4.9. Data Analysis
5. Conclusions
Supplementary Materials
ijms-13-01209-s001.pdfAcknowledgments
References
- Galluzzi, L.; Vicencio, J.M.; Kepp, O.; Tasdemir, E.; Maiuri, M.C.; Kroemer, G. To die or not to die: That is the autophagic question. Curr. Mol. Med 2008, 8, 78–91. [Google Scholar]
- Spano, A.; Monaco, G.; Barni, S.; Sciola, L. Cisplatin treatment of NIH/3T3 cultures induces a form of autophagic death in polyploid cells. Histol. Histopathol 2008, 23, 717–730. [Google Scholar]
- Periyasamy-Thandavan, S.; Jiang, M.; Wei, Q.; Smith, R.; Yin, X.M.; Dong, Z. Autophagy is cytoprotective during cisplatin injury of renal proximal tubular cells. Kidney Int 2008, 74, 631–640. [Google Scholar]
- Abedin, M.J.; Wang, D.; McDonnell, M.A.; Lehmann, U.; Kelekar, A. Autophagy delays apoptotic death in breast cancer cells following DNA damage. Cell Death Differ 2007, 14, 500–510. [Google Scholar]
- Zhou, W.J.; Deng, R.; Zhang, X.Y.; Feng, G.K.; Gu, L.Q.; Zhu, X.F. G-quadruplex ligand SYUIQ-5 induces autophagy by telomere damage and TRF2 delocalization in cancer cells. Mol. Cancer Ther 2009, 8, 3203–3213. [Google Scholar]
- Suzuki, K.; Kodama, S.; Watanabe, M. Recruitment of ATM protein to double strand DNA irradiated with ionizing radiation. J. Biol. Chem 1999, 274, 25571–25575. [Google Scholar]
- Cimprich, K.A.; Cortez, D. ATR: An essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol 2008, 9, 616–627. [Google Scholar]
- Brown, E.J.; Baltimore, D. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev 2000, 14, 397–402. [Google Scholar]
- O’Driscoll, M. Mouse models for ATR deficiency. DNA Repair (Amst) 2009, 8, 1333–1337. [Google Scholar]
- Cortez, D.; Guntuku, S.; Qin, J.; Elledge, S.J. ATR and ATRIP: Partners in checkpoint signaling. Science 2001, 294, 1713–1716. [Google Scholar]
- Unsal-Kacmaz, K.; Makhov, A.M.; Griffith, J.D.; Sancar, A. Preferential binding of ATR protein to UV-damaged DNA. Proc. Natl. Acad. Sci. USA 2002, 99, 6673–6678. [Google Scholar]
- Lowndes, N.F.; Murguia, J.R. Sensing and responding to DNA damage. Curr. Opin. Genet. Dev 2000, 10, 17–25. [Google Scholar]
- Rieber, M.; Rieber, M.S. Sensitization to radiation-induced DNA damage accelerates loss of bcl-2 and increases apoptosis and autophagy. Cancer Biol. Ther 2008, 7, 1561–1566. [Google Scholar]
- Chen, L.H.; Loong, C.C.; Su, T.L.; Lee, Y.J.; Chu, P.M.; Tsai, M.L.; Tsai, P.H.; Tu, P.H.; Chi, C.W.; Lee, H.C.; et al. Autophagy inhibition enhances apoptosis triggered by BO-1051, an N-mustard derivative, and involves the ATM signaling pathway. Biochem. Pharmacol 2011, 81, 594–605. [Google Scholar]
- Alexander, A.; Cai, S.L.; Kim, J.; Nanez, A.; Sahin, M.; MacLean, K.H.; Inoki, K.; Guan, K.L.; Shen, J.; Person, M.D.; et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc. Natl. Acad. Sci. USA 2010, 107, 4153–4158. [Google Scholar]
- Klionsky, D.J.; Abeliovich, H.; Agostinis, P.; Agrawal, D.K.; Aliev, G.; Askew, D.S.; Baba, M.; Baehrecke, E.H.; Bahr, B.A.; Ballabio, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008, 4, 151–175. [Google Scholar]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol 2007, 8, 741–752. [Google Scholar]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death by apoptosis. Trends Mol. Med 2006, 12, 440–450. [Google Scholar]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar]
- Mizushima, N.; Yamamoto, A.; Hatano, M.; Kobayashi, Y.; Kabeya, Y.; Suzuki, K.; Tokuhisa, T.; Ohsumi, Y.; Yoshimori, T. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J. Cell Biol 2001, 152, 657–668. [Google Scholar]
- Al-Khalaf, H.H.; Hendrayani, S.F.; Aboussekhra, A. ATR controls the p21WAF1/Cip1 protein up-regulation and apoptosis in response to low UV fluences. Mol. Carcinog 2011. [Google Scholar] [CrossRef]
- Heffernan, T.P.; Kawasumi, M.; Blasina, A.; Anderes, K.; Conney, A.H.; Nghiem, P. ATR-Chk1 pathway inhibition promotes apoptosis after UV treatment in primary human keratinocytes: Potential basis for the UV protective effects of caffeine. J. Invest. Dermatol 2009, 129, 1805–1815. [Google Scholar]
- Galluzzi, L.; Morselli, E.; Vicencio, J.M.; Kepp, O.; Joza, N.; Tajeddine, N.; Kroemer, G. Life, death and burial: Multifaceted impact of autophagy. Biochem. Soc. Trans 2008, 36, 786–790. [Google Scholar]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar]
- Wang, Y.; Singh, R.; Massey, A.C.; Kane, S.S.; Kaushik, S.; Grant, T.; Xiang, Y.; Cuervo, A.M.; Czaja, M.J. Loss of macroautophagy promotes or prevents fibroblast apoptosis depending on the death stimulus. J. Biol. Chem 2008, 283, 4766–4777. [Google Scholar]
- Singh, R.; Czaja, M.J. Compensatory mechanisms and the type of injury determine the fate of cells with impaired macroautophagy. Autophagy 2008, 4, 516–518. [Google Scholar]
- Buron, N.; Micheau, O.; Cathelin, S.; Lafontaine, P.O.; Creuzot-Garcher, C.; Solary, E. Differential mechanisms of conjunctival cell death induction by ultraviolet irradiation and benzalkonium chloride. Invest. Ophthalmol. Vis. Sci 2006, 47, 4221–4230. [Google Scholar]
- Codogno, P.; Meijer, A.J. Autophagy and signaling: their role in cell survival and cell death. Cell Death Differ 2005, 12, 1509–1518. [Google Scholar]
- Shiloh, Y. ATM and ATR: Networking cellular responses to DNA damage. Curr. Opin. Genet. Dev 2001, 11, 71–77. [Google Scholar]
- Helt, C.E.; Cliby, W.A.; Keng, P.C.; Bambara, R.A.; O’Reilly, M.A. Ataxia telangiectasia mutated (ATM) and ATM and Rad3-related protein exhibit selective target specificities in response to different forms of DNA damage. J. Biol. Chem 2005, 280, 1186–1192. [Google Scholar]
- Stiff, T.; Walker, S.A.; Cerosaletti, K.; Goodarzi, A.A.; Petermann, E.; Concannon, P.; O’Driscoll, M.; Jeggo, P.A. ATR-dependent phosphorylation and activation of ATM in response to UV treatment or replication fork stalling. EMBO J 2006, 25, 5775–5782. [Google Scholar]
- Jazayeri, A.; Falck, J.; Lukas, C.; Bartek, J.; Smith, G.C.; Lukas, J.; Jackson, S.P. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat. Cell Biol 2006, 8, 37–45. [Google Scholar]
- Adams, K.E.; Medhurst, A.L.; Dart, D.A.; Lakin, N.D. Recruitment of ATR to sites of ionising radiation-induced DNA damage requires ATM and components of the MRN protein complex. Oncogene 2006, 25, 3894–3904. [Google Scholar]





© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chen, L.-H.; Chu, P.-M.; Lee, Y.-J.; Tu, P.-H.; Chi, C.-W.; Lee, H.-C.; Chiou, S.-H. Targeting Protective Autophagy Exacerbates UV-Triggered Apoptotic Cell Death. Int. J. Mol. Sci. 2012, 13, 1209-1224. https://doi.org/10.3390/ijms13011209
Chen L-H, Chu P-M, Lee Y-J, Tu P-H, Chi C-W, Lee H-C, Chiou S-H. Targeting Protective Autophagy Exacerbates UV-Triggered Apoptotic Cell Death. International Journal of Molecular Sciences. 2012; 13(1):1209-1224. https://doi.org/10.3390/ijms13011209
Chicago/Turabian StyleChen, Li-Hsin, Pei-Ming Chu, Yi-Jang Lee, Pang-Hsien Tu, Chin-Wen Chi, Hsin-Chen Lee, and Shih-Hwa Chiou. 2012. "Targeting Protective Autophagy Exacerbates UV-Triggered Apoptotic Cell Death" International Journal of Molecular Sciences 13, no. 1: 1209-1224. https://doi.org/10.3390/ijms13011209