Protection against Ischemia-Induced Oxidative Stress Conferred by Vagal Stimulation in the Rat Heart: Involvement of the AMPK-PKC Pathway

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
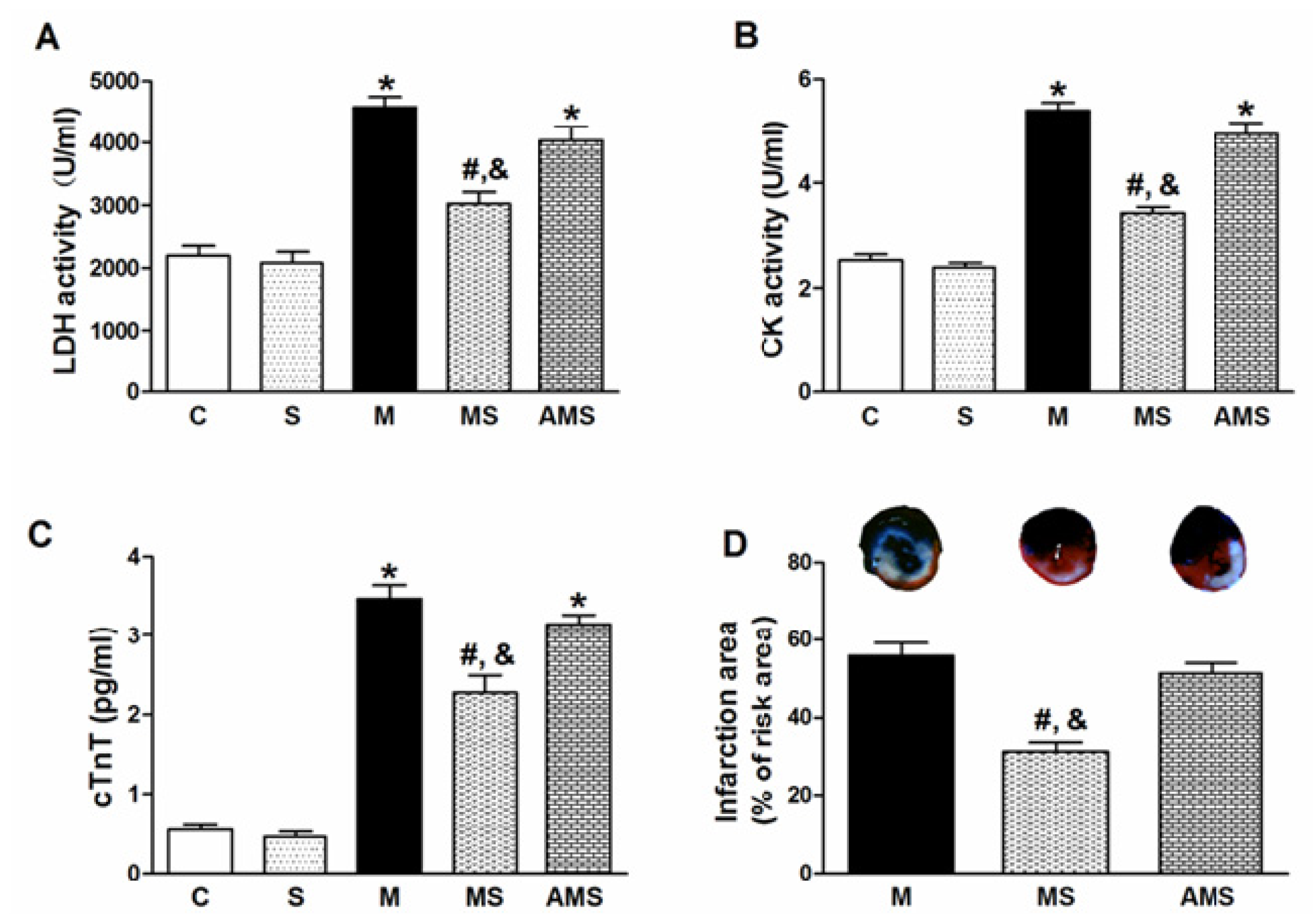
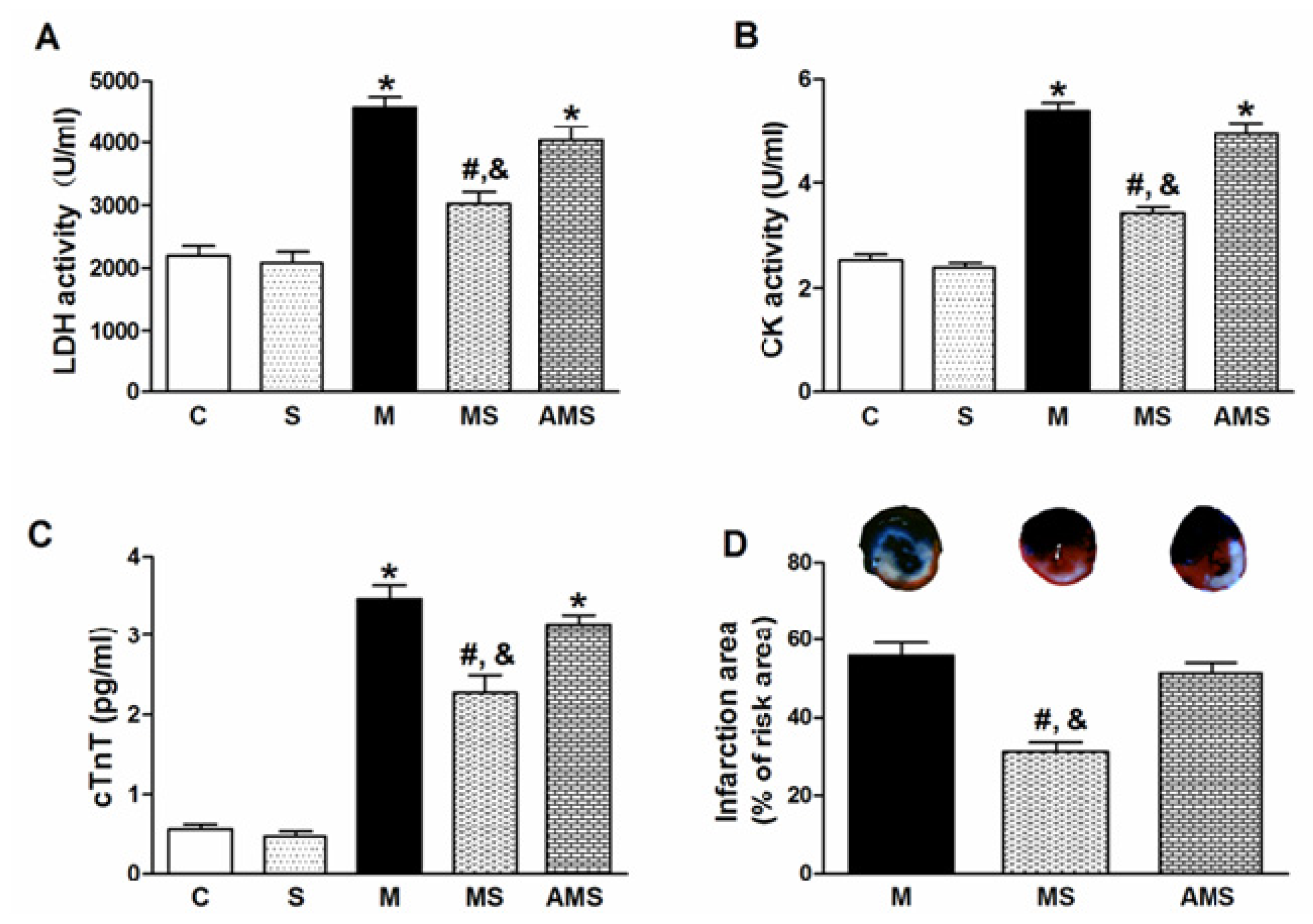
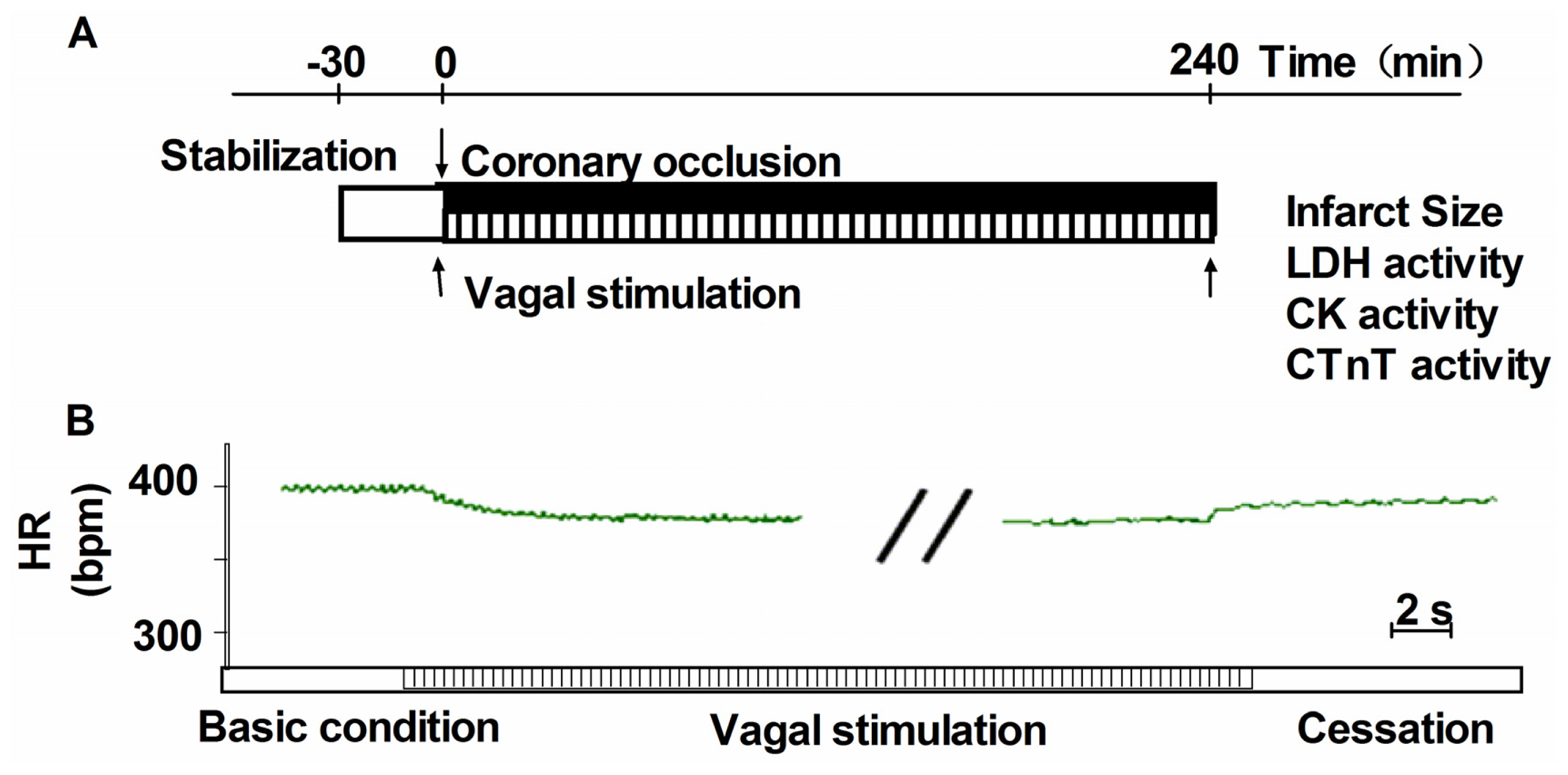
2.1. Vagal Stimulation Attenuated Myocardial Injury and Improved Cardiac Function
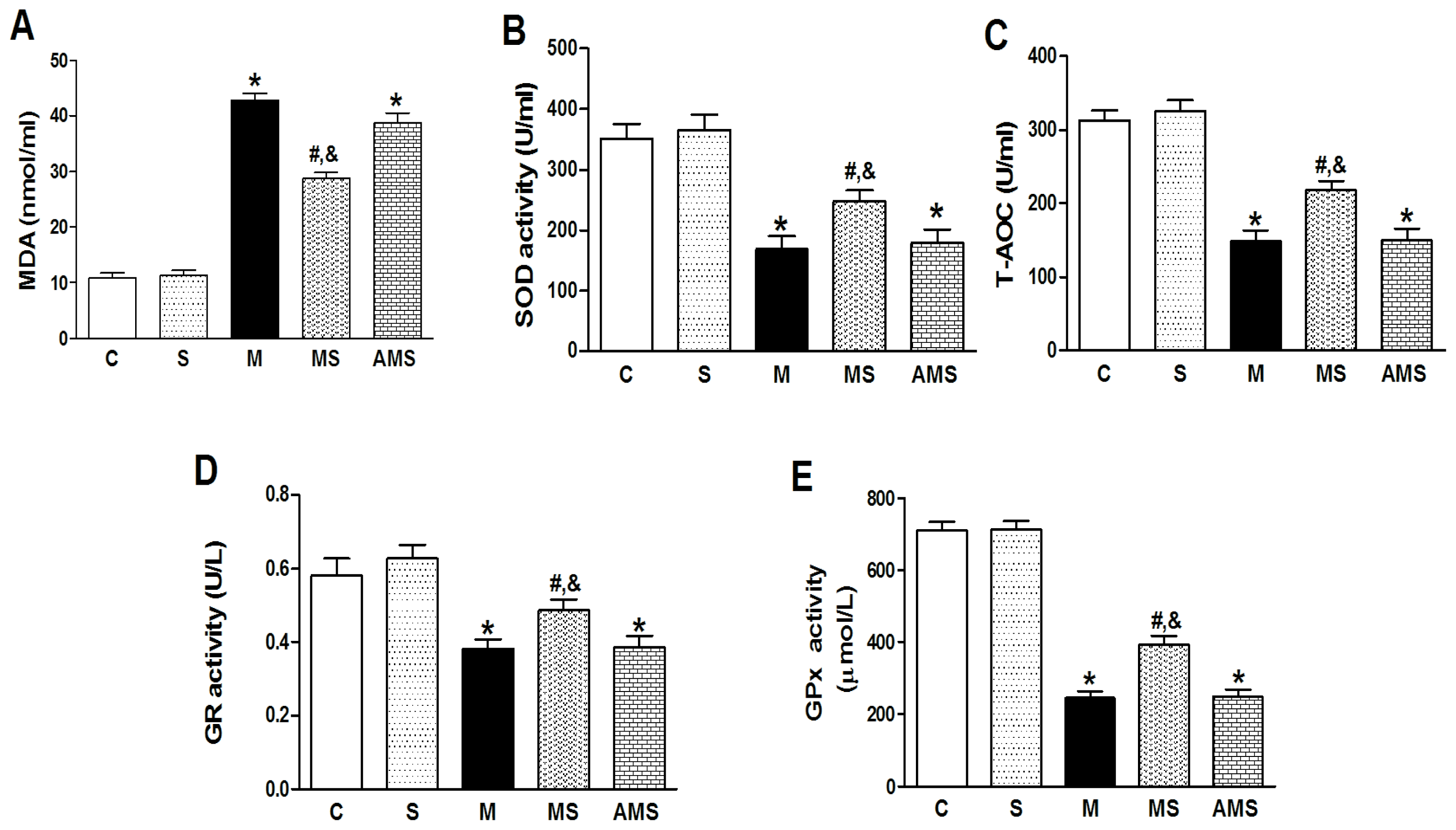
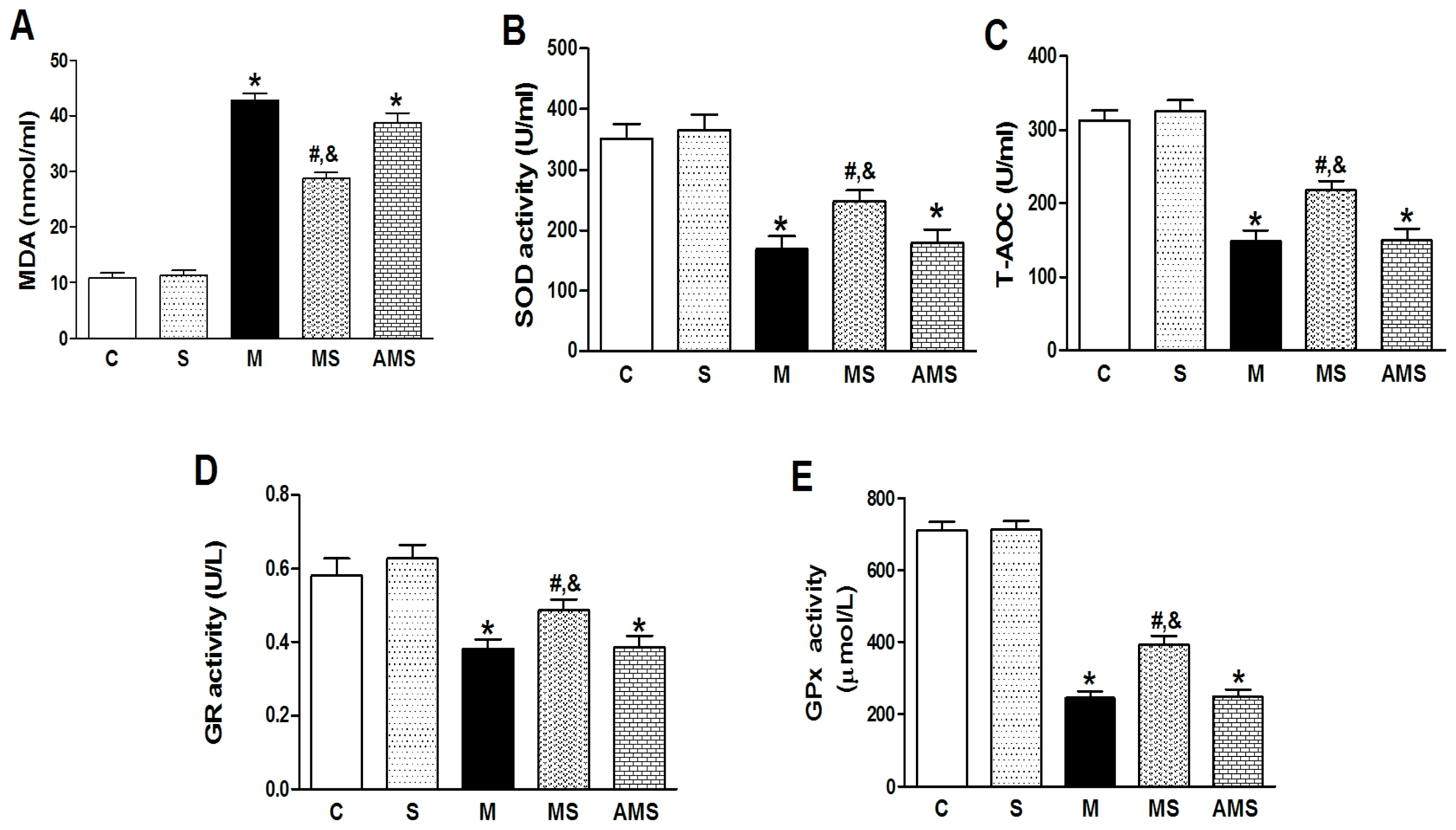
2.2. Changes of Serum Oxidant and Antioxidant Enzyme Activities in Each Group
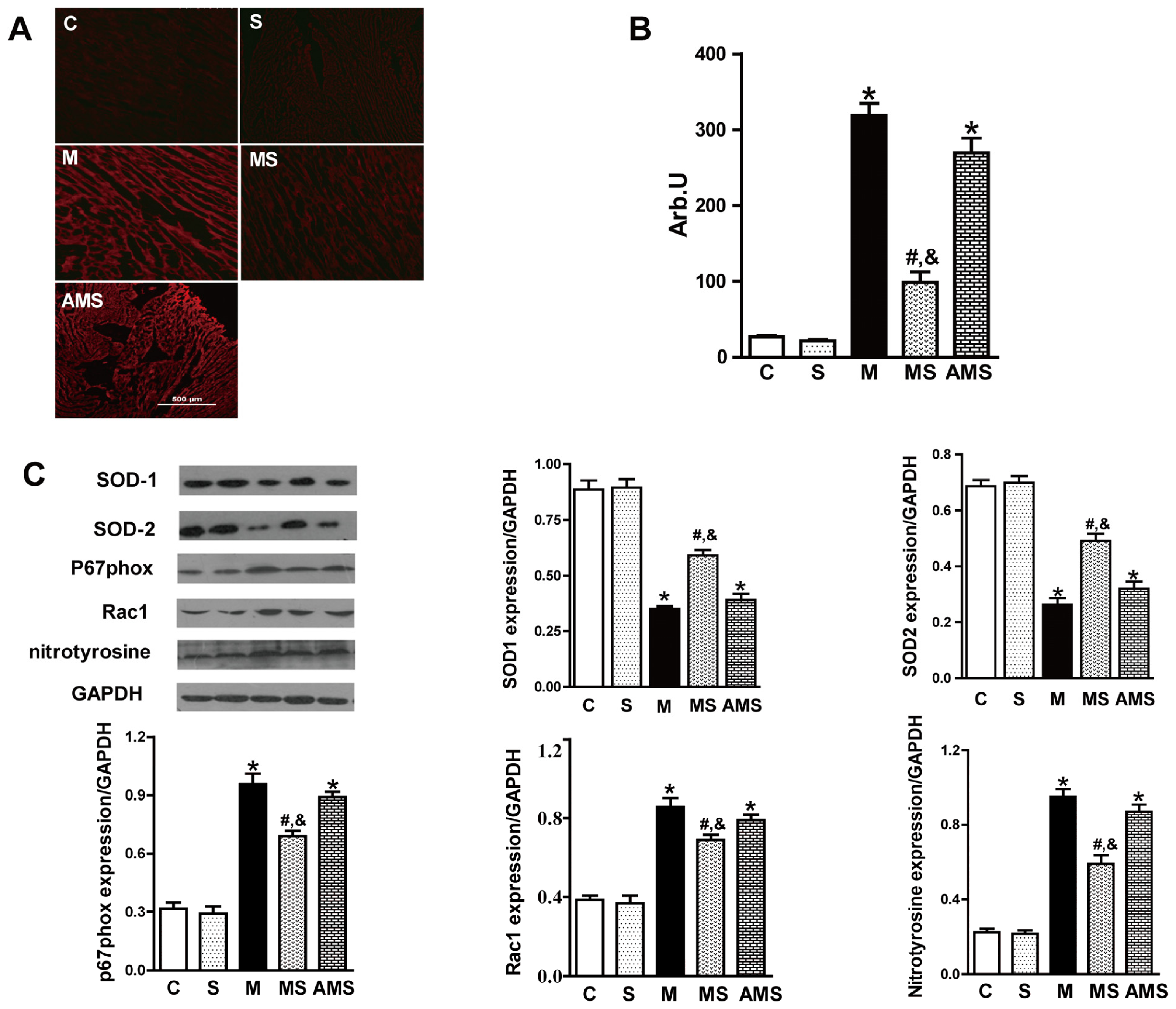
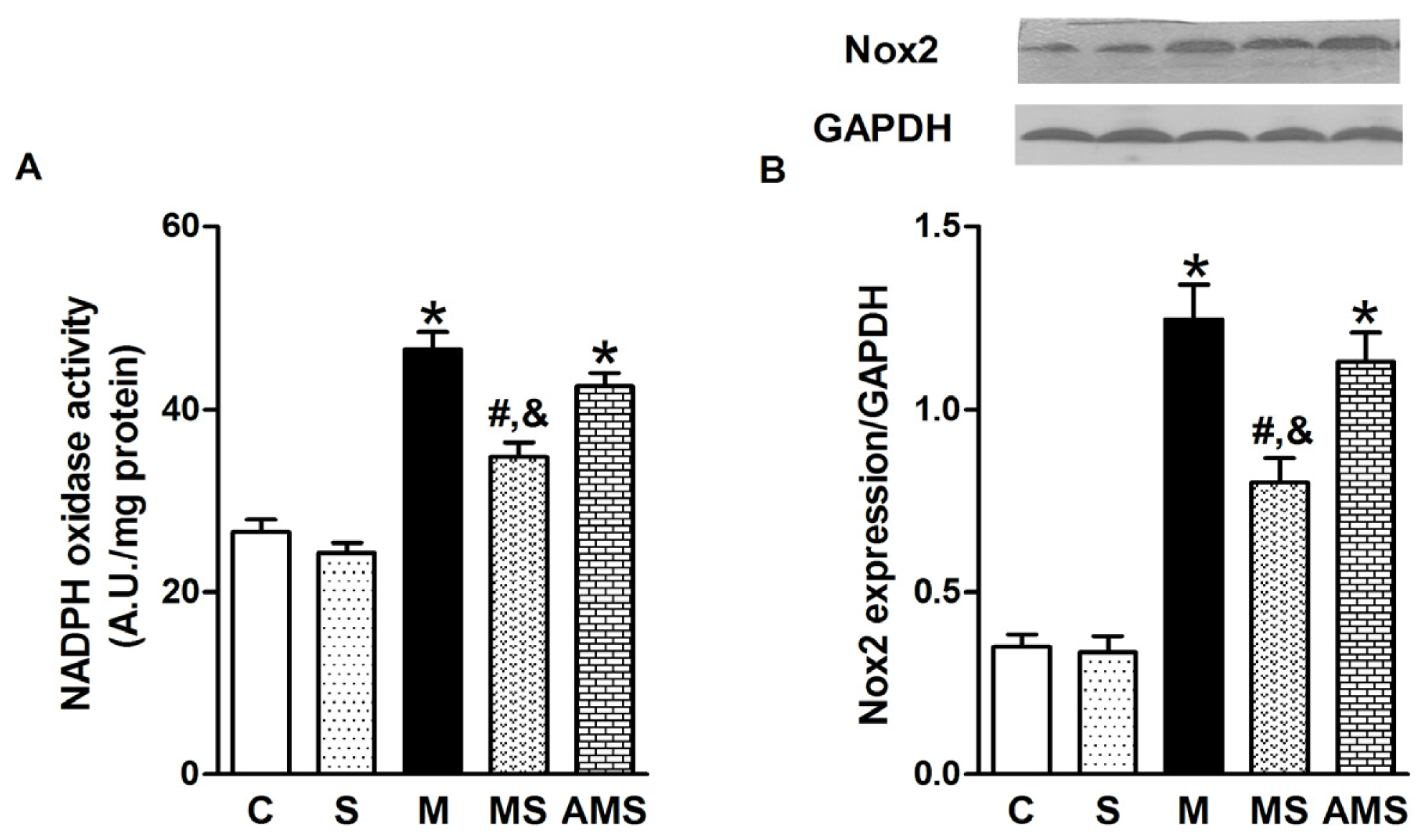
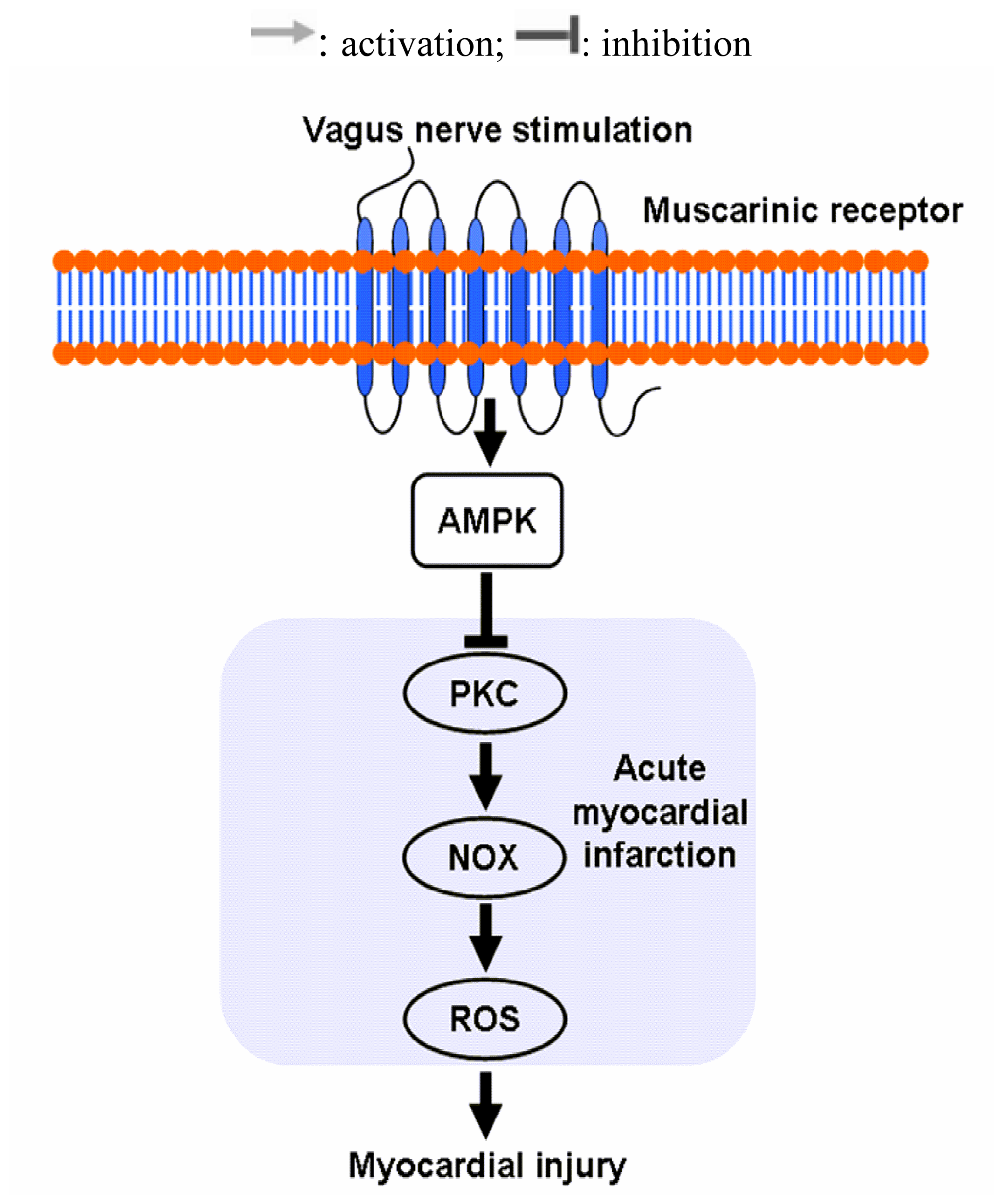
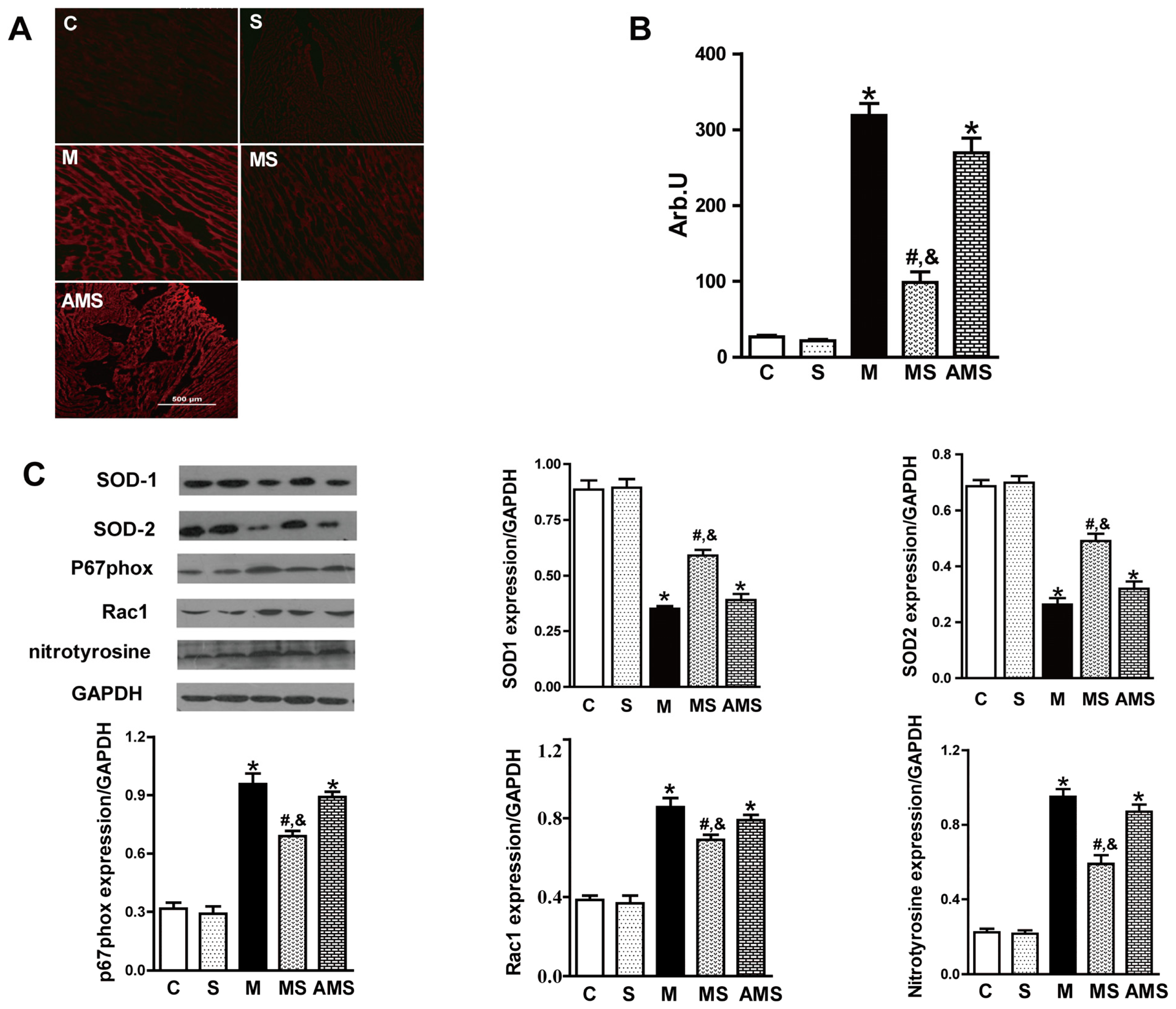
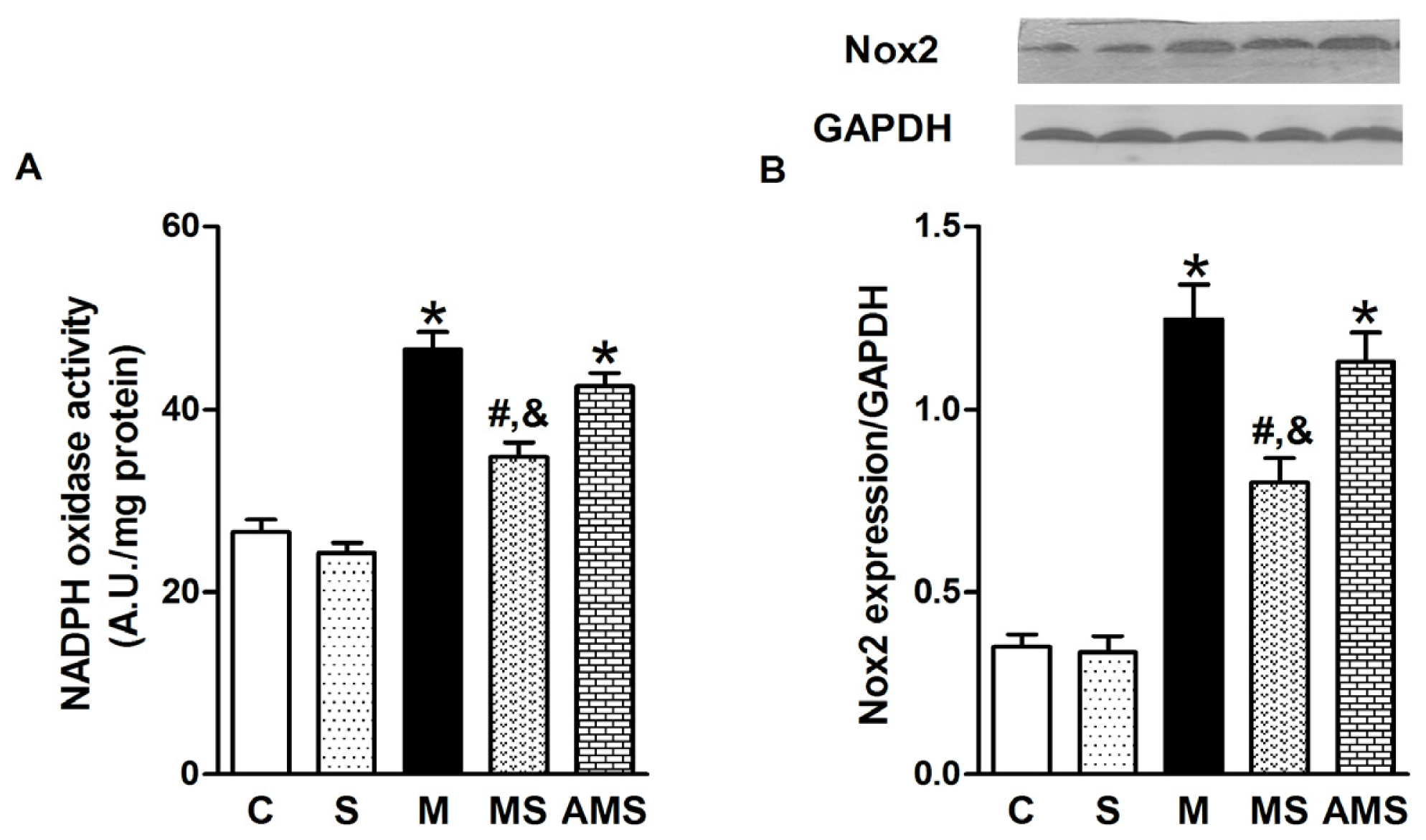
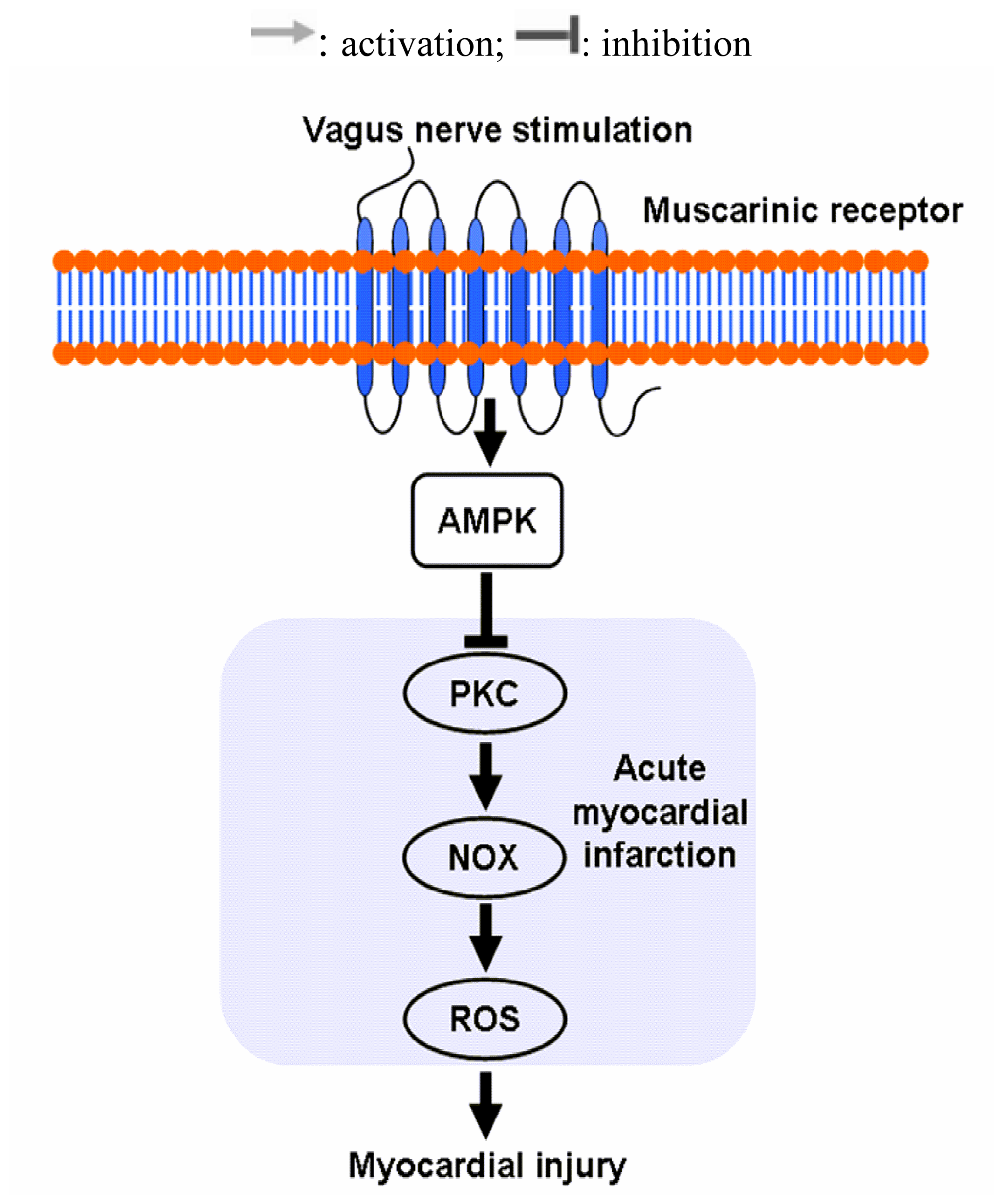
2.3. Vagal Stimulation Inhibited Oxidative Stress in Rats with AMI Mainly via the NADPH Pathway
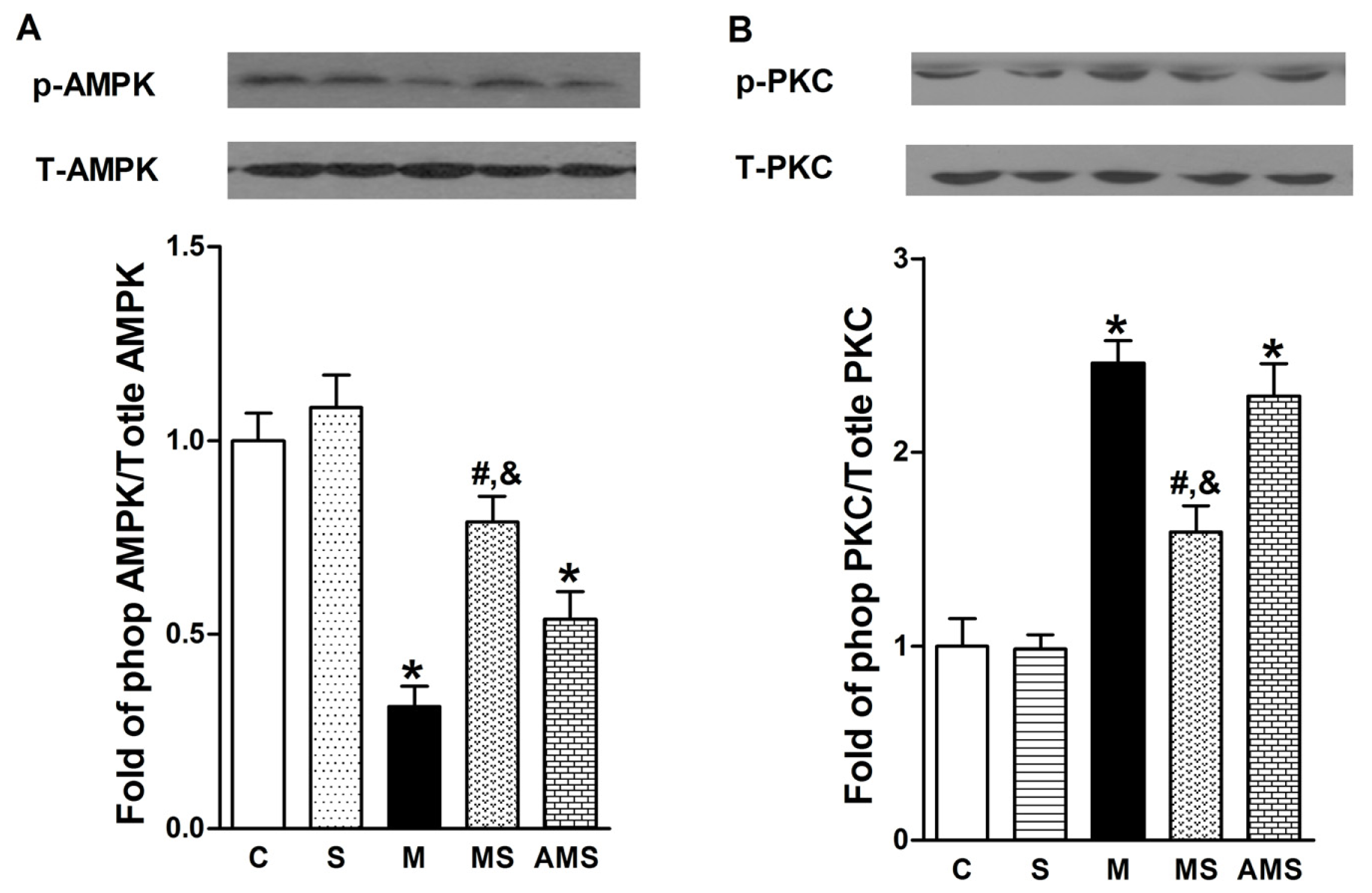
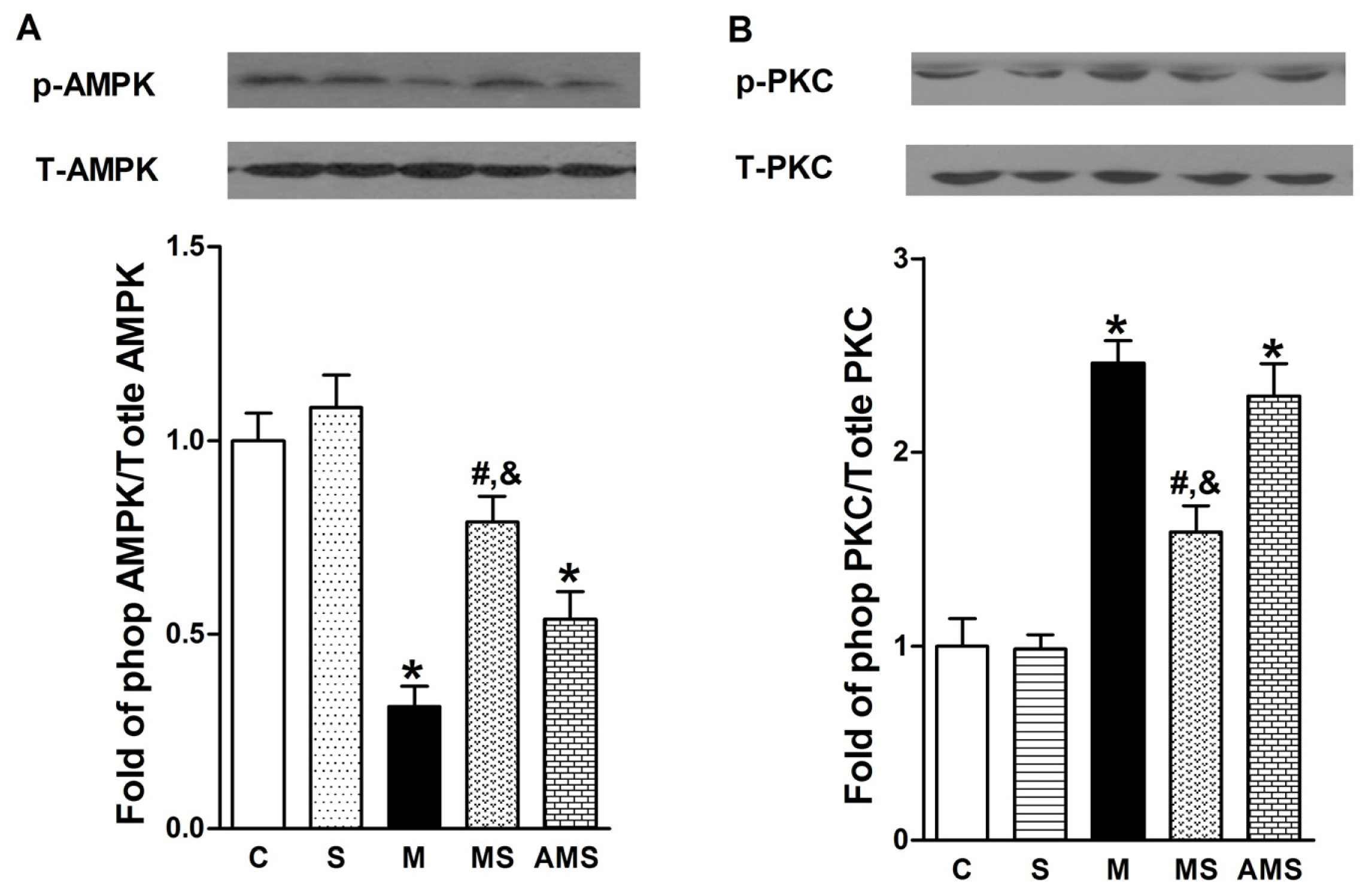
2.4 Effects of Vagal Stimulation on AMPK-PKC Pathway
3. Experimental Section
3.1. Animals
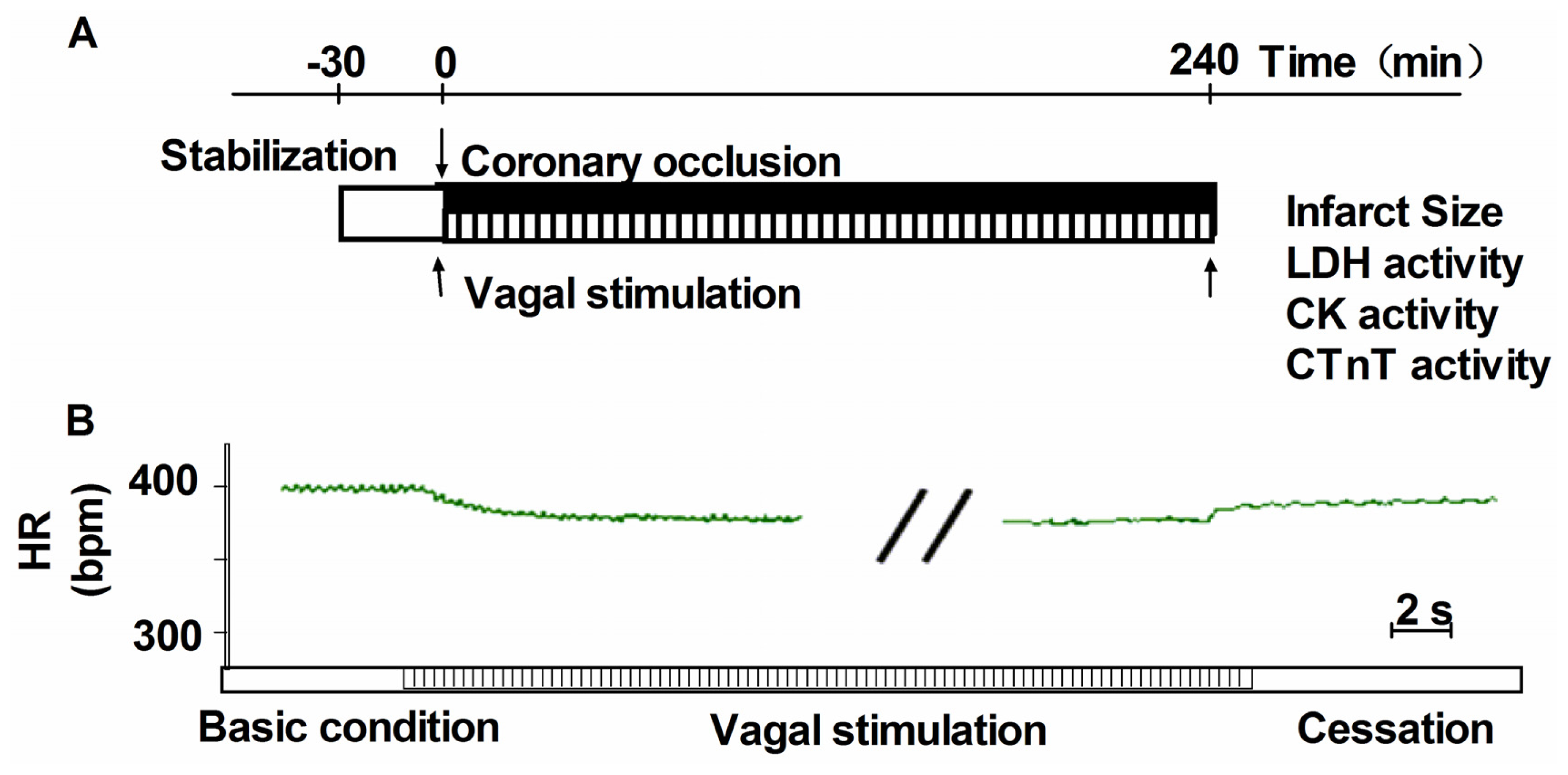
3.2. Heamodynamics Measurements and Induction of Acute Myocardial Infarction
3.3. Vagus Nerve Stimulation
3.4. Exclusion Criteria
3.5. Blood Sampling and Tissue Preparation
3.6. Cardiac Specific Injury Markers Measurement
3.7. Assessment of Infarction Size
3.8. Determination of Oxidant and Antioxidant Enzyme Activities
3.9. NADPH Oxidase Activity
3.10. Measurement of Superoxide Generation
3.11. Western Blotting Analysis
3.12. Statistical Analysis
4. Conclusions
Acknowledgments
- Conflict of InterestThe authors declare no conflict of interest.
References
- Bagatini, M.D.; Martins, C.C.; Battisti, V.; Gasparetto, D.; Da, R.C.; Spanevello, R.M.; Ahmed, M.; Schmatz, R.; Schetinger, M.R.; Morsch, V.M. Oxidative stress versus antioxidant defenses in patients with acute myocardial infarction. Heart Vessels 2011, 26, 55–63. [Google Scholar]
- Vanden, H.T.; Li, C.; Shao, Z.; Schumacker, P.T.; Becker, L.B. Significant levels of oxidants are generated by isolated cardiomyocytes during ischemia prior to reperfusion. J. Mol. Cell. Cardiol 1997, 29, 2571–2583. [Google Scholar]
- Sun, B.; Sun, G.B.; Xiao, J.; Chen, R.C.; Wang, X.; Wu, Y.; Cao, L.; Yang, Z.H.; Sun, X.B. Isorhamnetin inhibits H2O2-induced activation of the intrinsic apoptotic pathway in H9c2 cardiomyocytes through scavenging reactive oxygen species and ERK inactivation. J. Cell. Biochem 2012, 113, 473–485. [Google Scholar]
- Murdoch, C.E.; Grieve, D.J.; Cave, A.C.; Looi, Y.H.; Shah, A.M. NADPH oxidase and heart failure. Curr. Opin. Pharmacol 2006, 6, 148–153. [Google Scholar]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev 2007, 87, 245–313. [Google Scholar]
- Akki, A.; Zhang, M.; Murdoch, C.; Brewer, A.; Shah, A.M. NADPH oxidase signaling and cardiac myocyte function. J. Mol. Cell. Cardiol 2009, 47, 15–22. [Google Scholar]
- Meischl, C.; Krijnen, P.A.; Sipkens, J.A.; Cillessen, S.A.; Munoz, I.G.; Okroj, M.; Ramska, M.; Muller, A.; Visser, C.A.; Musters, R.J.; et al. Ischemia induces nuclear NOX2 expression in cardiomyocytes and subsequently activates apoptosis. Apoptosis 2006, 11, 913–921. [Google Scholar]
- Krijnen, P.A.; Meischl, C.; Hack, C.E.; Meijer, C.J.; Visser, C.A.; Roos, D.; Niessen, H.W. Increased Nox2 expression in human cardiomyocytes after acute myocardial infarction. J. Clin. Pathol 2003, 56, 194–199. [Google Scholar]
- Heymes, C.; Bendall, J.K.; Ratajczak, P.; Cave, A.C.; Samuel, J.L.; Hasenfuss, G.; Shah, A.M. Increased myocardial NADPH oxidase activity in human heart failure. J. Am. Coll. Cardiol 2003, 41, 2164–2171. [Google Scholar]
- Byrne, J.A.; Grieve, D.J.; Bendall, J.K.; Li, J.M.; Gove, C.; Lambeth, J.D.; Cave, A.C.; Shah, A.M. Contrasting roles of NADPH oxidase isoforms in pressure-overload versus angiotensin II-induced cardiac hypertrophy. Circ. Res 2003, 93, 802–805. [Google Scholar]
- Sroka, K. On the genesis of myocardial ischemia. Z. Kardiol 2004, 93, 768–783. [Google Scholar]
- Jardine, D.L.; Charles, C.J.; Ashton, R.K.; Bennett, S.I.; Whitehead, M.; Frampton, C.M.; Nicholls, M.G. Increased cardiac sympathetic nerve activity following acute myocardial infarction in a sheep model. J. Physiol 2005, 565, 325–333. [Google Scholar]
- Jankowska, E.A.; Ponikowski, P.; Piepoli, M.F.; Banasiak, W.; Anker, S.D.; Poole-Wilson, P.A. Autonomic imbalance and immune activation in chronic heart failure—Pathophysiological links. Cardiovasc. Res 2006, 70, 434–445. [Google Scholar]
- Kong, S.S.; Liu, J.J.; Hwang, T.C.; Yu, X.J.; Lu, Y.; Zang, W.J. Tumour necrosis factor-alpha and its receptors in the beneficial effects of vagal stimulation after myocardial infarction in rats. Clin. Exp. Pharmacol. Physiol 2011, 38, 300–306. [Google Scholar]
- Li, M.; Zheng, C.; Sato, T.; Kawada, T.; Sugimachi, M.; Sunagawa, K. Vagal nerve stimulation markedly improves long-term survival after chronic heart failure in rats. Circulation 2004, 109, 120–124. [Google Scholar]
- Schwartz, P.J.; de Ferrari, G.M.; Sanzo, A.; Landolina, M.; Rordorf, R.; Raineri, C.; Campana, C.; Revera, M.; Ajmone-Marsan, N.; Tavazzi, L.; et al. Long term vagal stimulation in patients with advanced heart failure: First experience in man. Eur. J. Heart Fail 2008, 10, 884–891. [Google Scholar]
- Ando, M.; Katare, R.G.; Kakinuma, Y.; Zhang, D.; Yamasaki, F.; Muramoto, K.; Sato, T. Efferent vagal nerve stimulation protects heart against ischemia-induced arrhythmias by preserving connexin43 protein. Circulation 2005, 112, 164–170. [Google Scholar]
- Brack, K.E.; Patel, V.H.; Coote, J.H.; Ng, G.A. Nitric oxide mediates the vagal protective effect on ventricular fibrillation via effects on action potential duration restitution in the rabbit heart. J. Physiol 2007, 583, 695–704. [Google Scholar]
- Ceolotto, G.; Papparella, I.; Lenzini, L.; Sartori, M.; Mazzoni, M.; Iori, E.; Franco, L.; Gallo, A.; de Kreutzenberg, S.V.; Tiengo, A.; et al. Insulin generates free radicals in human fibroblasts ex vivo by a protein kinase C-dependent mechanism, which is inhibited by pravastatin. Free Radic. Biol. Med 2006, 41, 473–483. [Google Scholar]
- Tsai, K.L.; Chen, L.H.; Chiou, S.H.; Chiou, G.Y.; Chen, Y.C.; Chou, H.Y.; Chen, L.K.; Chen, H.Y.; Chiu, T.H.; Tsai, C.S.; et al. Coenzyme Q10 suppresses oxLDL-induced endothelial oxidative injuries by the modulation of LOX-1-mediated ROS generation via the AMPK/PKC/NADPH oxidase signaling pathway. Mol. Nutr. Food Res 2011, 55, S227–S240. [Google Scholar]
- Siwik, D.A.; Tzortzis, J.D.; Pimental, D.R.; Chang, D.L.; Pagano, P.J.; Singh, K.; Sawyer, D.B.; Colucci, W.S. Inhibition of copper-zinc superoxide dismutase induces cell growth, hypertrophic phenotype, and apoptosis in neonatal rat cardiac myocytes in vitro. Circ. Res 1999, 85, 147–153. [Google Scholar]
- Ookawara, T.; Imazeki, N.; Matsubara, O.; Kizaki, T.; Oh-Ishi, S.; Nakao, C.; Sato, Y.; Ohno, H. Tissue distribution of immunoreactive mouse extracellular superoxide dismutase. Am. J. Physiol 1998, 275, C840–C847. [Google Scholar]
- Roe, N.D.; Thomas, D.P.; Ren, J. Inhibition of NADPH oxidase alleviates experimental diabetes-induced myocardial contractile dysfunction. Diabetes Obes. Metab 2011, 13, 465–473. [Google Scholar]
- Rothwell, S.E.; Richards, A.M.; Pemberton, C.J. Resistin worsens cardiac ischaemia-reperfusion injury. Biochem. Biophys. Res. Commun 2006, 349, 400–407. [Google Scholar]
- Hardie, D.G.; Carling, D. The AMP-activated protein kinase—Fuel gauge of the mammalian cell? Eur. J. Biochem 1997, 246, 259–273. [Google Scholar]
- Bey, E.A.; Xu, B.; Bhattacharjee, A.; Oldfield, C.M.; Zhao, X.; Li, Q.; Subbulakshmi, V.; Feldman, G.M.; Wientjes, F.B.; Cathcart, M.K. Protein kinase C delta is required for p47phox phosphorylation and translocation in activated human monocytes. J. Immunol 2004, 173, 5730–5738. [Google Scholar]
- Ceolotto, G.; Gallo, A.; Papparella, I.; Franco, L.; Murphy, E.; Iori, E.; Pagnin, E.; Fadini, G.P.; Albiero, M.; Semplicini, A.; et al. Rosiglitazone reduces glucose-induced oxidative stress mediated by NAD(P)H oxidase via AMPK-dependent mechanism. Arterioscler. Thromb. Vasc. Biol 2007, 27, 2627–2633. [Google Scholar]
- Institute of Laboratory Animal Resources, Guide for the Care and Use of Laboratory Animals; National Institutes of Health publication No. 85–23; National Academies Press: Washington, DC, USA, 1996.
- Krishnamurthy, P.; Rajasingh, J.; Lambers, E.; Qin, G.; Losordo, D.W.; Kishore, R. IL-10 inhibits inflammation and attenuates left ventricular remodeling after myocardial infarction via activation of STAT3 and suppression of HuR. Circ. Res 2009, 104, e9–e18. [Google Scholar]
- Yao, Y.Y.; Yin, H.; Shen, B.; Chao, L.; Chao, J. Tissue kallikrein and kinin infusion rescues failing myocardium after myocardial infarction. J. Card Fail 2007, 13, 588–596. [Google Scholar]
- Nakahara, T.; Kawada, T.; Sugimachi, M.; Miyano, H.; Sato, T.; Shishido, T.; Yoshimura, R.; Miyashita, H.; Inagaki, M.; Alexander, J.J.; et al. Accumulation of cAMP augments dynamic vagal control of heart rate. Am. J. Physiol 1998, 275, H562–H567. [Google Scholar]
- Kawada, T.; Yamazaki, T.; Akiyama, T.; Li, M.; Ariumi, H.; Mori, H.; Sunagawa, K.; Sugimachi, M. Vagal stimulation suppresses ischemia-induced myocardial interstitial norepinephrine release. Life Sci 2006, 78, 882–887. [Google Scholar]
- Lee, T.M.; Chen, C.C.; Hsu, Y.J. Differential effects of NADPH oxidase and xanthine oxidase inhibition on sympathetic reinnervation in postinfarct rat hearts. Free Radic. Biol. Med 2011, 50, 1461–1470. [Google Scholar]

© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kong, S.-S.; Liu, J.-J.; Yu, X.-J.; Lu, Y.; Zang, W.-J. Protection against Ischemia-Induced Oxidative Stress Conferred by Vagal Stimulation in the Rat Heart: Involvement of the AMPK-PKC Pathway. Int. J. Mol. Sci. 2012, 13, 14311-14325. https://doi.org/10.3390/ijms131114311
Kong S-S, Liu J-J, Yu X-J, Lu Y, Zang W-J. Protection against Ischemia-Induced Oxidative Stress Conferred by Vagal Stimulation in the Rat Heart: Involvement of the AMPK-PKC Pathway. International Journal of Molecular Sciences. 2012; 13(11):14311-14325. https://doi.org/10.3390/ijms131114311
Chicago/Turabian StyleKong, Shan-Shan, Jin-Jun Liu, Xiao-Jiang Yu, Yi Lu, and Wei-Jin Zang. 2012. "Protection against Ischemia-Induced Oxidative Stress Conferred by Vagal Stimulation in the Rat Heart: Involvement of the AMPK-PKC Pathway" International Journal of Molecular Sciences 13, no. 11: 14311-14325. https://doi.org/10.3390/ijms131114311