Photoluminescence Intermittency from Single Quantum Dots to Organic Molecules: Emerging Themes

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction and Overview
2. Photoluminescence Intermittency in Quantum Dots
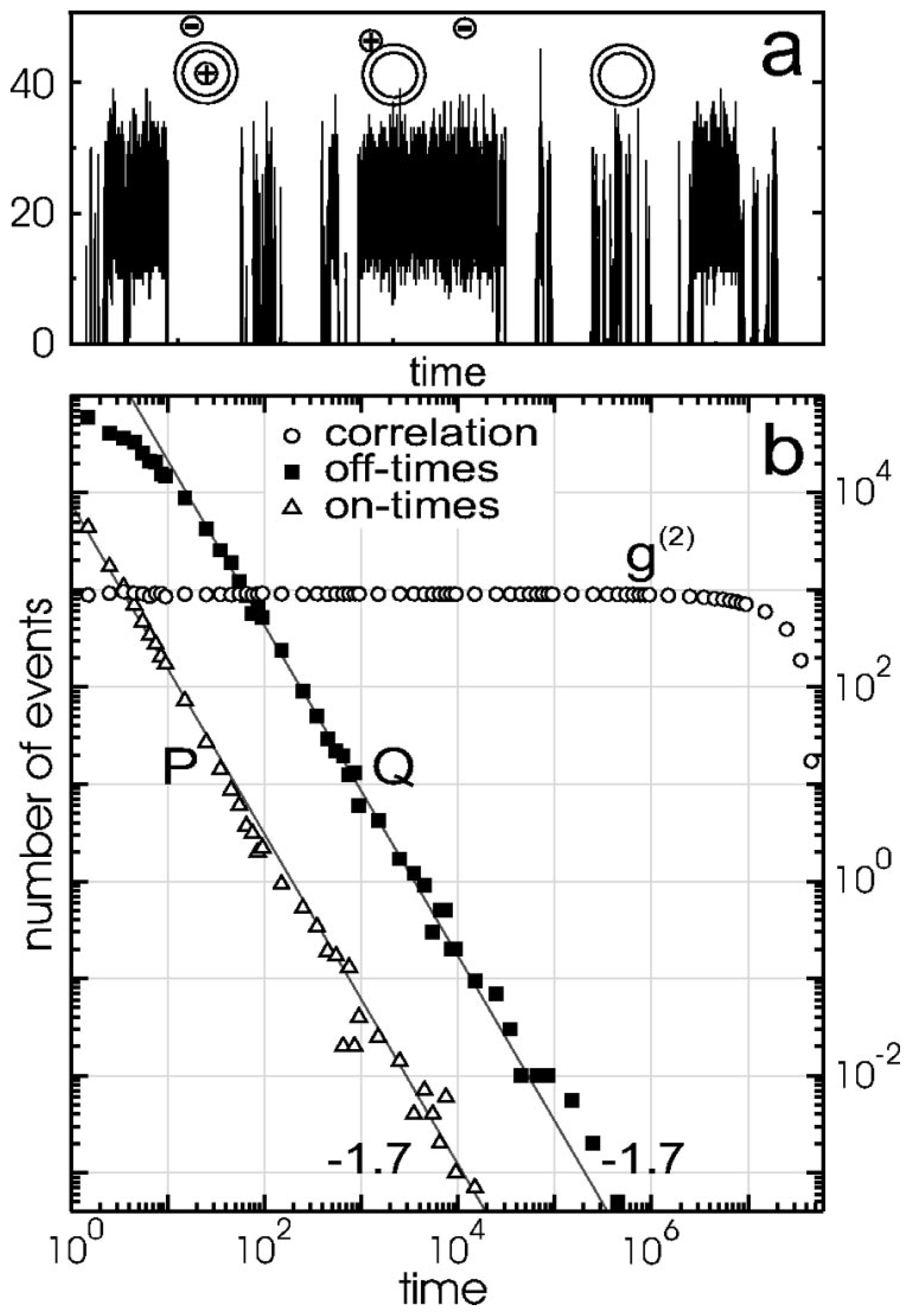
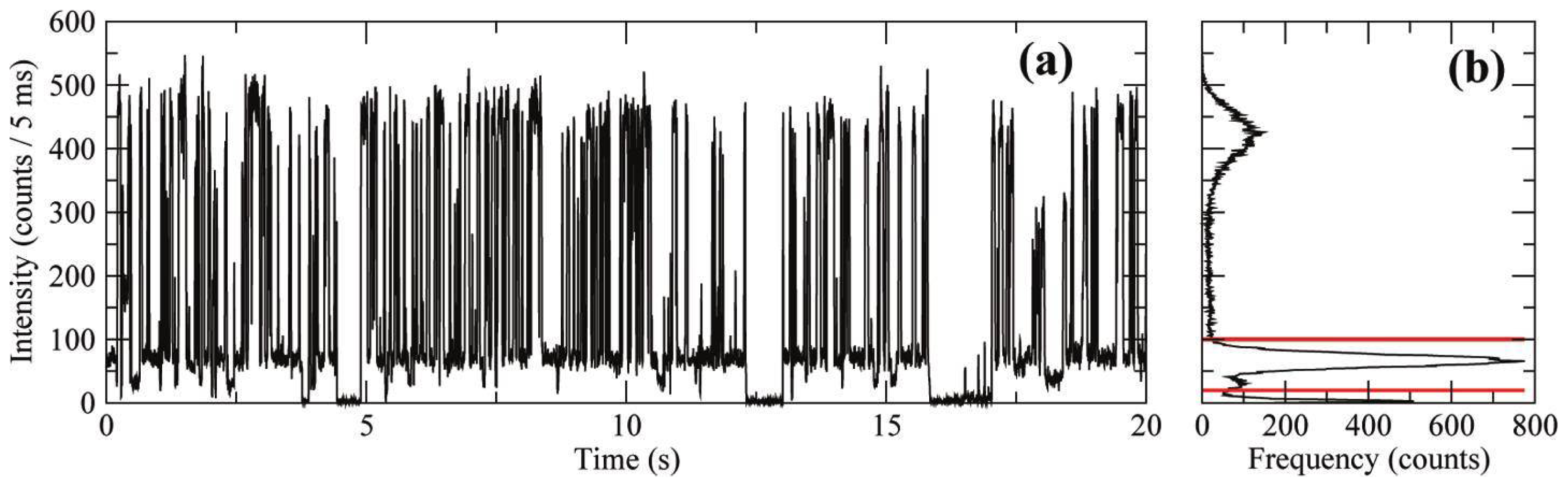
2.1. Early Work
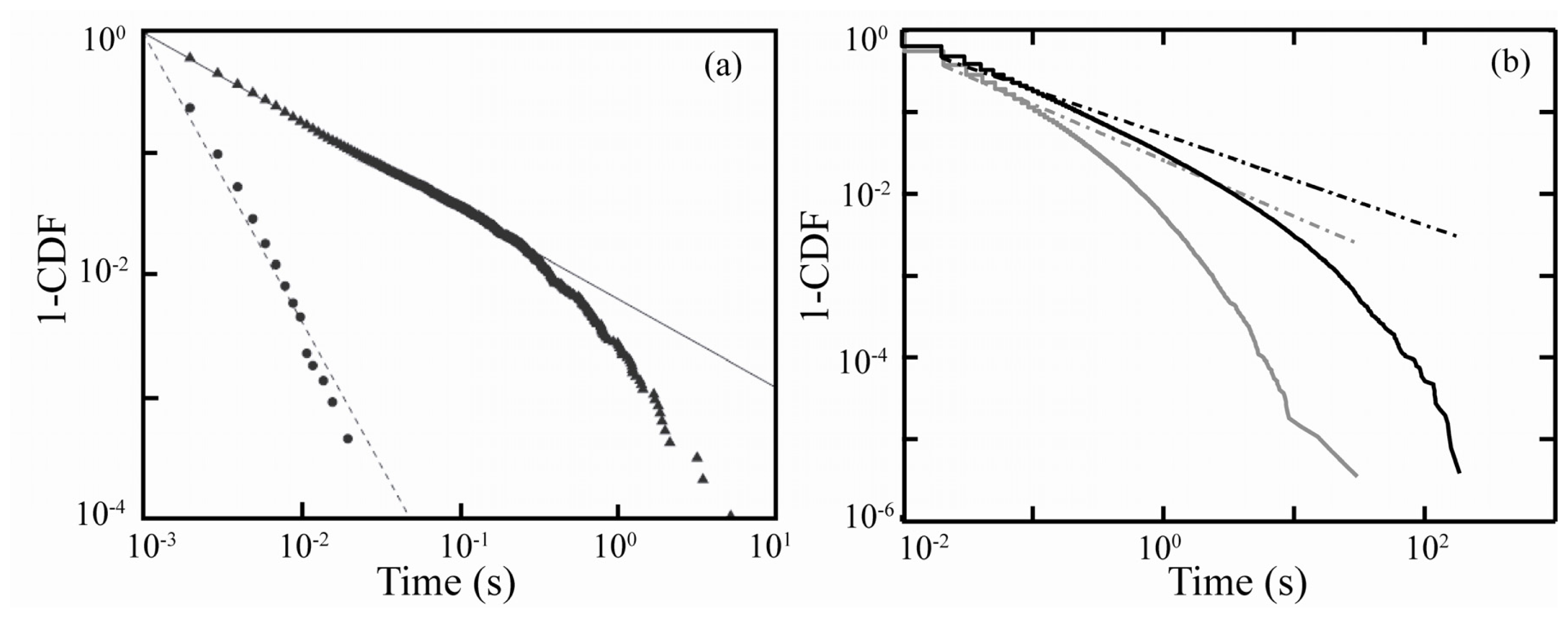
- If electron transfer occurs through tunneling, then both the on- and off-event duration distributions should be temperature invariant.
- Depending on the location of the hole relative to the electron, multiple emission intensity or “gray” states may be observed.
- The power-law exponent depends on the potential energy of the electron in the trap site; therefore, blinking statistics should demonstrate dependence on the dielectric constant of the surrounding environment.
- Shell quality and thickness should impact blinking statistics.
- The elimination of charge trapping sites should result in an absence of PI.
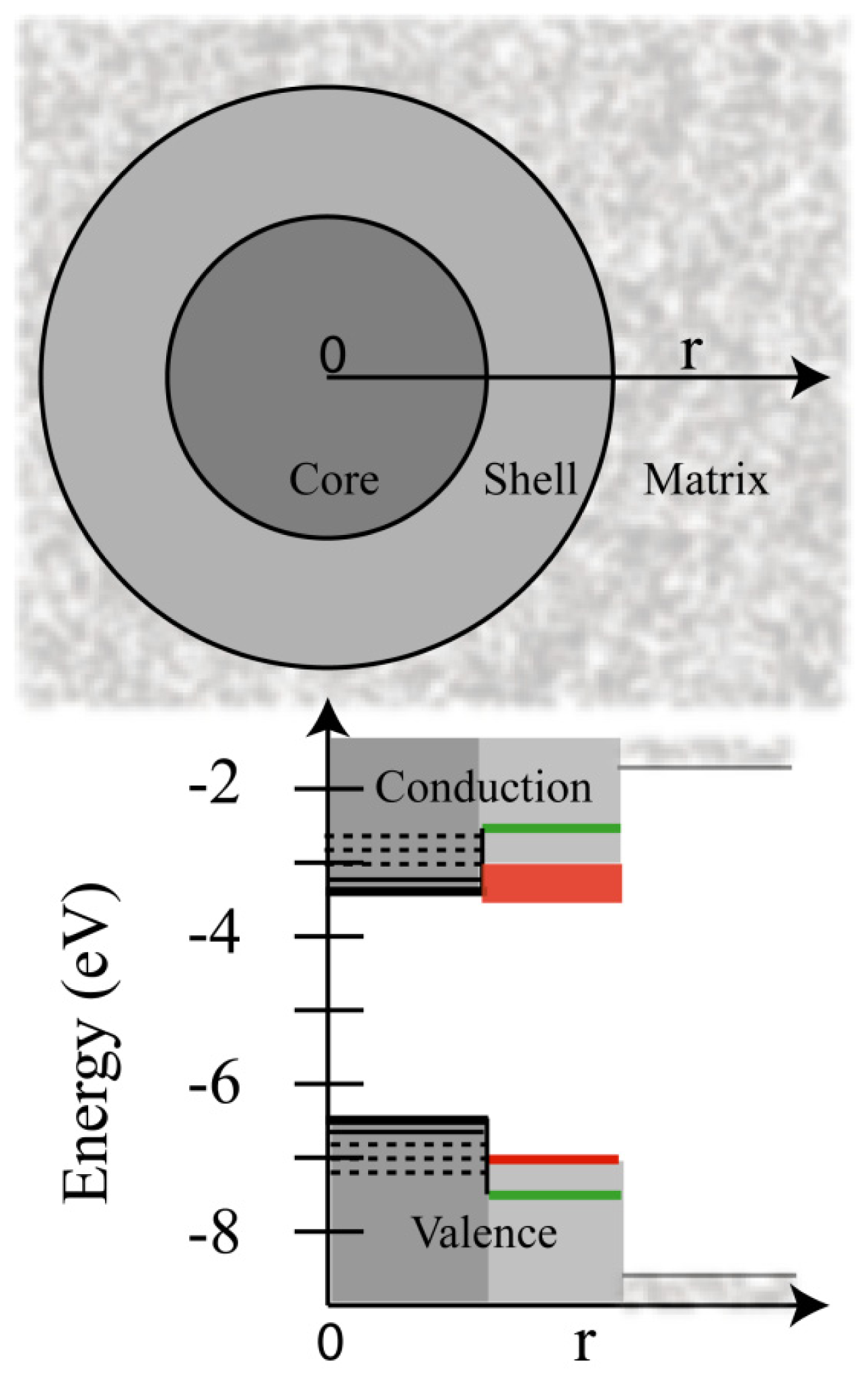
2.2. Blinking Suppression, Gray States, and Broken Power-Laws in Giant QDs
2.3. Blinking Suppression and Exciton Lifetimes
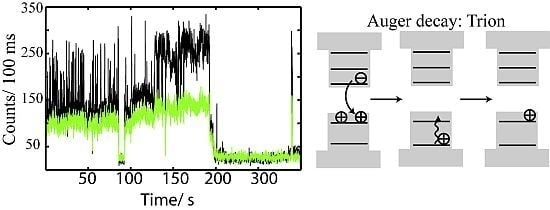
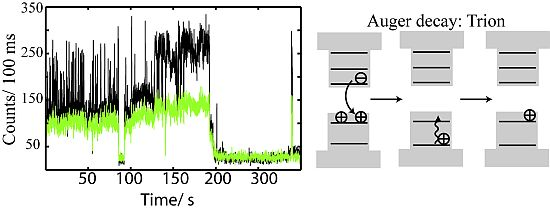
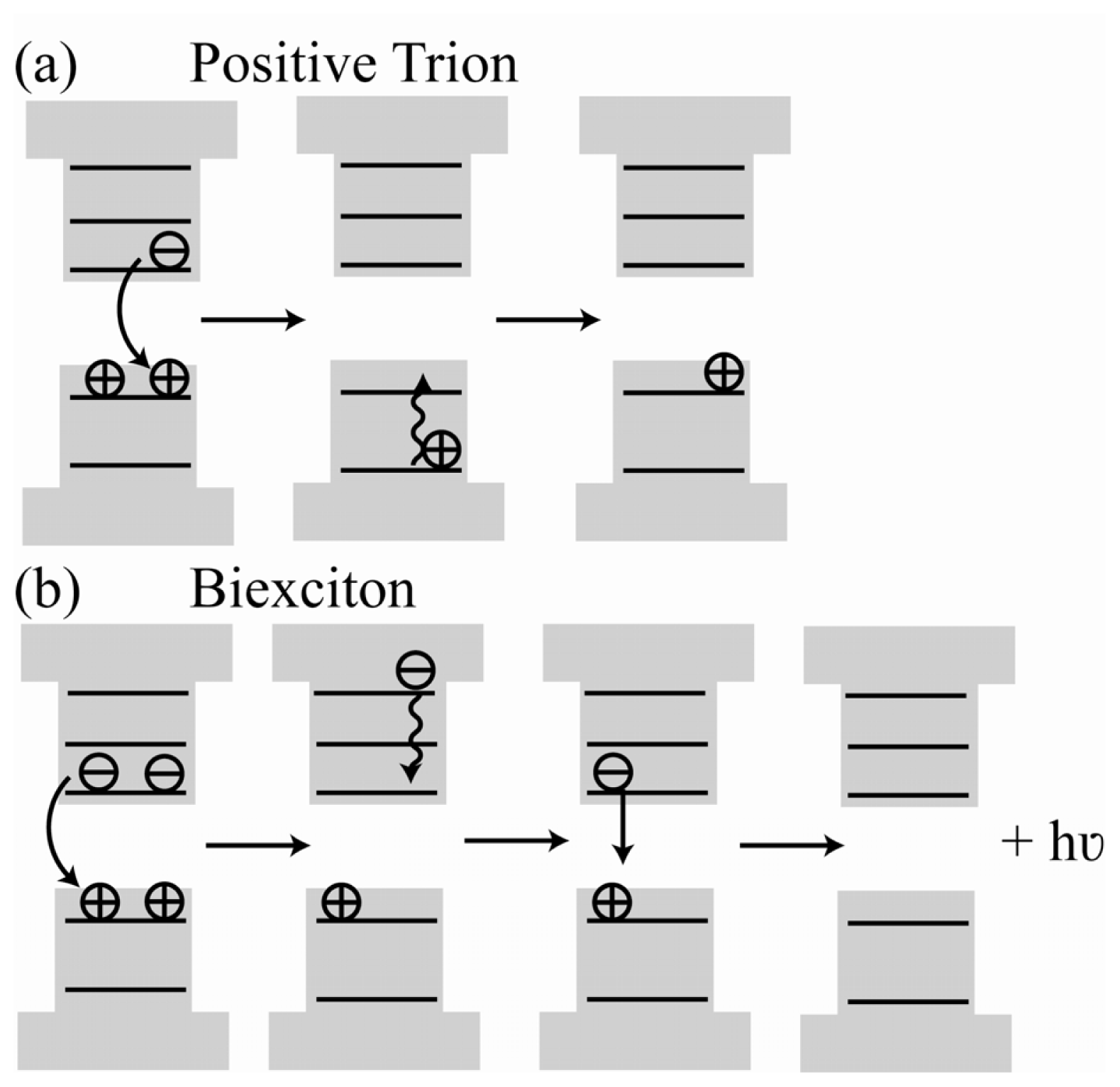
2.4. Auger Decay in Quantum Dots
2.4.1. Auger Decay and the Gray State
2.4.2. Alloying and Volume Effects
2.5. Challenges to the Charging Model for QD PI
2.6. New Mechanisms
2.6.1. Multiple Charging Mechanism
2.6.2. Multiple Recombination Centers
3. Photoluminescence Intermittency in Organic Luminophores
3.1. Electron Transfer
3.2. Proton Transfer
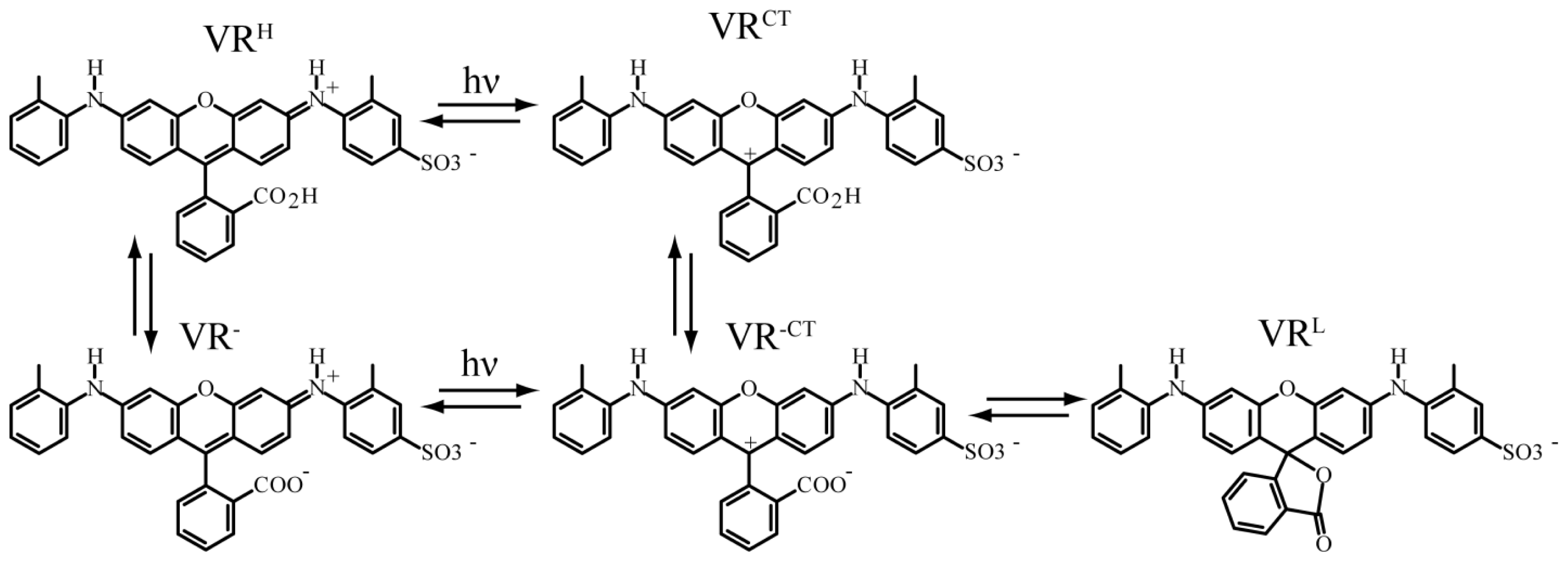
3.3. Spectral Diffusion and Environmental Effects
3.4. Conformational Relaxation
4. Conclusions and Outlook
Acknowledgments
References and Notes
- Moerner, W.E.; Kador, L. Optical-detection and spectroscopy of single molecules in a solid. Phys. Rev. Lett 1989, 62, 2535–2538. [Google Scholar]
- Orrit, M.; Bernard, J. Single pentacene molecules detected by fluorescence excitation in a p-terphenyl crystal. Phys. Rev. Lett 1990, 65, 2716–2719. [Google Scholar]
- Cang, H.; Shan Xu, C.; Yang, H. Progress in single-molecule tracking spectroscopy. Chem. Phys. Lett 2008, 457, 285–291. [Google Scholar]
- Smith, M.B.; Karatekin, E.; Gohlke, A.; Mizuno, H.; Watanabe, N.; Vavylonis, D. Interactive, computer-assisted tracking of speckle trajectories in fluorescence microscopy: Application to actin polymerization and membrane fusion. Biophys. J 2011, 101, 1794–1804. [Google Scholar]
- Rolfe, D.J.; McLachlan, C.I.; Hirsch, M.; Needham, S.R.; Tynan, C.J.; Webb, S.E.D.; Martin-Fernandez, M.L.; Hobson, M.P. Automated multidimensional single molecule fluorescence microscopy feature detection and tracking. Eur. Biophys. J. Biophy 2011, 40, 1167–1186. [Google Scholar]
- Andersson, S.B. A nonlinear controller for three-dimensional tracking of a fluorescent particle in a confocal microscope. Appl. Phys. B: Lasers Opt 2011, 104, 161–173. [Google Scholar]
- Vogelsang, J.; Steinhauer, C.; Forthmann, C.; Stein, I.H.; Person-Skegro, B.; Cordes, T.; Tinnefeld, P. Make them blink: Probes for super-resolution microscopy. ChemPhysChem 2010, 11, 2475–2490. [Google Scholar]
- Biteen, J. Moving toward the future of single-molecule-based super-resolution imaging. Biopolymers 2011, 95, 287–289. [Google Scholar]
- Fukaminato, T. Single-molecule fluorescence photoswitching: Design and synthesis of photoswitchable fluorescent molecules. J. Photochem. Photobiol. C: Photochem. Rev 2011, 12, 177–208. [Google Scholar]
- Tian, Y.; Martinez, M.M.; Pappas, D. Fluorescence correlation spectroscopy: A review of biochemical and microfluidic applications. Appl. Spectrosc 2011, 65, 115A–124A. [Google Scholar]
- Heuff, R.F.; Swift, J.L.; Cramb, D.T. Fluorescence correlation spectroscopy using quantum dots: Advances, challenges and opportunities. Phys. Chem. Chem. Phys 2007, 9, 1870–1880. [Google Scholar]
- Elson, E.L. Fluorescence correlation spectroscopy: Past, present, future. Biophys. J 2011, 101, 2855–2870. [Google Scholar]
- Yahiatene, I.; Doose, S.; Huser, T.; Sauer, M. Correlation-matrix analysis of two-color coincidence events in single-molecule fluorescence experiments. Anal. Chem 2012, 84, 2729–2736. [Google Scholar]
- Sobhy, M.A.; Elshenawy, M.M.; Takahashi, M.; Whitman, B.H.; Walter, N.G.; Hamdan, S.M. Versatile single-molecule multi-color excitation and detection fluorescence setup for studying biomolecular dynamics. Rev. Sci. Instrum 2011, 82, 113702. [Google Scholar]
- Haga, T.; Takahashi, S.; Sonehara, T.; Kumazaki, N.; Anazawa, T. Dual-view imaging system using a wide-range dichroic mirror for simultaneous four-color single-molecule detection. Anal. Chem 2011, 83, 6948–6955. [Google Scholar]
- Hou, X.; Cheng, W. Single-molecule detection using continuous wave excitation of two-photon fluorescence. Opt. Lett 2011, 36, 3185–3187. [Google Scholar]
- Cialla, D.; März, A.; Böhme, R.; Theil, F.; Weber, K.; Schmitt, M.; Popp, J. Surface-enhanced raman spectroscopy (sers): Progress and trends. Anal. Bioanal. Chem 2012, 403, 27–54. [Google Scholar]
- Lohmüller, T.; Iversen, L.; Schmidt, M.; Rhodes, C.; Tu, H.L.; Lin, W.C.; Groves, J.T. Single molecule tracking on supported membranes with arrays of optical nanoantennas. Nano Lett 2012, 12, 1717–1721. [Google Scholar]
- Kuno, M.; Fromm, D.P.; Hamann, H.F.; Gallagher, A.; Nesbitt, D.J. “On”/“off” fluorescence intermittency of single semiconductor quantum dots. J. Chem. Phys 2001, 115, 1028–1040. [Google Scholar]
- Ambrose, W.P.; Basche, T.; Moerner, W.E. Detection and spectroscopy of single pentacene molecules in a para-terphenyl crystal by means of fluorescence excitation. J. Chem. Phys 1991, 95, 7150–7163. [Google Scholar]
- Haase, M.; Hübner, C.G.; Reuther, E.; Herrmann, A.; Müllen, K.; Basché, T. Exponential and power-law kinetics in single-molecule fluorescence intermittency. J. Phys. Chem. B 2004, 108, 10445–10450. [Google Scholar]
- Vogel, M.; Gruber, A.; Wrachtrup, J.; von Borczyskowski, C. Determination of intersystem crossing parameters via observation of quantum jumps on single molecules. J. Phys. Chem 1995, 99, 14915–14917. [Google Scholar]
- Sher, P.H.; Smith, J.M.; Dalgarno, P.A.; Warburton, R.J.; Chen, X.; Dobson, P.J.; Daniels, S.M.; Pickett, N.L.; O’Brien, P. Power law carrier dynamics in semiconductor nanocrystals at nanosecond timescales. Appl. Phys. Lett 2008, 92, 101111. [Google Scholar]
- Kuno, M.; Fromm, D.P.; Hamann, H.F.; Gallagher, A.; Nesbitt, D.J. Nonexponential “blinking” kinetics of single cdse quantum dots: A universal power law behavior. J. Chem. Phys 2000, 112, 3117–3120. [Google Scholar]
- Cichos, F.; von Borczyskowski, C.; Orrit, M. Power-law intermittency of single emitters. Curr. Opin. Colloid Interface Sci 2007, 12, 272–284. [Google Scholar]
- Andresen, M.; Stiel, A.C.; Trowitzsch, S.; Weber, G.; Eggeling, C.; Wahl, M.C.; Hell, S.W.; Jakobs, S. Structural basis for reversible photoswitching in dronpa. Proc. Natl. Acad. Sci. USA 2007, 104, 13005–13009. [Google Scholar]
- Dickson, R.M.; Cubitt, A.B.; Tsien, R.Y.; Moerner, W.E. On/off blinking and switching behaviour of single molecules of green fluorescent protein. Nature 1997, 388, 355–358. [Google Scholar]
- Knappenberger, K.L., Jr; Wong, D.B.; Xu, W.; Schwartzberg, A.M.; Wolcott, A.; Zhang, J.Z.; Leone, S.R. Excitation-wavelength dependence of fluorescence intermittency in cdse nanorods. Acs Nano 2008, 2, 2143–2153. [Google Scholar]
- Ai, N.; Walden-Newman, W.; Song, Q.; Kalliakos, S.; Strauf, S. Suppression of blinking and enhanced exciton emission from individual carbon nanotubes. Acs Nano 2011, 5, 2664–2670. [Google Scholar]
- Lin, H.; Tabaei, S.R.; Thomsson, D.; Mirzov, O.; Larsson, P.-O.; Scheblykin, I.G. Fluorescence blinking, exciton dynamics, and energy transfer domains in single conjugated polymer chains. J. Am. Chem. Soc 2008, 130, 7042–7051. [Google Scholar]
- Hoogenboom, J.P.; Hernando, J.; van Dijk, E.M.H.R.; van Hulst, N.F.; Garcia-Parajo, M.F. Power-law blinking in the fluorescence of single organic molecules. ChemPhysChem 2007, 8, 823–833. [Google Scholar]
- Frantsuzov, P.; Kuno, M.; Janko, B.; Marcus, R.A. Universal emission intermittency in quantum dots, nanorods and nanowires. Nat. Phys 2008, 4, 519–522. [Google Scholar]
- Orrit, M. Chemical and physical aspects of charge transfer in the fluorescence intermittency of single molecules and quantum dots. Photochem. Photobiol. Sci 2010, 9, 637–642. [Google Scholar]
- Kulzer, F.; Xia, T.; Orrit, M. Single molecules as optical nanoprobes for soft and complex matter. Angew. Chem. Int. Ed 2010, 49, 854–866. [Google Scholar]
- Efros, A.L.; Rosen, M. Random telegraph signal in the photoluminescence intensity of a single quantum dot. Phys. Rev. Lett 1997, 78, 1110–1113. [Google Scholar]
- Verberk, R.; van Oijen, A.M.; Orrit, M. Simple model for the power-law blinking of single semiconductor nanocrystals. Phys. Rev. B 2002, 66, 233202. [Google Scholar]
- Ko, H.C.; Yuan, C.T.; Lin, S.H.; Tang, J. Blinking suppression of single quantum dots in agarose gel. Appl. Phys. Lett 2010, 96, 012104. [Google Scholar]
- Durisic, N.; Godin, A.G.; Walters, D.; Gruetter, P.; Wiseman, P.W.; Heyes, C.D. Probing the “dark” fraction of core-shell quantum dots by ensemble and single particle ph-dependent spectroscopy. ACS Nano 2011, 5, 9062–9073. [Google Scholar]
- Krasselt, C.; Schuster, J.; von Borczyskowski, C. Photoinduced hole trapping in single semiconductor quantum dots at specific sites at silicon oxide interfaces. Phys. Chem. Chem. Phys 2011, 13, 17084–17092. [Google Scholar]
- Roy, S.; Aguirre, A.; Higgins, D.A.; Chikan, V. Investigation of charge transfer interactions in cdse nanorod p3ht/pmma blends by optical microscopy. J. Phys. Chem. C 2012, 116, 3153–3160. [Google Scholar]
- Fujiwara, H.; Ohta, H.; Chiba, T.; Sasaki, K. Temporal response analysis of trap states of single cdse/zns quantum dots on a thin metal substrate. J. Photochem. Photobiol. A Chem 2011, 221, 160–163. [Google Scholar]
- Mandal, A.; Tamai, N. Suppressed blinking behavior of thioglycolic acid capped cdte quantum dot by amine functionalization. Appl. Phys. Lett 2011, 99, 263111. [Google Scholar]
- Fomenko, V.; Nesbitt, D.J. Solution control of radiative and nonradiative lifetimes: A novel contribution to quantum dot blinking suppression. Nano Lett 2008, 8, 287–293. [Google Scholar]
- Tang, J.; Marcus, R.A. Photoinduced spectral diffusion and diffusion-controlled electron transfer reactions in fluorescence intermittency of quantum dots. J. Chin. Chem. Soc 2006, 53, 1–13. [Google Scholar]
- Issac, A.; Krasselt, C.; Cichos, F.; von Borczyskowski, C. Influence of the dielectric environment on the photoluminescence intermittency of cdse quantum dots. ChemPhysChem 2012, 13, 3223–3230. [Google Scholar]
- Hartmann, T.; Reineker, P.; Yudson, V.I. Auger release of a deeply trapped carrier in a quantum dot. Phys. Rev. B 2011, 84, 245317. [Google Scholar]
- Hartmann, T.; Yudson, V.I.; Reineker, P. Model for the off-time distribution in blinking quantum dots. J. Lumin 2011, 131, 379–381. [Google Scholar]
- Tang, J.; Marcus, R.A. Mechanisms of fluorescence blinking in semiconductor nanocrystal quantum dots. J. Chem. Phys 2005, 123, 054704. [Google Scholar]
- Tang, J.; Marcus, R.A. Single particle versus ensemble average: From power-law intermittency of a single quantum dot to quasistretched exponential fluorescence decay of an ensemble. J. Chem. Phys 2005, 123, 204511. [Google Scholar]
- Spinicelli, P.; Buil, S.; Quelin, X.; Mahler, B.; Dubertret, B.; Hermier, J.P. Bright and grey states in CdSe-CdS nanocrystals exhibiting strongly reduced blinking. Phys. Rev. Lett 2009, 102, 136801. [Google Scholar]
- Mahler, B.; Spinicelli, P.; Buil, S.; Quelin, X.; Hermier, J.-P.; Dubertret, B. Towards non-blinking colloidal quantum dots. Nat. Mater 2008, 7, 659–664. [Google Scholar]
- Gómez, D.E.; van Embden, J.; Mulvaney, P.; Fernée, M.J.; Rubinsztein-Dunlop, H. Exciton–trion transitions in single cdse–cds core–shell nanocrystals. Acs Nano 2009, 3, 2281–2287. [Google Scholar]
- Zhang, K.; Chang, H.Y.; Fu, A.H.; Alivisatos, A.P.; Yang, H. Continuous distribution of emission states from single cdse/zns quantum dots. Nano Lett 2006, 6, 843–847. [Google Scholar]
- Montiel, D.; Yang, H. Observation of correlated emission intensity and polarization fluctuations in single cdse/zns quantum dots. J. Phys. Chem. A 2008, 112, 9352–9355. [Google Scholar]
- Similar elimination of long off -durations was reported (almost simultaneously) in giant multishell CdSe quantum dots by Chen et al. in 2008. See reference 64.
- Riley, E.A.; Hess, C.M.; Whitham, P.J.; Reid, P.J. Beyond power laws: A new approach for analyzing single molecule photoluminescence intermittency. J. Chem. Phys 2012, 136, 184508. [Google Scholar]
- Amecke, N.; Cichos, F. Intermediate intensity levels during the emission intermittency of single cdse/zns quantum dots. J. Lumin 2011, 131, 375–378. [Google Scholar]
- Reiss, P.; Protière, M.; Li, L. Core/shell semiconductor nanocrystals. Small 2009, 5, 154–168. [Google Scholar]
- Hummon, M.R.; Stollenwerk, A.J.; Narayanamurti, V.; Anikeeva, P.O.; Panzer, M.J.; Wood, V.; Bulović, V. Measuring charge trap occupation and energy level in cdse/zns quantum dots using a scanning tunneling microscope. Phys. Rev. B 2010, 81, 115439. [Google Scholar]
- Steiner, D.; Dorfs, D.; Banin, U.; Della Sala, F.; Manna, L.; Millo, O. Determination of band offsets in heterostructured colloidal nanorods using scanning tunneling spectroscopy. Nano Lett 2008, 8, 2954–2958. [Google Scholar]
- Malko, A.V.; Park, Y.-S.; Sampat, S.; Galland, C.; Vela, J.; Chen, Y.; Hollingsworth, J.A.; Klimov, V.I.; Htoon, H. Pump-intensity- and shell-thickness-dependent evolution of photoluminescence blinking in individual core/shell cdse/cds nanocrystals. Nano Lett 2011, 11, 5213–5218. [Google Scholar]
- García-Santamaría, F.; Chen, Y.; Vela, J.; Schaller, R.D.; Hollingsworth, J.A.; Klimov, V.I. Suppressed auger recombination in “giant” nanocrystals boosts optical gain performance. Nano Lett 2009, 9, 3482–3488. [Google Scholar]
- Wang, X.; Ren, X.; Kahen, K.; Hahn, M.A.; Rajeswaran, M.; Maccagnano-Zacher, S.; Silcox, J.; Cragg, G.E.; Efros, A.L.; Krauss, T.D. Non-blinking semiconductor nanocrystals. Nature 2009, 459, 686–689. [Google Scholar]
- Chen, Y.; Vela, J.; Htoon, H.; Casson, J.L.; Werder, D.J.; Bussian, D.A.; Klimov, V.I.; Hollingsworth, J.A. “Giant” multishell cdse nanocrystal quantum dots with suppressed blinking. J. Am. Chem. Soc 2008, 130, 5026–5027. [Google Scholar]
- Cragg, G.E.; Efros, A.L. Suppression of auger processes in confined structures. Nano Lett 2010, 10, 313–317. [Google Scholar]
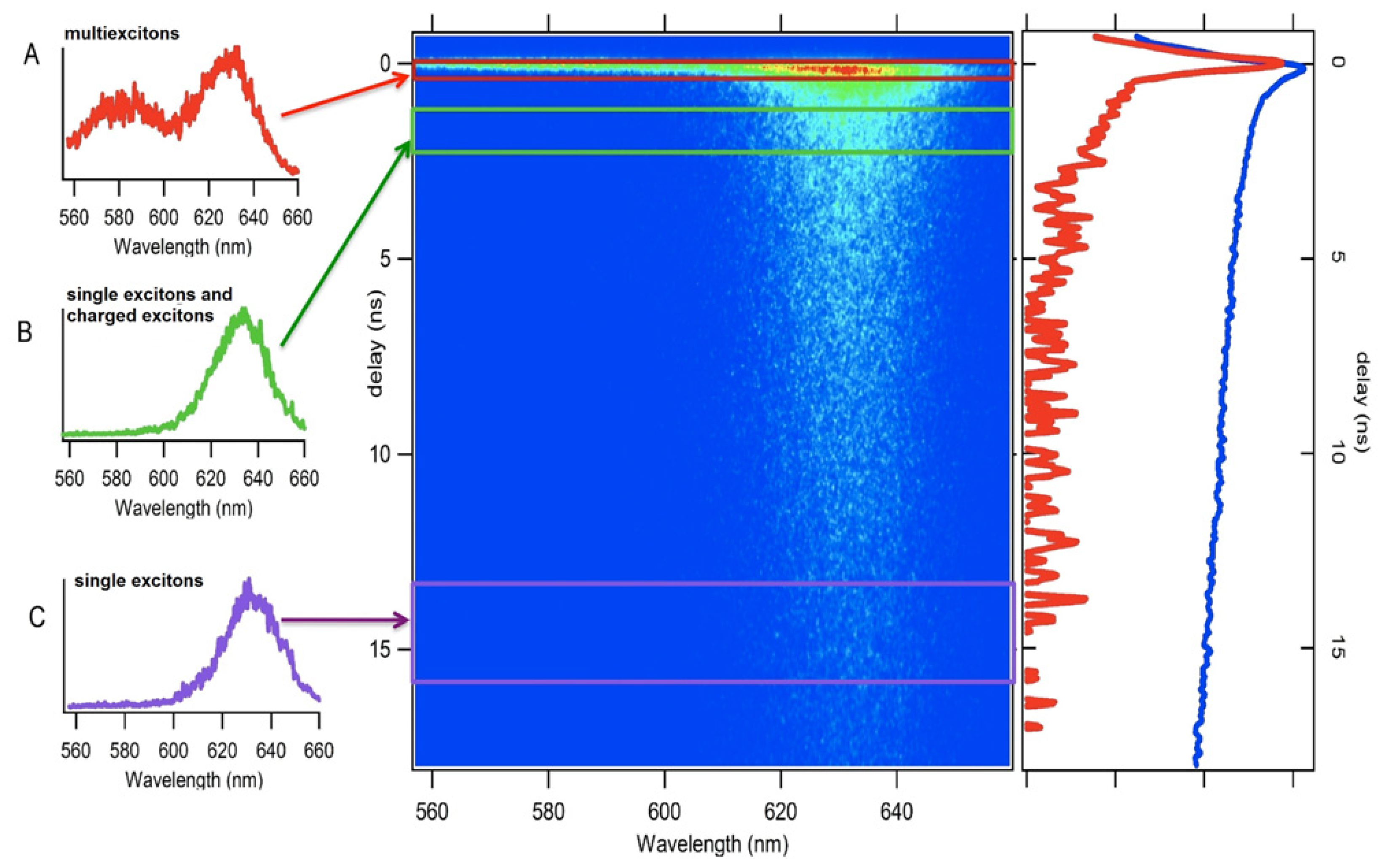
- Htoon, H.; Malko, A.V.; Bussian, D.; Vela, J.; Chen, Y.; Hollingsworth, J.A.; Klimov, V.I. Highly emissive multiexcitons in steady-state photoluminescence of individual “giant” cdse/cds core/shell nanocrystals. Nano Lett 2010, 10, 2401–2407. [Google Scholar]
- Park, Y.S.; Malko, A.V.; Vela, J.; Chen, Y.; Ghosh, Y.; Garcia-Santamaria, F.; Hollingsworth, J.A.; Klimov, V.I.; Htoon, H. Near-unity quantum yields of biexciton emission from cdse/cds nanocrystals measured using single-particle spectroscopy. Phys. Rev. Lett 2011, 106, 187401. [Google Scholar]
- Zhao, J.; Nair, G.; Fisher, B.R.; Bawendi, M.G. Challenge to the charging model of semiconductor-nanocrystal fluorescence intermittency from off-state quantum yields and multiexciton blinking. Phys. Rev. Lett 2010, 104, 157403. [Google Scholar]
- Marceddu, M.; Saba, M.; Quochi, F.; Lai, A.; Huang, J.; Talapin, D.V.; Mura, A.; Bongiovanni, G. Charged excitons, auger recombination and optical gain in cdse/cds nanocrystals. Nanotechnology 2012, 23, 015201. [Google Scholar]
- Deutsch, Z.; Avidan, A.; Pinkas, I.; Oron, D. Energetics and dynamics of exciton-exciton interactions in compound colloidal semiconductor quantum dots. Phys. Chem. Chem. Phys 2011, 13, 3210–3219. [Google Scholar]
- Garcia-Santamaria et al. state the opposite relationship which appears to be in error.
- Louyer, Y.; Biadala, L.; Trebbia, J.B.; Fernee, M.J.; Tamarat, P.; Lounis, B. Efficient biexciton emission in elongated cdse/zns nanocrystals. Nano Lett 2011, 11, 4370–4375. [Google Scholar]
- Rainò, G.; Stöferle, T.; Moreels, I.; Gomes, R.; Kamal, J.S.; Hens, Z.; Mahrt, R.F. Probing the wave function delocalization in cdse/cds dot-in-rod nanocrystals by time- and temperature-resolved spectroscopy. Acs Nano 2011, 5, 4031–4036. [Google Scholar]
- Zavelani-Rossi, M.; Lupo, M.G.; Tassone, F.; Manna, L.; Lanzani, G. Suppression of biexciton auger recombination in cdse/cds dot/rods: Role of the electronic structure in the carrier dynamics. Nano Lett 2010, 10, 3142–3150. [Google Scholar]
- Ghosh, Y.; Mangum, B.D.; Casson, J.L.; Williams, D.J.; Htoon, H.; Hollingsworth, J.A. New insights into the complexities of shell growth and the strong influence of particle volume in nonblinking “giant” core/shell nanocrystal quantum dots. J. Am. Chem. Soc 2012, 134, 9634–9643. [Google Scholar]
- Krauss, T.D.; Peterson, J.J. Bright future for fluorescence blinking in semiconductor nanocrystals. J. Phys. Chem. Lett 2010, 1, 1377–1382. [Google Scholar]
- García-Santamaría, F.; Brovelli, S.; Viswanatha, R.; Hollingsworth, J.A.; Htoon, H.; Crooker, S.A.; Klimov, V.I. Breakdown of volume scaling in auger recombination in cdse/cds heteronanocrystals: The role of the core-shell interface. Nano Lett 2011, 11, 687–693. [Google Scholar]
- Vela, J.; Htoon, H.; Chen, Y.; Park, Y.-S.; Ghosh, Y.; Goodwin, P.M.; Werner, J.H.; Wells, N.P.; Casson, J.L.; Hollingsworth, J.A. Effect of shell thickness and composition on blinking suppression and the blinking mechanism in ‘giant’ CdSe/CdS nanocrystal quantum dots. J. Biophoton 2010, 3, 706–717. [Google Scholar]
- She, C.; Demortiere, A.; Shevchenko, E.V.; Pelton, M. Using shape to control photoluminescence from cdse/cds core/shell nanorods. J. Phys. Chem. Lett 2011, 2, 1469–1475. [Google Scholar]
- Fisher, B.; Caruge, J.M.; Zehnder, D.; Bawendi, M. Room-temperature ordered photon emission from multiexciton states in single cdse core-shell nanocrystals. Phys. Rev. Lett 2005, 94, 087403. [Google Scholar]
- Nair, G.; Zhao, J.; Bawendi, M.G. Biexciton quantum yield of single semiconductor nanocrystals from photon statistics. Nano Lett 2011, 11, 1136–1140. [Google Scholar]
- Cordones, A.A.; Bixby, T.J.; Leone, S.R. Direct measurement of off-state trapping rate fluctuations in single quantum dot fluorescence. Nano Lett 2011, 11, 3366–3369. [Google Scholar]
- Rosen, S.; Schwartz, O.; Oron, D. Transient fluorescence of the off state in blinking cdse/cds/zns semiconductor nanocrystals is not governed by auger recombination. Phys. Rev. Lett 2010, 104, 157404. [Google Scholar]
- Jha, P.P.; Guyot-Sionnest, P. Trion decay in colloidal quantum dots. ACS Nano 2009, 3, 1011–1015. [Google Scholar]
- Jha, P.P.; Guyot-Sionnest, P. Electrochemical switching of the photoluminescence of single quantum dots. J. Phys. Chem. C 2010, 114, 21138–21141. [Google Scholar]
- Galland, C.; Ghosh, Y.; Steinbrück, A.; Sykora, M.; Hollingsworth, J.A.; Klimov, V.I.; Htoon, H. Two types of luminescence blinking revealed by spectroelectrochemistry of single quantum dots. Nature 2011, 479, 203–207. [Google Scholar]
- Qin, W.; Shah, R.A.; Guyot-Sionnest, P. Cdses/zns alloyed nanocrystal lifetime and blinking studies under electrochemical control. Acs Nano 2012, 6, 912–918. [Google Scholar]
- Baker, T.A.; Rouge, J.L.; Nesbitt, D.J. Single molecule studies of quantum dot fluorescence intermittency: Evidence for both dark and light-assisted blinking dynamics. Mol. Phys 2009, 107, 1867–1878. [Google Scholar]
- Osborne, M.A.; Lees, S.F. Quantum dot photoluminescence activation and decay: Dark, bright, and reversible populations in ZnS-capped CdSe nanocrystals. Acs Nano 2011, 5, 8295–8304. [Google Scholar]
- Califano, M. Off-state quantum yields in the presence of surface trap states in cdse nanocrystals: The inadequacy of the charging model to explain blinking. J. Phys. Chem. C 2011, 115, 18051–18054. [Google Scholar]
- Frantsuzov, P.A.; Volkán-Kacsó, S.; Jankó, B. Model of fluorescence intermittency of single colloidal semiconductor quantum dots using multiple recombination centers. Phys. Rev. Lett 2009, 103, 207402. [Google Scholar]
- Frantsuzov, P.A.; Marcus, R.A. Explanation of quantum dot blinking without the long-lived trap hypothesis. Phys. Rev. B 2005, 72, 155321. [Google Scholar]
- Volkán-Kacsó, S.; Frantsuzov, P.A.; Jankó, B. Correlations between subsequent blinking events in single quantum dots. Nano Lett 2010, 10, 2761–2765. [Google Scholar]
- Plakhotnik, T.; Fernée, M.J.; Littleton, B.; Rubinsztein-Dunlop, H.; Potzner, C.; Mulvaney, P. Anomalous power laws of spectral diffusion in quantum dots: A connection to luminescence intermittency. Phys. Rev. Lett 2010, 105, 167402. [Google Scholar]
- Bernard, J.; Fleury, L.; Talon, H.; Orrit, M. Photon bunching in the fluorescence from single molecules: A probe for intersystem crossing. J. Chem. Phys 1993, 98, 850–859. [Google Scholar]
- Basché, T.; Kummer, S.; Bräuchle, C. Direct spectroscopic observation of quantum jumps of a single-molecule. Nature 1995, 373, 132–134. [Google Scholar]
- Yip, W.T.; Hu, D.H.; Yu, J.; Vanden Bout, D.A.; Barbara, P.F. Classifying the photophysical dynamics of single- and multiple-chromophoric molecules by single molecule spectroscopy. J. Phys. Chem. A 1998, 102, 7564–7575. [Google Scholar]
- Weston, K.D.; Carson, P.J.; DeAro, J.A.; Buratto, S.K. Single-molecule detection fluorescence of surface-bound species in vacuum. Chem. Phys. Lett 1999, 308, 58–64. [Google Scholar]
- Ha, T.; Enderle, T.; Chemla, D.S.; Selvin, P.R.; Weiss, S. Quantum jumps of single molecules at room temperature. Chem. Phys. Lett 1997, 271, 1–5. [Google Scholar]
- Zondervan, R.; Kulzer, F.; Orlinskii, S.B.; Orrit, M. Photoblinking of rhodamine 6g in poly(vinyl alcohol): Radical dark state formed through the triplet. J. Phys. Chem. A 2003, 107, 6770–6776. [Google Scholar]
- Yeow, E.K.L.; Melnikov, S.M.; Bell, T.D.M.; De Schryver, F.C.; Hofkens, J. Characterizing the fluorescence intermittency and photobleaching kinetics of dye molecules immobilized on a glass surface. J. Phys. Chem. A 2006, 110, 1726–1734. [Google Scholar]
- Barbon, A.; Bott, E.D.; Brustolon, M.; Fabris, M.; Kahr, B.; Kaminsky, W.; Reid, P.J.; Wong, S.M.; Wustholz, K.L.; Zanre, R. Triplet states of the nonlinear optical chromophore dcm in single crystals of potassium hydrogen phthalate and their relationship to single-molecule dark states. J. Am. Chem. Soc 2009, 131, 11548–11557. [Google Scholar]
- Haase, M.; Hübner, C.G.; Nolde, F.; Müllen, K.; Basché, T. Photoblinking and photobleaching of rylene diimide dyes. Phys. Chem. Chem. Phys 2011, 13, 1776–1785. [Google Scholar]
- Clifford, J.N.; Bell, T.D.M.; Tinnefeld, P.; Heilemann, M.; Melnikov, S.M.; Hotta, J.; Sliwa, M.; Dedecker, P.; Sauer, M.; Hofkens, J.; et al. Fluorescence of single molecules in polymer films: Sensitivity of blinking to local environment. J. Phys. Chem. B 2007, 111, 6987–6991. [Google Scholar]
- Yasuda, M.; Iida, A.; Ito, S.; Miyasaka, H. Fluorescence detection of single guest molecules in ultrasmall droplets of nonpolar solvent. Phys. Chem. Chem. Phys 2012, 14, 345–352. [Google Scholar]
- Kawai, K.; Matsutani, E.; Maruyama, A.; Majima, T. Probing the charge-transfer dynamics in DNA at the single-molecule level. J. Am. Chem. Soc 2011, 133, 15568–15577. [Google Scholar]
- Stein, I.H.; Capone, S.; Smit, J.H.; Baumann, F.; Cordes, T.; Tinnefeld, P. Linking single-molecule blinking to chromophore structure and redox potentials. Chemphyschem 2012, 13, 931–937. [Google Scholar]
- Issac, A.; Hildner, R.; Ernst, D.; Hippius, C.; Wurthner, F.; Kohler, J. Single molecule studies of calix[4]arene-linked perylene bisimide dimers: Relationship between blinking, lifetime and/or spectral fluctuations. Phys. Chem. Chem. Phys 2012, 14, 10789–10798. [Google Scholar]
- Bott, E.D.; Riley, E.A.; Kahr, B.; Reid, P.J. Unraveling the dispersed kinetics of dichlorofluorescein in potassium hydrogen phthalate crystals. J. Phys. Chem. A 2010, 114, 7331–7337. [Google Scholar]
- Bott, E.D.; Riley, E.A.; Kahr, B.; Reid, P.J. Proton-transfer mechanism for dispersed decay kinetics of single molecules isolated in potassium hydrogen phthalate. ACS Nano 2009, 3, 2403–2411. [Google Scholar]
- Wustholz, K.L.; Bott, E.D.; Isborn, C.M.; Li, X.S.; Kahr, B.; Reid, P.J. Dispersive kinetics from single molecules oriented in single crystals of potassium acid phthalate. J. Phys. Chem. C 2007, 111, 9146–9156. [Google Scholar]
- Wustholz, K.L.; Bott, E.D.; Kahr, B.; Reid, P.J. Memory and spectral diffusion in single-molecule emission. J. Phys. Chem. C 2008, 112, 7877–7885. [Google Scholar]
- Bullard, T.; Wustholz, K.L.; Bott, E.D.; Robertson, M.; Reid, P.J.; Kahr, B. Role of kinks in dyeing crystals: Confocal luminescence microscopy from single molecules to square centimeters. Cryst. Growth Des 2009, 9, 982–990. [Google Scholar]
- Sluss, D.; Bingham, C.; Burr, M.; Bott, E.D.; Riley, E.A.; Reid, P.J. Temperature-dependent fluorescence intermittency for single molecules of violamine r in poly(vinyl alcohol). J. Mater. Chem 2009, 19, 7561–7566. [Google Scholar]
- Riley, E.A.; Bingham, C.; Bott, E.D.; Kahr, B.; Reid, P.J. Two mechanisms for fluorescence intermittency of single violamine r molecules. Phys. Chem. Chem. Phys 2011, 13, 1879–1887. [Google Scholar]
- Riley, E.A.; Hess, C.M.; Pioquinto, J.R.L.; Kaminsky, W.; Kahr, B.; Reid, P.J. Proton transfer and photoluminescence intermittency of single emitters in dyed crystals. J. Phys. Chem. B 2012. [Google Scholar] [CrossRef]
- Krause, S.; Kowerko, D.; Börner, R.; Hübner, C.G.; von Borczyskowski, C. Spectral diffusion of single molecules in a hierarchical energy landscape. ChemPhysChem 2011, 12, 303–312. [Google Scholar]
- Masuo, S.; Yamane, Y.; Machida, S.; Itaya, A. Fluorescence behavior of individual charge-transfer complexes revealed by single-molecule fluorescence spectroscopy: Influence of the host polymer matrix. J. Photochem. Photobiol. A Chem 2012, 227, 65–70. [Google Scholar]
- Suzuki, K.; Habuchi, S.; Vacha, M. Blinking of single dye molecules in a polymer matrix is correlated with free volume in polymers. Chem. Phys. Lett 2011, 505, 157–160. [Google Scholar]
- Yoo, H.; Bahng, H.W.; Wasielewski, M.R.; Kim, D. Polymer matrix dependence of conformational dynamics within a pi-stacked perylenediimide dimer and trimer revealed by single molecule fluorescence spectroscopy. Phys. Chem. Chem. Phys 2012, 14, 2001–2007. [Google Scholar]
- Lee, J.-E.; Yang, J.; Kim, D. Single-molecule fluorescence dynamics of a butadiyne-linked porphyrin dimer: The effect of conformational flexibility in host polymers. Faraday Discuss 2012, 155, 277–288. [Google Scholar]
- Kruger, T.P.J.; Ilioaia, C.; Valkunas, L.; van Grondelle, R. Fluorescence intermittency from the main plant light-harvesting complex: Sensitivity to the local environment. J. Phys. Chem. B 2011, 115, 5083–5095. [Google Scholar]
- Kobayashi, H.; Onda, S.; Furumaki, S.; Habuchi, S.; Vacha, M. A single-molecule approach to conformation and photophysics of conjugated polymers. Chem. Phys. Lett 2012, 528, 1–6. [Google Scholar]
- Moerner, W.E. Single-molecule optical spectroscopy of autofluorescent proteins. J. Chem. Phys 2002, 117, 10925–10937. [Google Scholar]
- Voityuk, A.A.; Michel-Beyerle, M.E.; Rösch, N. Structure and rotation barriers for ground and excited states of the isolated chromophore of the green fluorescent protein. Chem. Phys. Lett 1998, 296, 269–276. [Google Scholar]
- Weber, W.; Helms, V.; McCammon, J.A.; Langhoff, P.W. Shedding light on the dark and weakly fluorescent states of green fluorescent proteins. Proc. Natl. Acad. Sci. USA 1999, 96, 6177–6182. [Google Scholar]
- Abbandonato, G.; Signore, G.; Nifosí, R.; Voliani, V.; Bizzarri, R.; Beltram, F. Cis-trans photoisomerization properties of gfp chromophore analogs. Eur. Biophys. J. Biophy 2011, 40, 1205–1214. [Google Scholar]










© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Riley, E.A.; Hess, C.M.; Reid, P.J. Photoluminescence Intermittency from Single Quantum Dots to Organic Molecules: Emerging Themes. Int. J. Mol. Sci. 2012, 13, 12487-12518. https://doi.org/10.3390/ijms131012487
Riley EA, Hess CM, Reid PJ. Photoluminescence Intermittency from Single Quantum Dots to Organic Molecules: Emerging Themes. International Journal of Molecular Sciences. 2012; 13(10):12487-12518. https://doi.org/10.3390/ijms131012487
Chicago/Turabian StyleRiley, Erin A., Chelsea M. Hess, and Philip J. Reid. 2012. "Photoluminescence Intermittency from Single Quantum Dots to Organic Molecules: Emerging Themes" International Journal of Molecular Sciences 13, no. 10: 12487-12518. https://doi.org/10.3390/ijms131012487