Probing Kinetic Mechanisms of Protein Function and Folding with Time-Resolved Natural and Magnetic Chiroptical Spectroscopies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
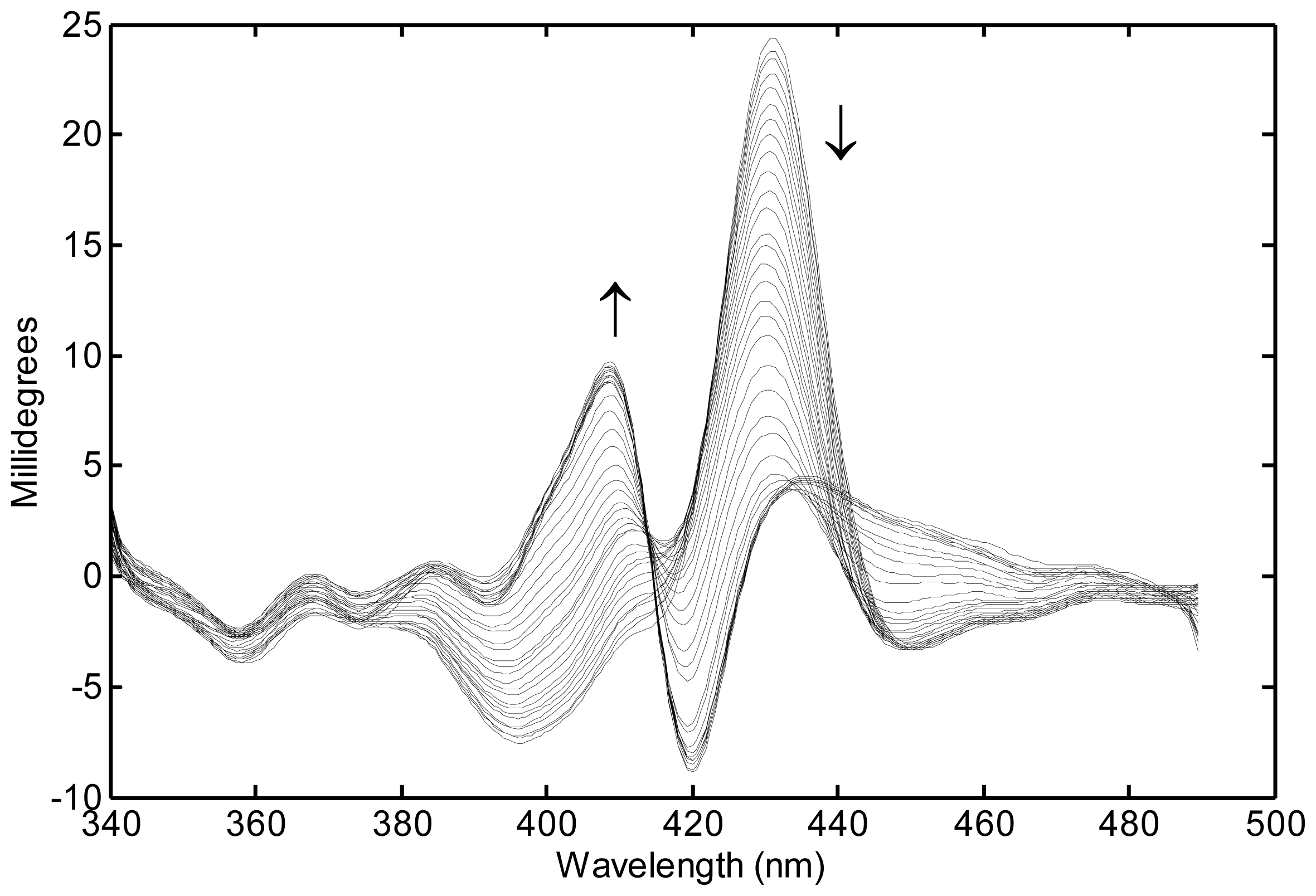{kind=link}
Abstract
:1. Introduction
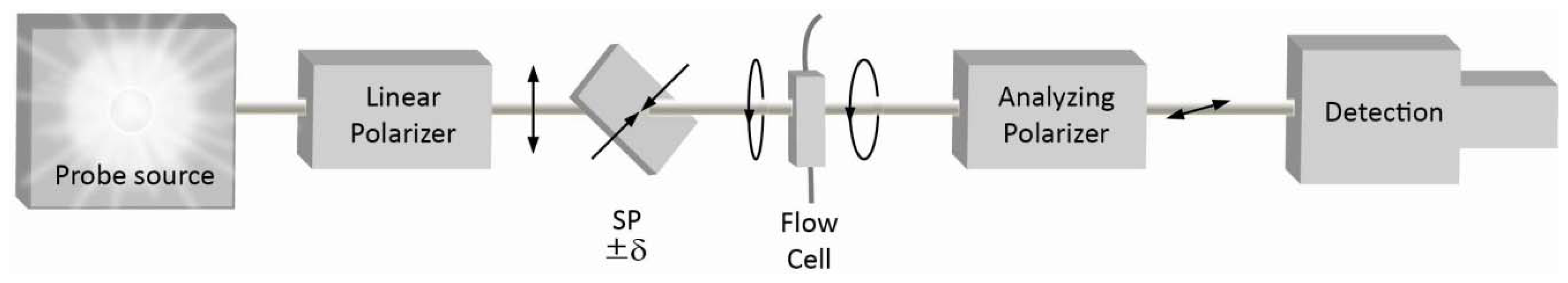
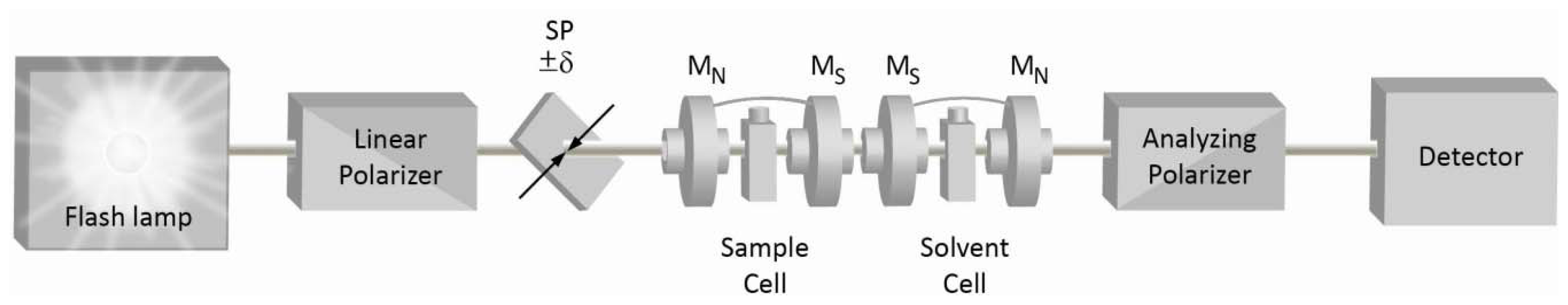
2. Quasi-Null Polarization Spectroscopy
3. Kinetics of Protein Structural Changes
3.1. Protein Function
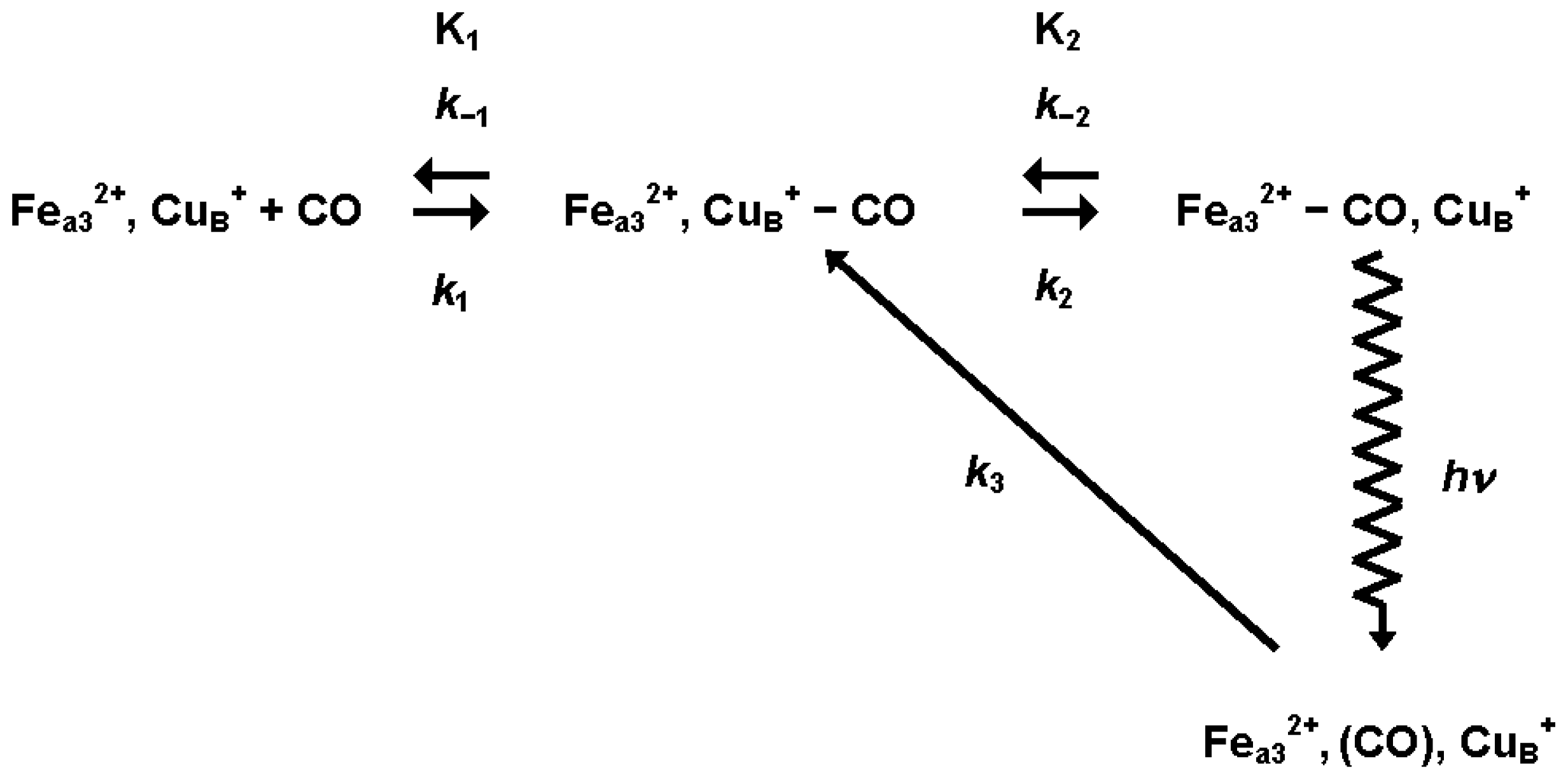
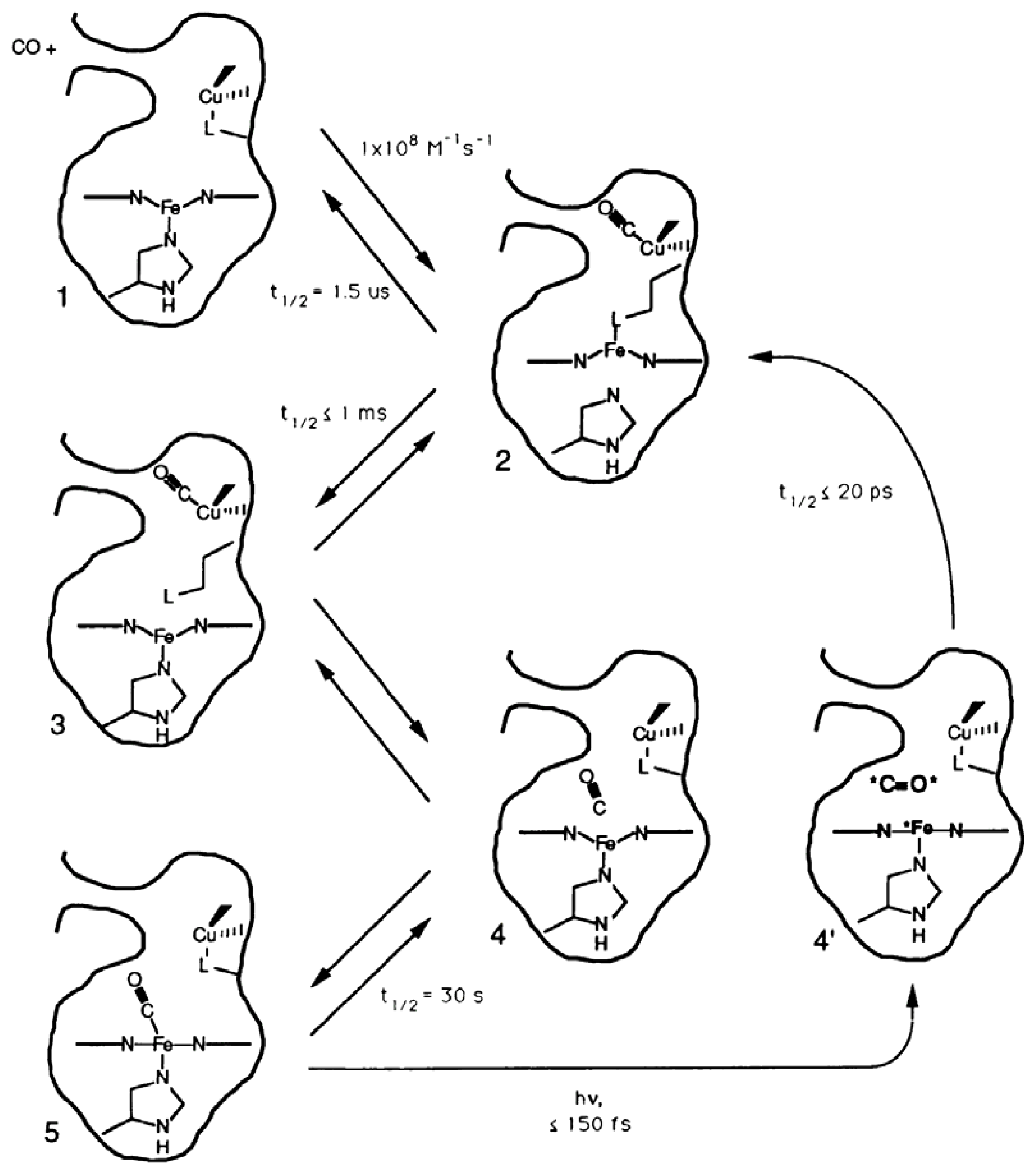
3.1.1. Ligand Photolysis in Cytochrome c Oxidase
3.1.2. Light Activation and De-Activation of Phytochrome
3.2. Protein Folding
3.2.1. Rapid Folding Triggers
3.2.2. Monitoring Protein Folding Reactions
4. Conclusions
Acknowledgments
References
- Anson, M.; Bayley, P.M. Measurement of circular dichroism at millisecond time resolution: Stopped-flow circular dichroism system. J. Phys. E Sci. Instrum 1974, 7. [Google Scholar] [CrossRef]
- Bayley, P.M.; Anson, M. Stopped-flow circular dichroism: New fast-kinetic system. Biopolymers 1974, 13, 401–405. [Google Scholar]
- Lewis, J.W.; Tilton, R.F.; Einterz, C.M.; Milder, S.J.; Kuntz, I.D.; Kliger, D.S. New technique for measuring circular dichroism changes on a nanosecond time scale: Application to (carbonmonoxy) myoglobin and (carbonmonoxy)hemoglobin. J. Phys. Chem 1985, 89, 289–294. [Google Scholar]
- Goldbeck, R.A.; Dawes, T.D.; Milder, S.J.; Lewis, J.W.; Kliger, D.S. Measurement of magnetic circular dichroism (MCD) on a nanosecond timescale. Chem. Phys. Lett 1989, 156, 545–549. [Google Scholar]
- Goldbeck, R.A.; Kliger, D.S. Nanosecond time-resolved absorption and polarization dichroism spectroscopies. Methods Enzymol 1993, 226, 147–177. [Google Scholar]
- Esquerra, R.M.; Goldbeck, R.A.; Kim-Shapiro, D.B.; Kliger, D.S. Fast time-resolved magnetic optical rotatory dispersion measurements. 1. Mueller analysis of optical and photoselection-induced artifacts. J. Phys. Chem. A 1998, 102, 8740–8748. [Google Scholar]
- Shapiro, D.B.; Goldbeck, R.A.; Che, D.; Esquerra, R.M.; Paquette, S.J.; Kliger, D.S. Nanosecond optical rotatory dispersion spectroscopy: Application to photolyzed hemoglobin-CO kinetics. Biophys. J 1995, 68, 326–334. [Google Scholar]
- Esquerra, R.M.; Goldbeck, R.A.; Kim-Shapiro, D.B.; Kliger, D.S. Fast time-resolved magnetic optical rotatory dispersion measurements. 2. Confirmation of Mueller analysis and application to myoglobin ligand photolysis. J. Phys. Chem. A 1998, 102, 8749–8758. [Google Scholar]
- Gold, J.S.; Milder, S.J.; Lewis, J.W.; Kliger, D.S. Transient circular dichroism of the luminescent state of Ru(bpy)32+. J. Am. Chem. Soc 1985, 107, 8285–8286. [Google Scholar]
- Milder, S.J.; Gold, J.S.; Kliger, D.S. Assignments of ground- and excited-state spectra from time-resolved absorption and circular dichroism measurements of the 2E State of (Δ)-Cr(bpy)33+. Inorg. Chem 1990, 29, 2506–2511. [Google Scholar]
- Milder, S.J.; Gold, J.S.; Kliger, D.S. Time-resolved circular dichroism of the lowest excited state of (Δ)-Ru(bpy)32+. Chem. Phys. Lett 1988, 144, 269–272. [Google Scholar]
- Milder, S.J.; Gold, J.S.; Kliger, D.S. Circular dichroism of a subnanosecond state: (Δ)-Fe(bpy)32+. J. Am. Chem. Soc 1986, 108, 8295–8296. [Google Scholar]
- Milder, S.J.; Weiss, P.S.; Kliger, D.S. Time-resolved absorption, circular dichroism, and emission of transfer-RNA: Evidence that the photo-cross-linking of 4-thiouridine in transfer-RNA occurs from the triplet state. Biochemistry 1989, 28, 2258–2264. [Google Scholar]
- Einterz, C.M.; Lewis, J.W.; Milder, S.J.; Kliger, D.S. Birefringence effects in transient circular-dichroism measurements with applications to the photolysis of carbonmonoxyhemoglobin and carbonmonoxymyoglobin. J. Phys. Chem 1985, 89, 3845–3853. [Google Scholar]
- Milder, S.J.; Bjorling, S.C.; Kuntz, I.D.; Kliger, D.S. Time-resolved circular dichroism and absorption studies of the photolysis reaction of (carbonmonoxy)myoglobin. Biophys. J 1988, 53, 659–664. [Google Scholar]
- Woodruff, W.H.; Einarsdottir, O.; Dyer, R.B.; Bagley, K.A.; Palmer, G.; Atherton, S.J.; Goldbeck, R.A.; Dawes, T.D.; Kliger, D.S. Nature and functional implications of the cytochrome-a3 transients after photodissociation of CO-cytochrome oxidase. Proc. Natl. Acad. Sci. USA 1991, 88, 2588–2592. [Google Scholar]
- Bjorling, S.C.; Goldbeck, R.A.; Paquette, S.J.; Milder, S.J.; Kliger, D.S. Allosteric intermediates in hemoglobin. 1. Nanosecond time-resolved circular dichroism spectroscopy. Biochemistry 1996, 35, 8619–8627. [Google Scholar]
- Chen, E.F.; Kliger, D.S. Time-resolved near UV circular dichroism and absorption studies of carbonmonoxymyoglobin photolysis intermediates. Inorg. Chim. Acta 1996, 242, 149–158. [Google Scholar]
- Goldbeck, R.A.; Dawes, T.D.; Einarsdottir, O.; Woodruff, W.H.; Kliger, D.S. Time-resolved magnetic circular dichroism spectroscopy of photolyzed carbonmonoxy cytochrome c oxidase (cytochrome-aa3). Biophys. J 1991, 60, 125–134. [Google Scholar]
- Goldbeck, R.A.; Einarsdottir, O.; Dawes, T.D.; Oconnor, D.B.; Surerus, K.K.; Fee, J.A.; Kliger, D.S. Magnetic circular dichroism study of cytochrome ba3 from Thermus thermophilus: Spectral contributions from cytochrome b and cytochrome a3 and nanosecond spectroscopy of CO photodissociation intermediates. Biochemistry 1992, 31, 9376–9387. [Google Scholar]
- O’Connor, D.B.; Goldbeck, R.A.; Hazzard, J.H.; Kliger, D.S.; Cusanovich, M.A. Time-resolved absorption and magnetic circular dichroism spectroscopy of cytochrome c3 from Desulfovibrio. Biophys. J 1993, 65, 1718–1726. [Google Scholar]
- Esquerra, R.M.; Goldbeck, R.A.; Kim-Shapiro, D.B.; Kliger, D.S. Spectroscopic evidence for nanosecond protein relaxation after photodissociation of myoglobin-CO. Biochemistry 1998, 37, 17527–17536. [Google Scholar]
- Goldbeck, R.A.; Esquerra, R.M.; Kliger, D.S. Hydrogen bonding to Trp β37 is the first step in a compound pathway for hemoglobin allostery. J. Am. Chem. Soc 2002, 124, 7646–7647. [Google Scholar]
- Vitale, D.J.; Goldbeck, R.A.; Kim-Shapiro, D.B.; Esquerra, R.M.; Parkhurst, L.J.; Kliger, D.S. Near-ultraviolet magnetic circular dichroism spectroscopy of protein conformational states: Correlation of tryptophan band position and intensity with hemoglobin allostery. Biochemistry 2000, 39, 7145–7152. [Google Scholar]
- Bjorling, S.C.; Zhang, C.F.; Farrens, D.L.; Song, P.S.; Kliger, D.S. Time-resolved circular dichroism of native oat phytochrome photointermediates. J. Am. Chem. Soc 1992, 114, 4581–4588. [Google Scholar]
- Zhang, C.F.; Farrens, D.L.; Bjorling, S.C.; Song, P.S.; Kliger, D.S. Time-resolved absorption studies of native etiolated oat phytochrome. J. Am. Chem. Soc 1992, 114, 4569–4580. [Google Scholar]
- Chen, E.F.; Lapko, V.N.; Lewis, J.W.; Song, P.S.; Kliger, D.S. Mechanism of native oat phytochrome photoreversion: A time-resolved absorption investigation. Biochemistry 1996, 35, 843–850. [Google Scholar]
- Chen, E.F.; Lapko, V.N.; Song, P.S.; Kliger, D.S. Dynamics of the N-terminal α-helix unfolding in the photoreversion reaction of phytochrome A. Biochemistry 1997, 36, 4903–4908. [Google Scholar]
- Chen, E.F.; Parker, W.; Lewis, J.W.; Song, P.S.; Kliger, D.S. Time-resolved UV circular dichroism of phytochrome A: Folding of the N-terminal region. J. Am. Chem. Soc 1993, 115, 9854–9855. [Google Scholar]
- Chen, E.F.; Gensch, T.; Gross, A.B.; Hendriks, J.; Hellingwerf, K.J.; Kliger, D.S. Dynamics of protein and chromophore structural changes in the photocycle of photoactive yellow protein monitored by time-resolved optical rotatory dispersion. Biochemistry 2003, 42, 2062–2071. [Google Scholar]
- Chen, E.F.; Swartz, T.E.; Bogomolni, R.A.; Kliger, D.S. A LOV story: The signaling state of the Phot1 LOV2 photocycle involves chromophore-triggered protein structure relaxation, as probed by far-UV time-resolved optical rotatory dispersion spectroscopy. Biochemistry 2007, 46, 4619–4624. [Google Scholar]
- Thomas, Y.G.; Szundi, I.; Lewis, J.W.; Kliger, D.S. Microsecond time-resolved circular dichroism of rhodopsin photointermediates. Biochemistry 2009, 48, 12283–12289. [Google Scholar]
- Woodruff, W.H.; Dyer, R.B.; Einarsdóttir, Ó.; Peterson, K.A.; Stoutland, P.O.; Bagley, K.A.; Palmer, G.; Schoonover, J.R.; Kliger, D.S.; Goldbeck, R.A.; et al. Ultrafast and not-so fast dynamics of cytochrome oxidase: The ligand shuttle and its possible functional significance. P. Soc. Photo-Opt. Instrum. Eng 1991, 1432. [Google Scholar] [CrossRef]
- Akiyama, S.; Takahashi, S.; Ishimori, K.; Morishima, I. Stepwise formation of α-helices during cytochrome c folding. Nat. Struct. Biol 2000, 7, 514–520. [Google Scholar]
- Knight, J.B.; Vishwanath, A.; Brody, J.P.; Austin, R.H. Hydrodynamic focusing on a silicon chip: Mixing nanoliters in microseconds. Phys. Rev. Lett 1998, 80, 3863–3866. [Google Scholar]
- Hertzog, D.E.; Michalet, X.; Jager, M.; Kong, X.X.; Santiago, J.G.; Weiss, S.; Bakajin, O. Femtomole mixer for microsecond kinetic studies of protein folding. Anal. Chem 2004, 76, 7169–7178. [Google Scholar]
- Jones, C.M.; Henry, E.R.; Hu, Y.; Chan, C.K.; Luck, S.D.; Bhuyan, A.; Roder, H.; Hofrichter, J.; Eaton, W.A. Fast events in protein folding initiated by nanosecond laser photolysis. Proc. Natl. Acad. Sci. USA 1993, 90, 11860–11864. [Google Scholar]
- Pascher, T.; Chesick, J.P.; Winkler, J.R.; Gray, H.B. Protein folding triggered by electron transfer. Science 1996, 271, 1558–1560. [Google Scholar]
- Chen, E.F.; Kumita, J.R.; Woolley, G.A.; Kliger, D.S. The kinetics of helix unfolding of an azobenzene cross-linked peptide probed by nanosecond time-resolved optical rotatory dispersion. J. Am. Chem. Soc 2003, 125, 12443–12449. [Google Scholar]
- Chen, E.F.; Wen, Y.X.; Lewis, J.W.; Goldbeck, R.A.; Kliger, D.S.; Strauss, C.E.M. Nanosecond laser temperature-jump optical rotatory dispersion: Application to early events in protein folding/ unfolding. Rev. Sci. Instrum 2005, 76, 083120:1–083120:7. [Google Scholar]
- Chen, E.F.; Wood, M.J.; Fink, A.L.; Kliger, D.S. Time-resolved circular dichroism studies of protein folding intermediates of cytochrome c. Biochemistry 1998, 37, 5589–5598. [Google Scholar]
- Bryngelson, J.D.; Wolynes, P.G. Spin glasses and the statistical mechanics of protein folding. Proc. Natl. Acad. Sci. USA 1987, 84, 7524–7528. [Google Scholar]
- Goldbeck, R.A.; Thomas, Y.G.; Chen, E.F.; Esquerra, R.M.; Kliger, D.S. Multiple pathways on a protein folding energy landscape: Kinetic evidence. Proc. Natl. Acad. Sci. USA 1999, 96, 2782–2787. [Google Scholar]
- Abel, C.J.; Goldbeck, R.A.; Latypov, R.F.; Roder, H.; Kliger, D.S. Conformational equilibration time of unfolded protein chains and the folding speed limit. Biochemistry 2007, 46, 4090–4099. [Google Scholar]
- Hagen, S.J.; Hofrichter, J.; Szabo, A.; Eaton, W.A. Diffusion-limited contact formation in unfolded cytochrome c: Estimating the maximum rate of protein folding. Proc. Natl. Acad. Sci. USA 1996, 93, 11615–11617. [Google Scholar]
- Chen, E.F.; Wittung-Stafshede, P.; Kliger, D.S. Far-UV time-resolved circular dichroism detection of electron-transfer-triggered cytochrome c folding. J. Am. Chem. Soc 1999, 121, 3811–3817. [Google Scholar]
- Chen, E.F.; Goldbeck, R.A.; Kliger, D.S. Earliest events in protein folding: Submicrosecond secondary structure formation in reduced cytochrome c. J. Phys. Chem. A 2003, 107, 8149–8155. [Google Scholar]
- Chen, E.F.; Goldbeck, R.A.; Kliger, D.S. The earliest events in protein folding: A structural requirement for ultrafast folding in cytochrome c. J. Am. Chem. Soc 2004, 126, 11175–11181. [Google Scholar]
- Chen, E.; Abel, C.J.; Goldbeck, R.A.; Kliger, D.S. Non-native heme-histidine ligation promotes microsecond time scale secondary structure formation in reduced horse heart cytochrome c. Biochemistry 2007, 46, 12463–12472. [Google Scholar]
- Chen, E.; van Vranken, V.; Kliger, D.S. The folding kinetics of the SDS-induced molten globule form of reduced cytochrome c. Biochemistry 2008, 47, 5450–5459. [Google Scholar]





© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kliger, D.S.; Chen, E.; Goldbeck, R.A. Probing Kinetic Mechanisms of Protein Function and Folding with Time-Resolved Natural and Magnetic Chiroptical Spectroscopies. Int. J. Mol. Sci. 2012, 13, 683-697. https://doi.org/10.3390/ijms13010683
Kliger DS, Chen E, Goldbeck RA. Probing Kinetic Mechanisms of Protein Function and Folding with Time-Resolved Natural and Magnetic Chiroptical Spectroscopies. International Journal of Molecular Sciences. 2012; 13(1):683-697. https://doi.org/10.3390/ijms13010683
Chicago/Turabian StyleKliger, David S., Eefei Chen, and Robert A. Goldbeck. 2012. "Probing Kinetic Mechanisms of Protein Function and Folding with Time-Resolved Natural and Magnetic Chiroptical Spectroscopies" International Journal of Molecular Sciences 13, no. 1: 683-697. https://doi.org/10.3390/ijms13010683