A Generic Force Field for Protein Coarse-Grained Molecular Dynamics Simulation

Abstract
:1. Introduction
2. Results
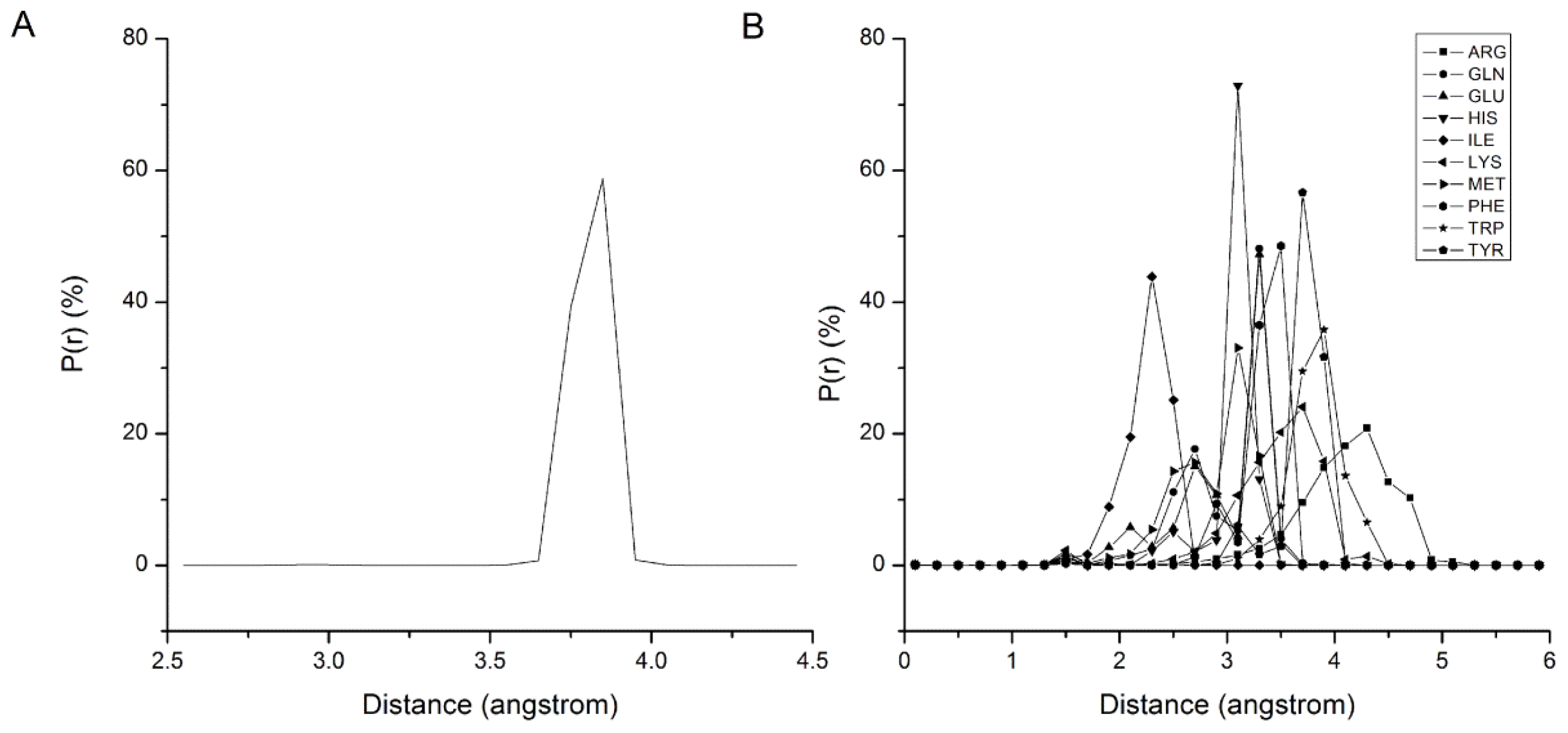
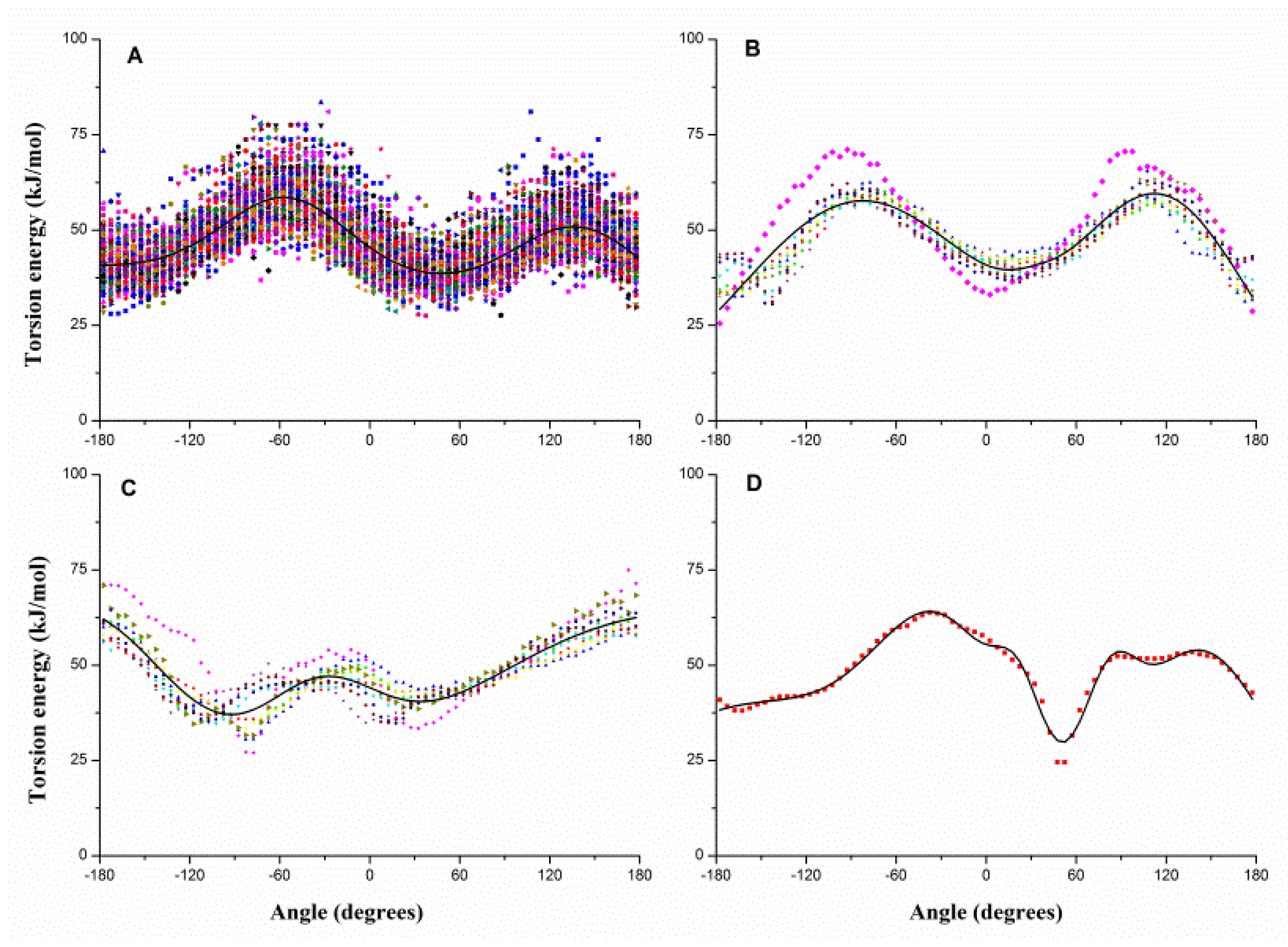
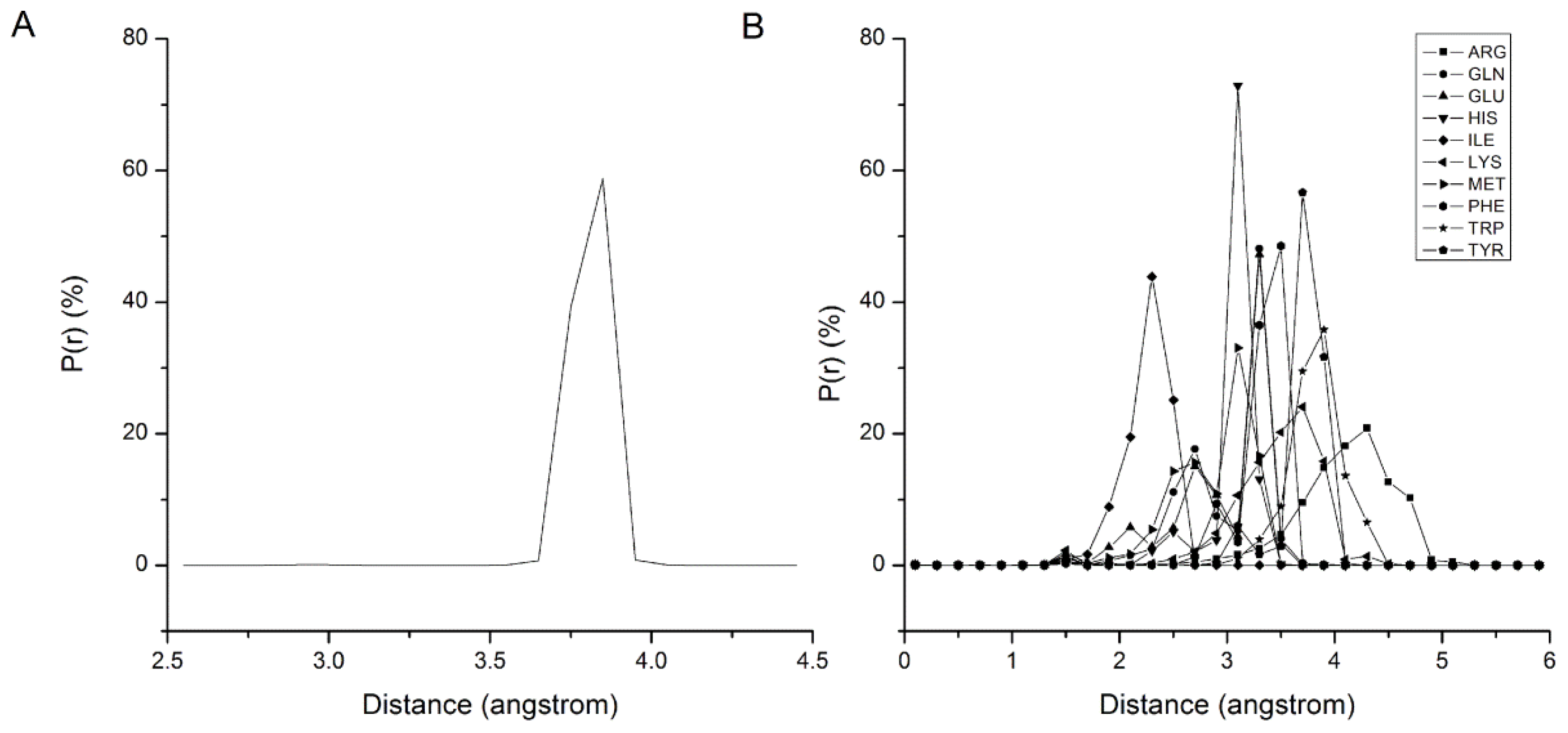
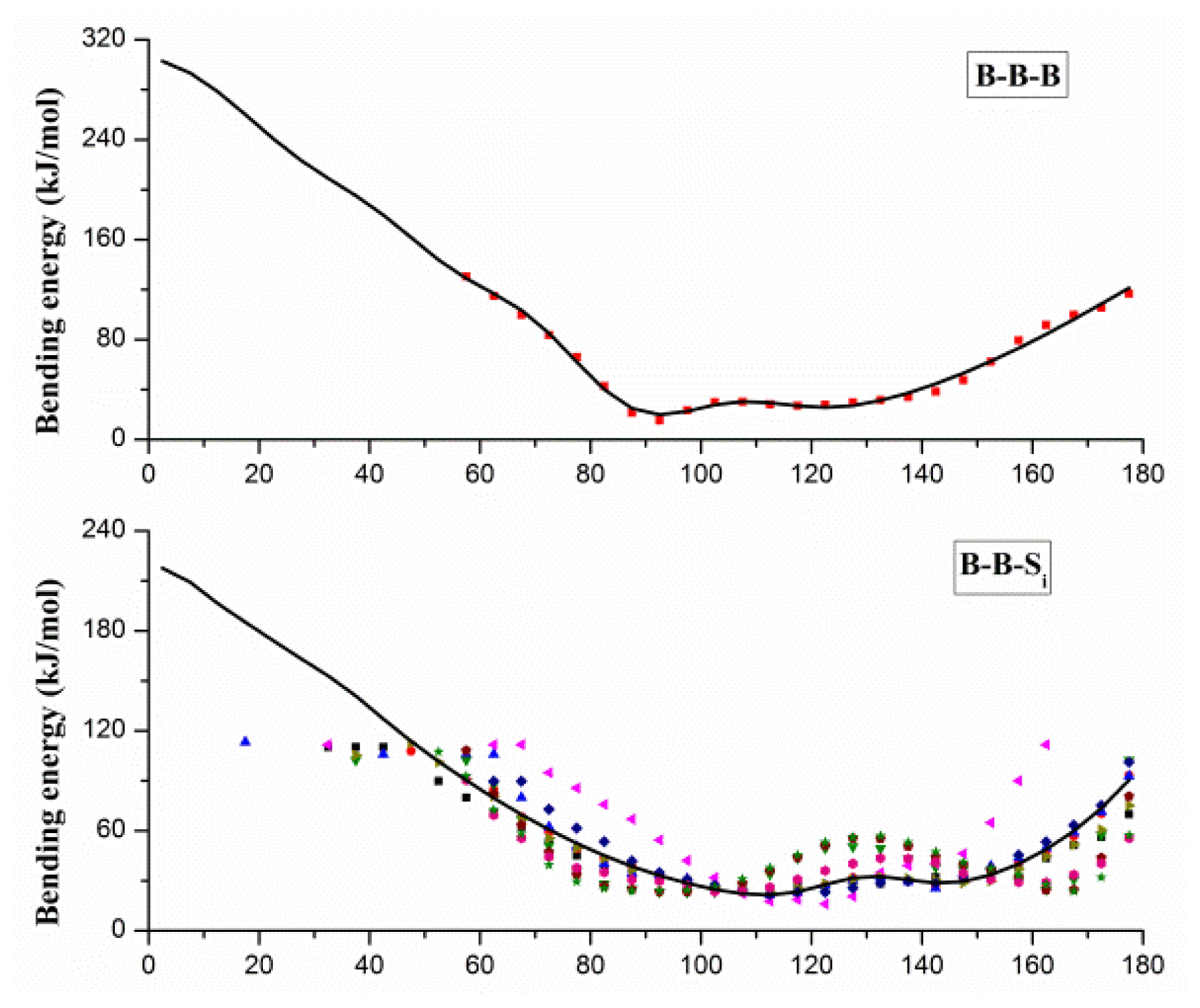
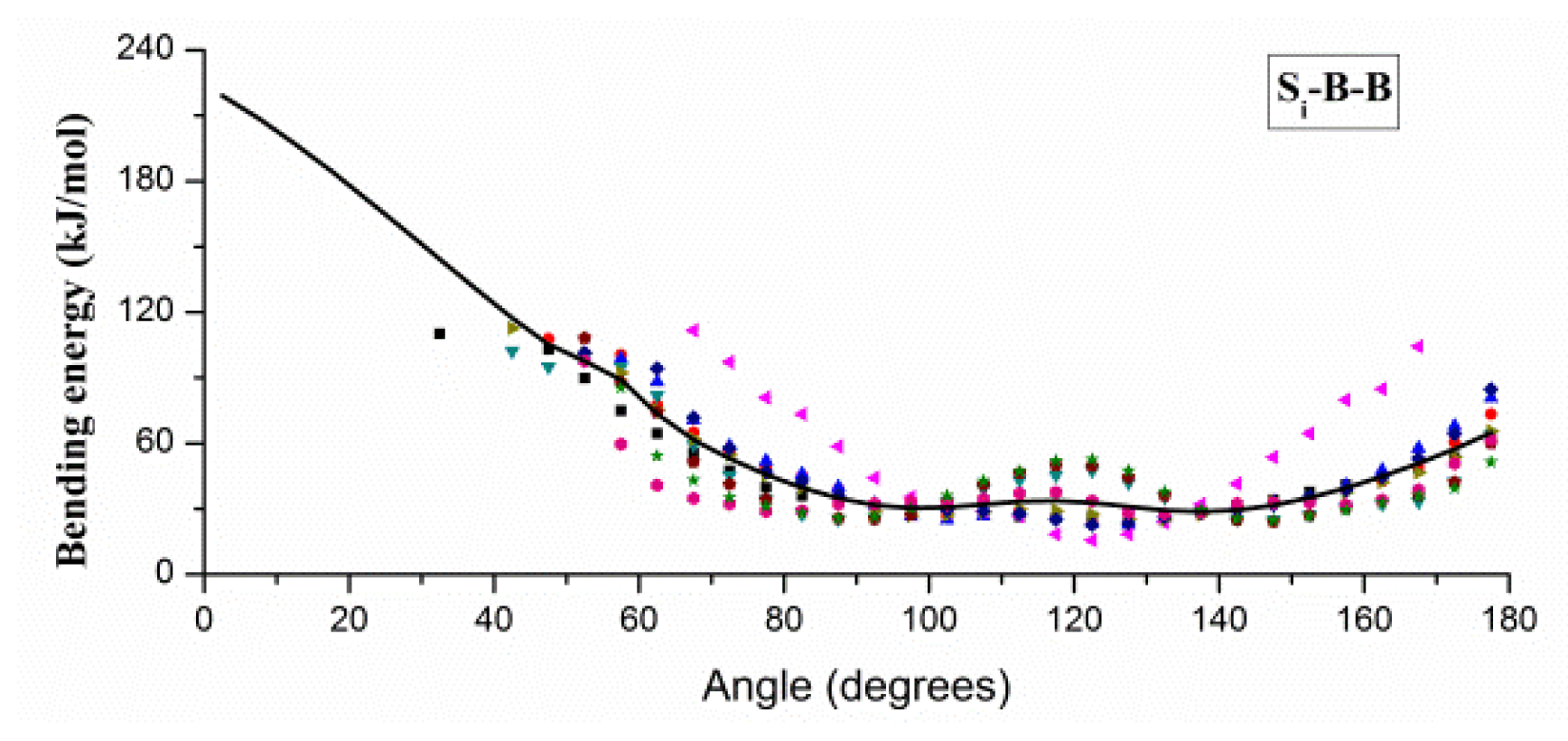
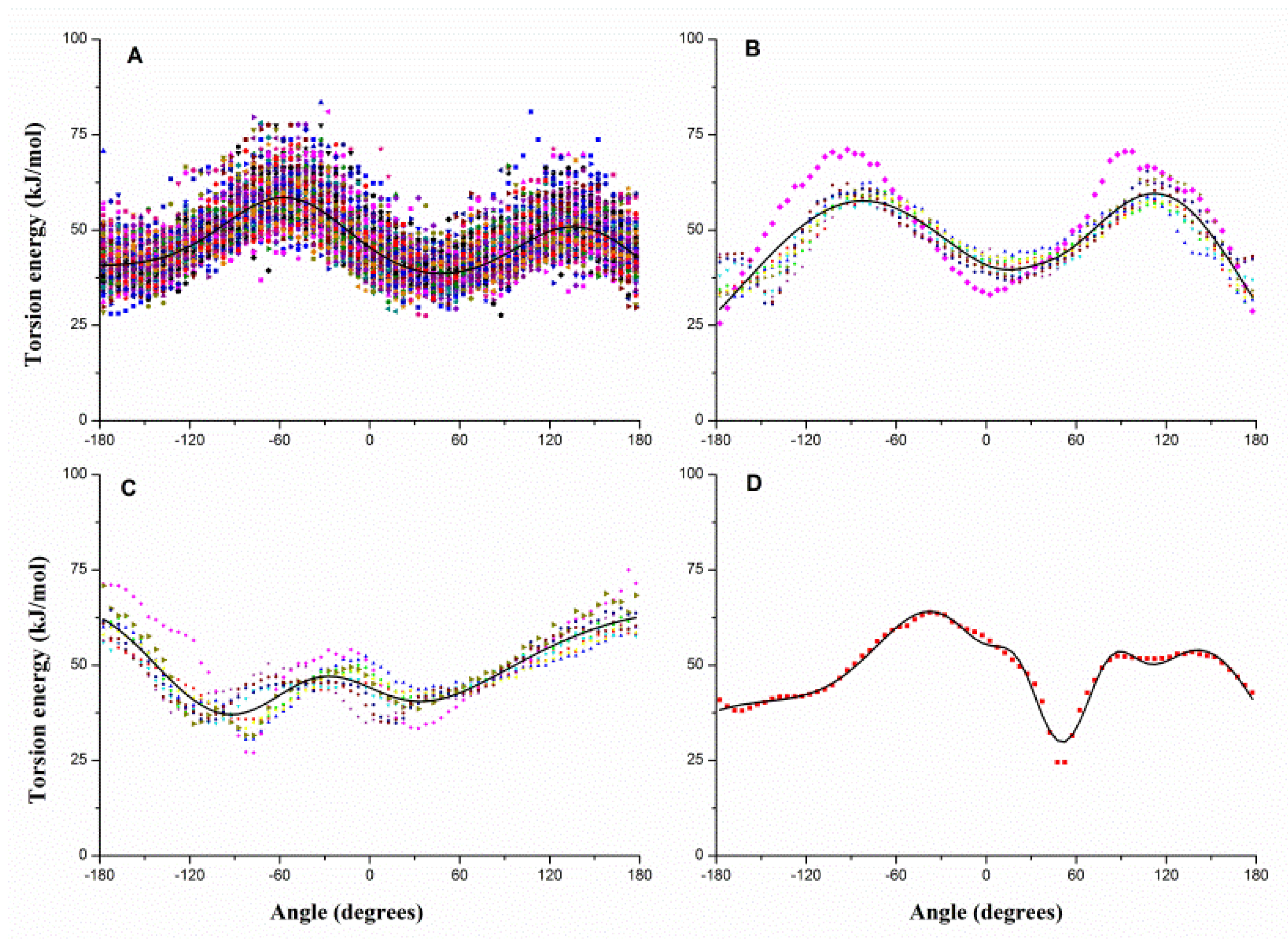
2.1. Results of Bonded Potential Parameterization
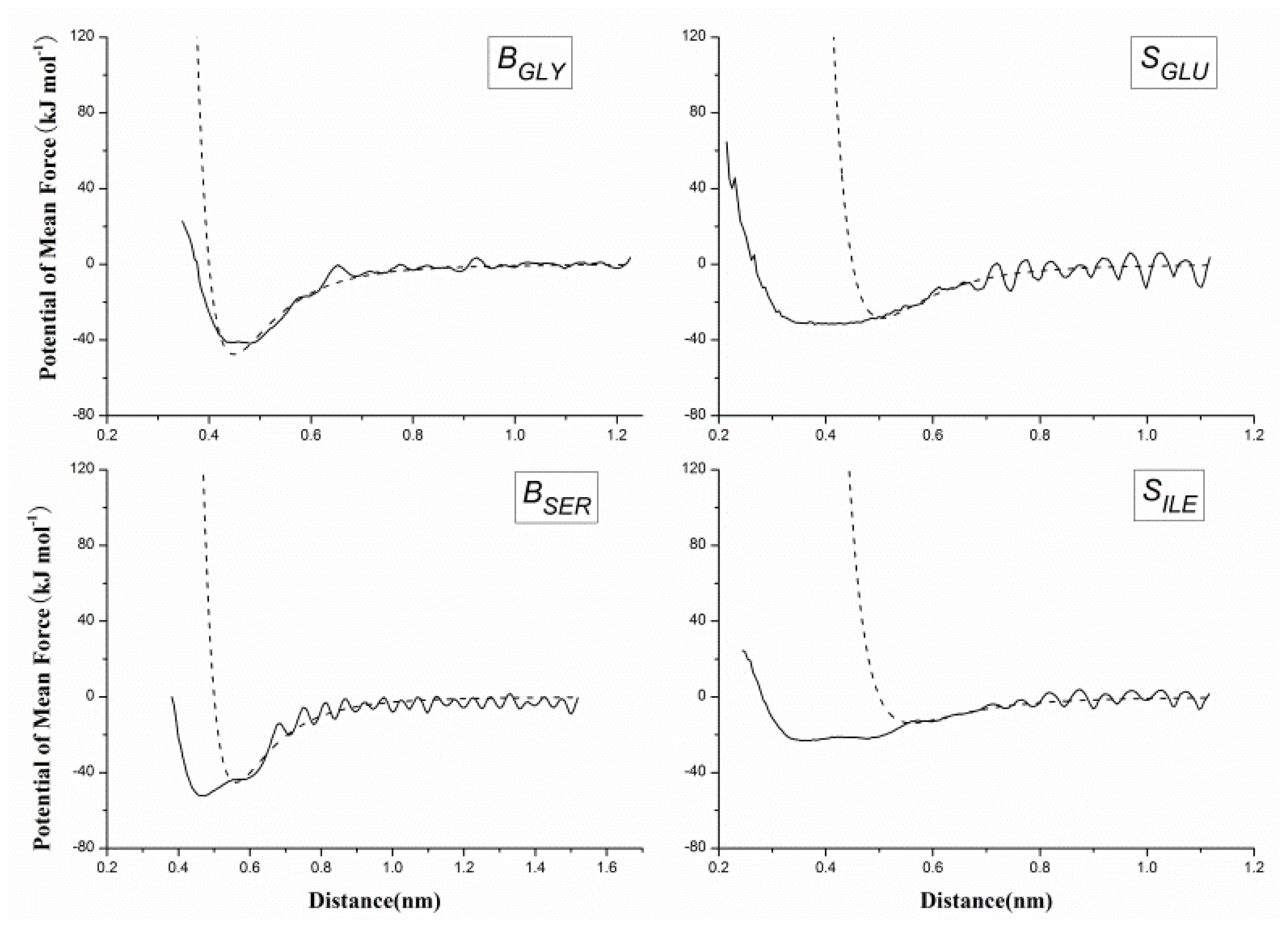
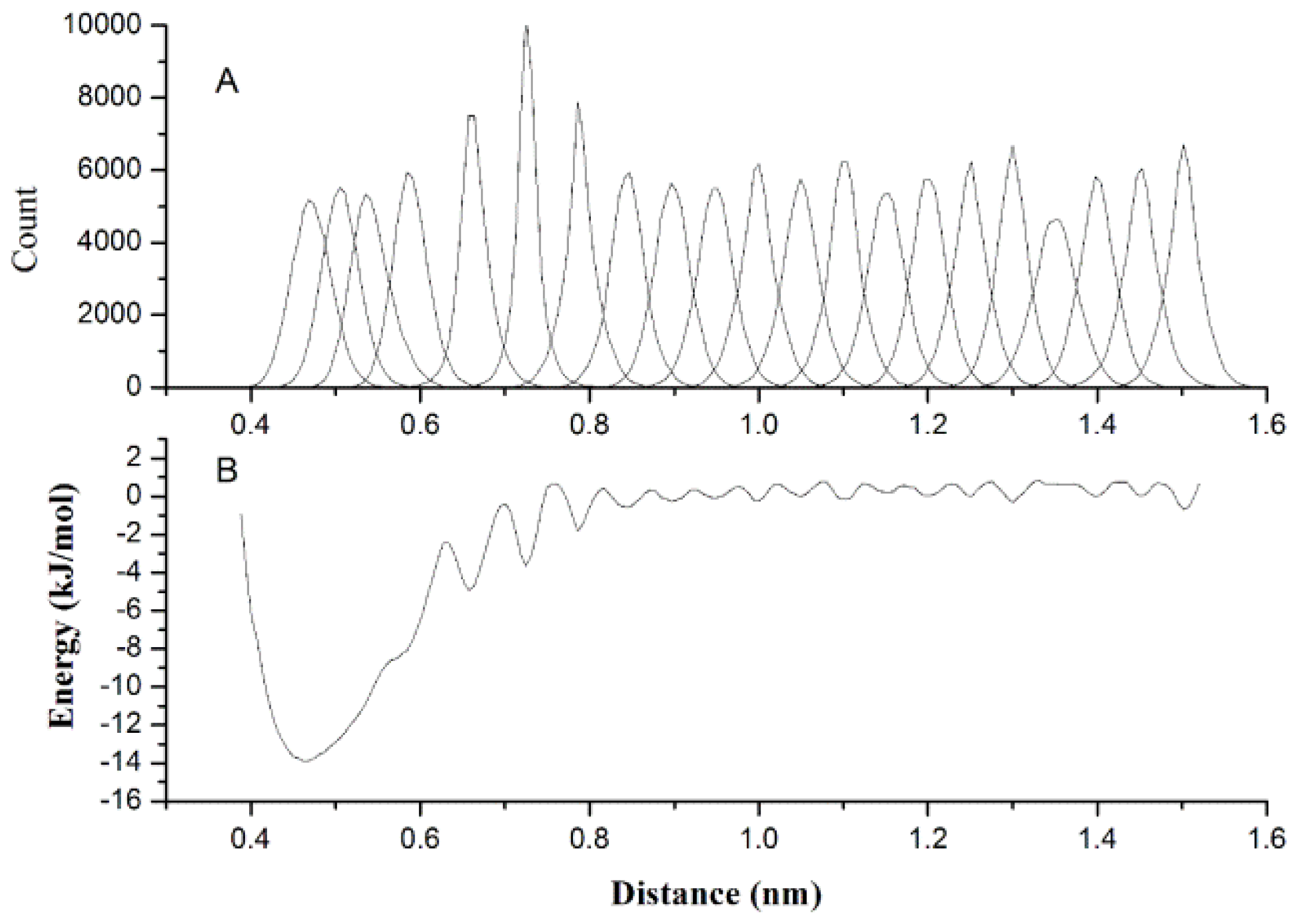
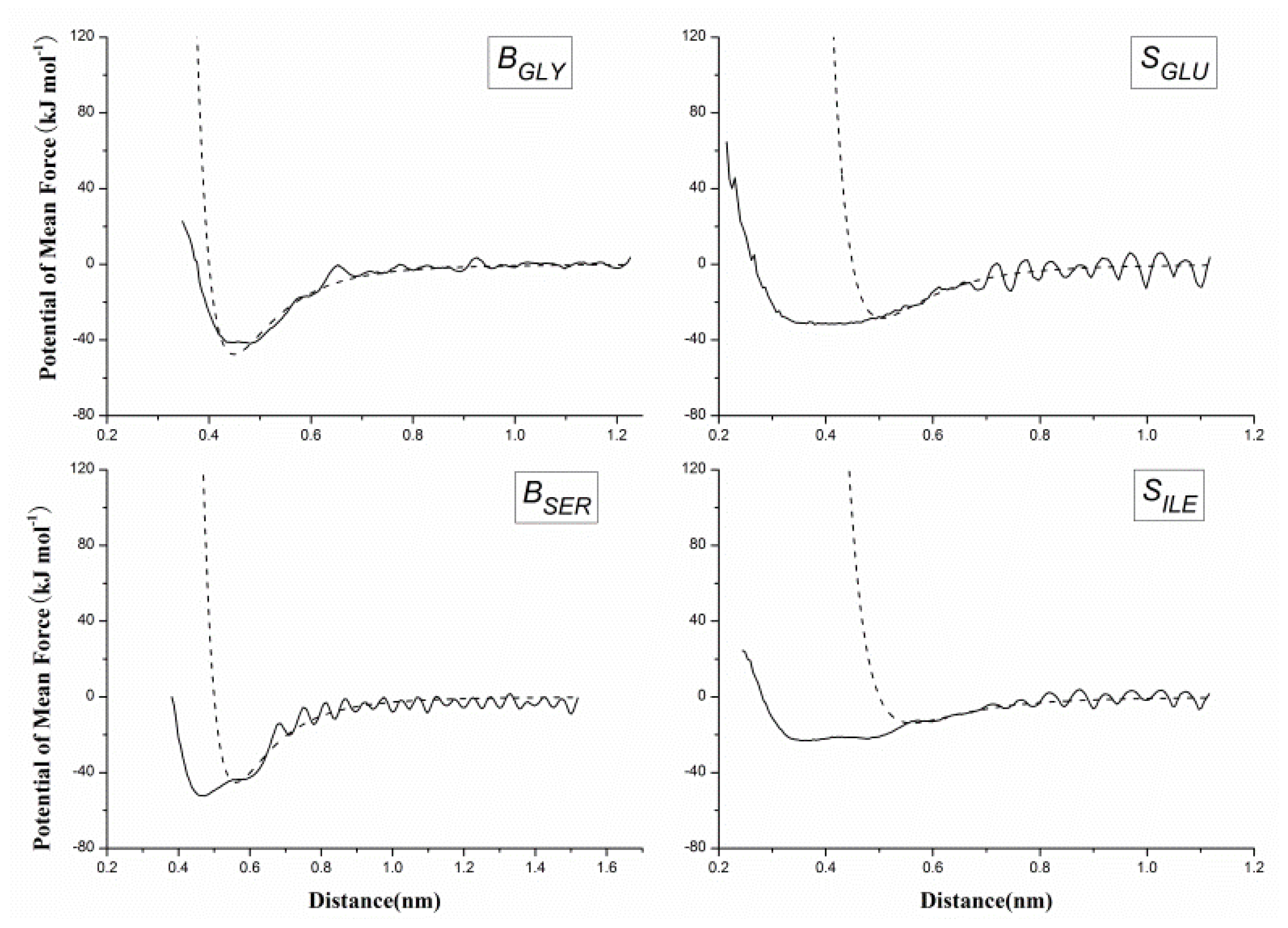
2.2. Results of Non-Bonded Potential Parameterization
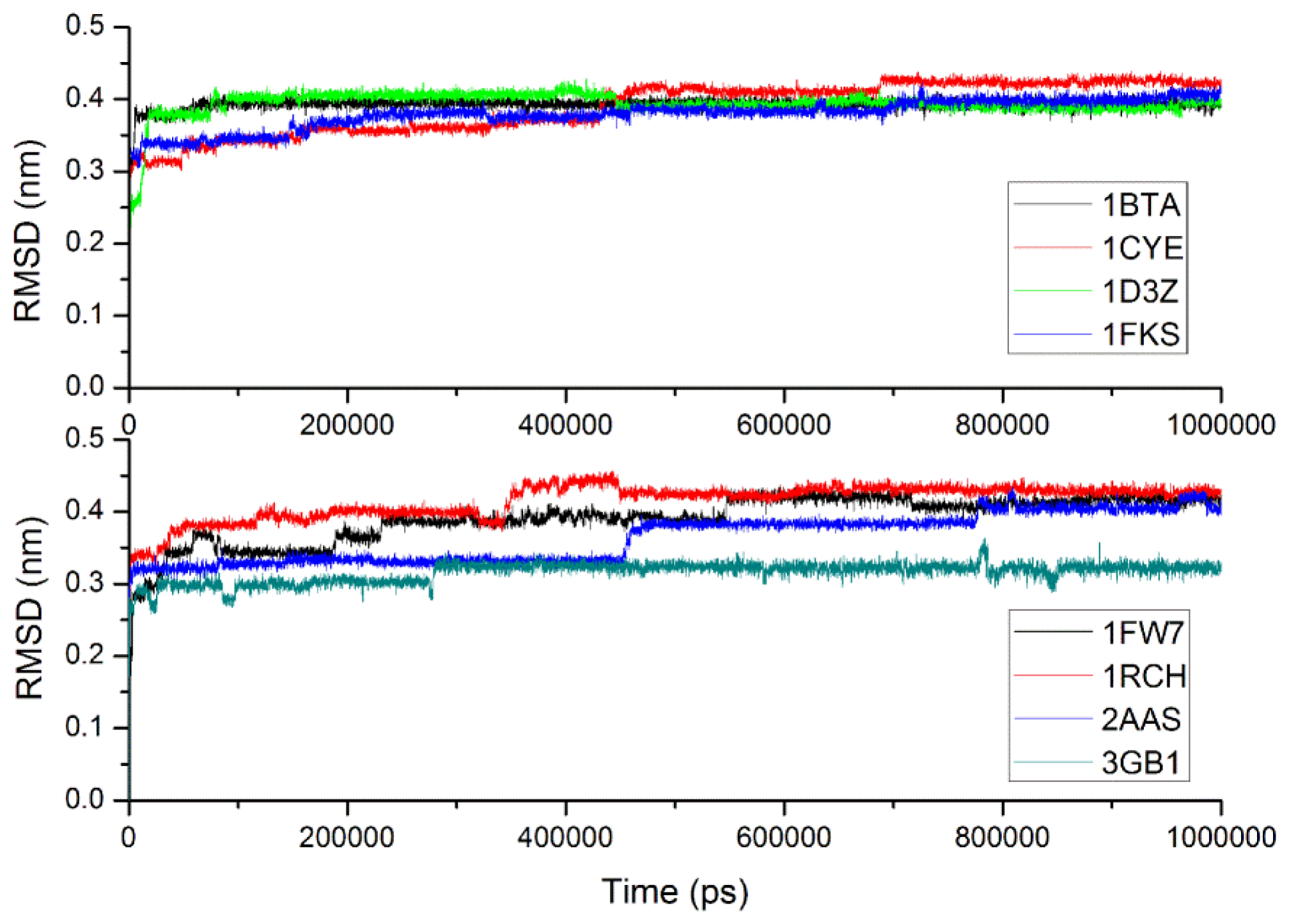
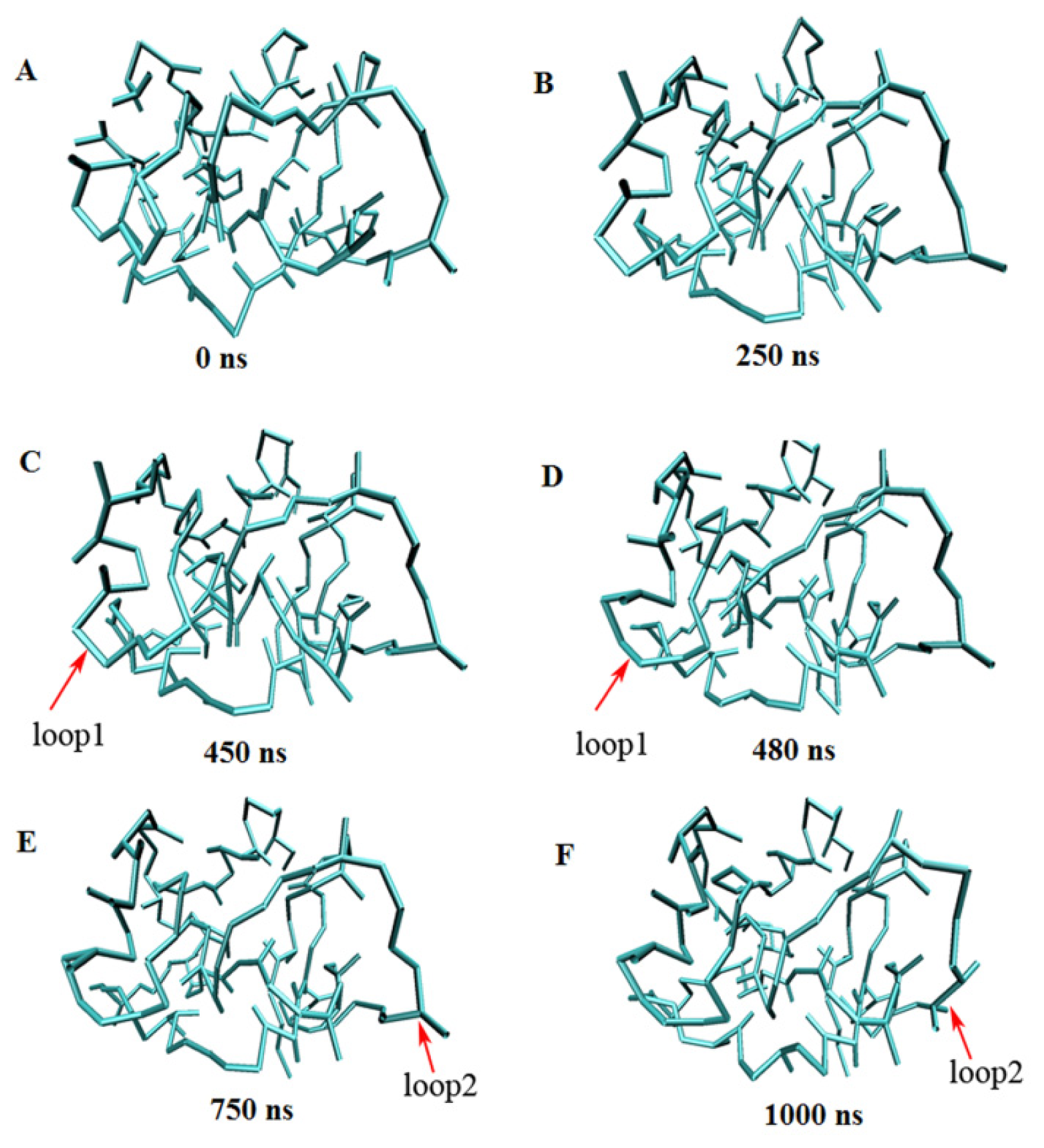
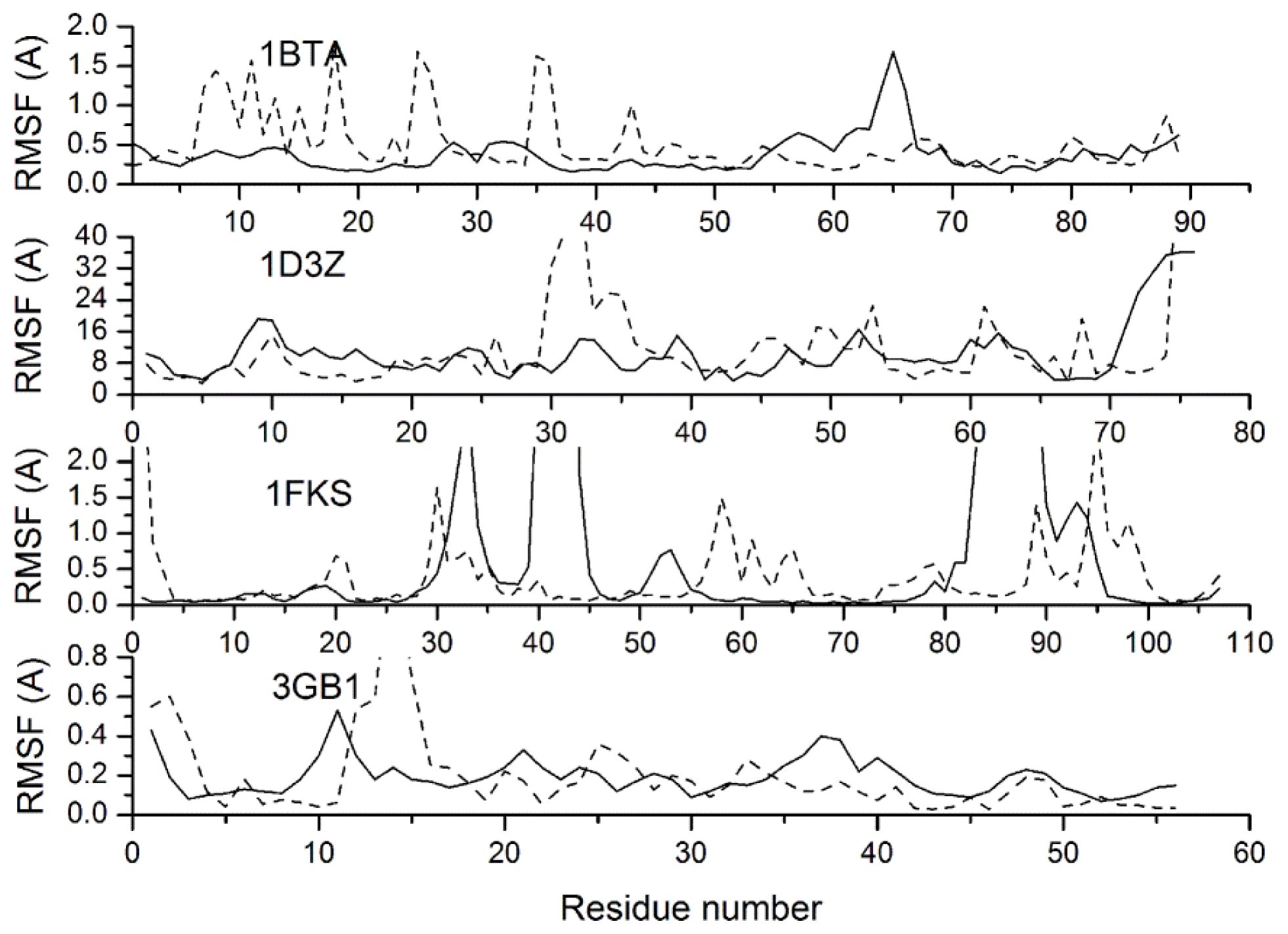
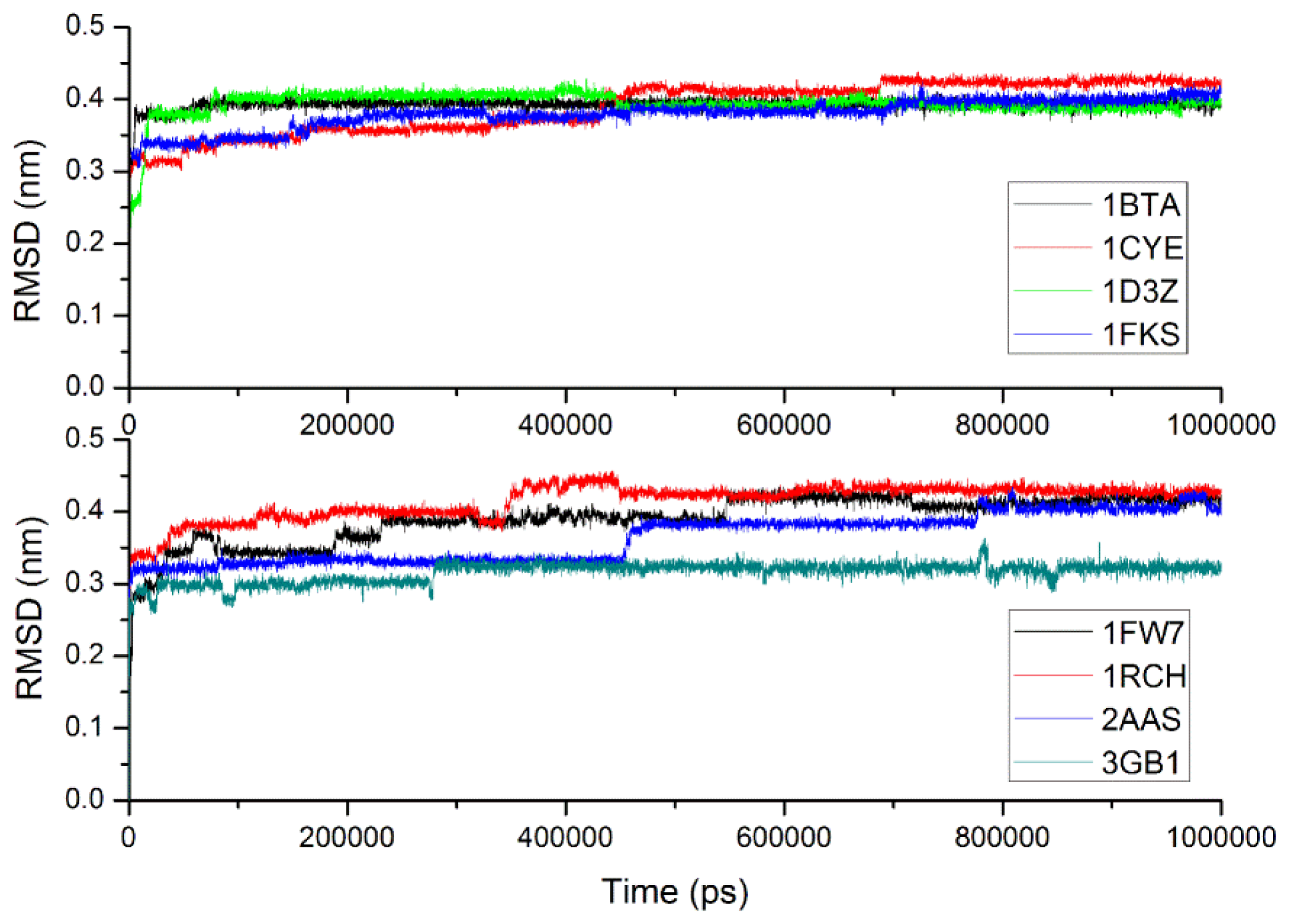
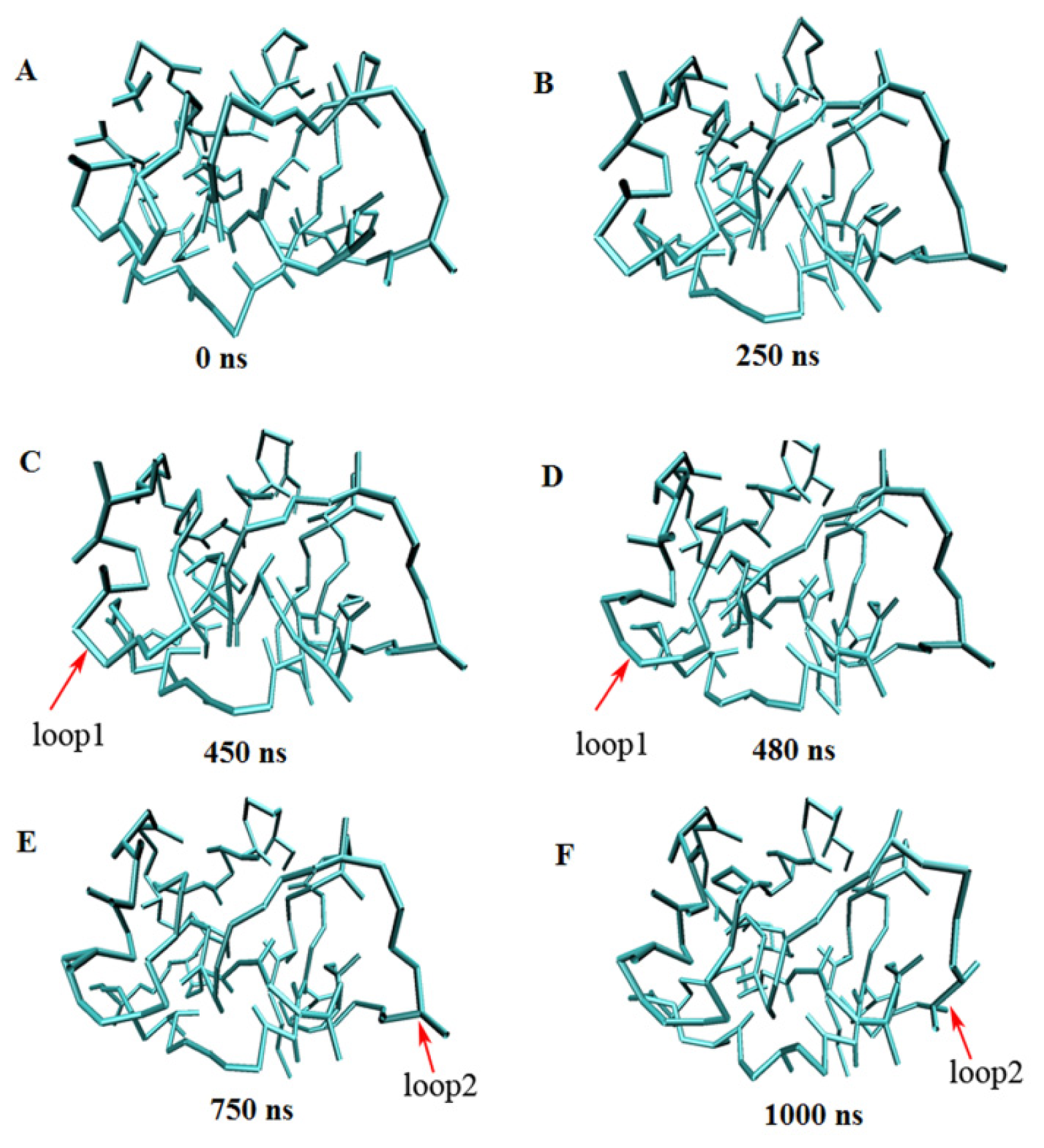
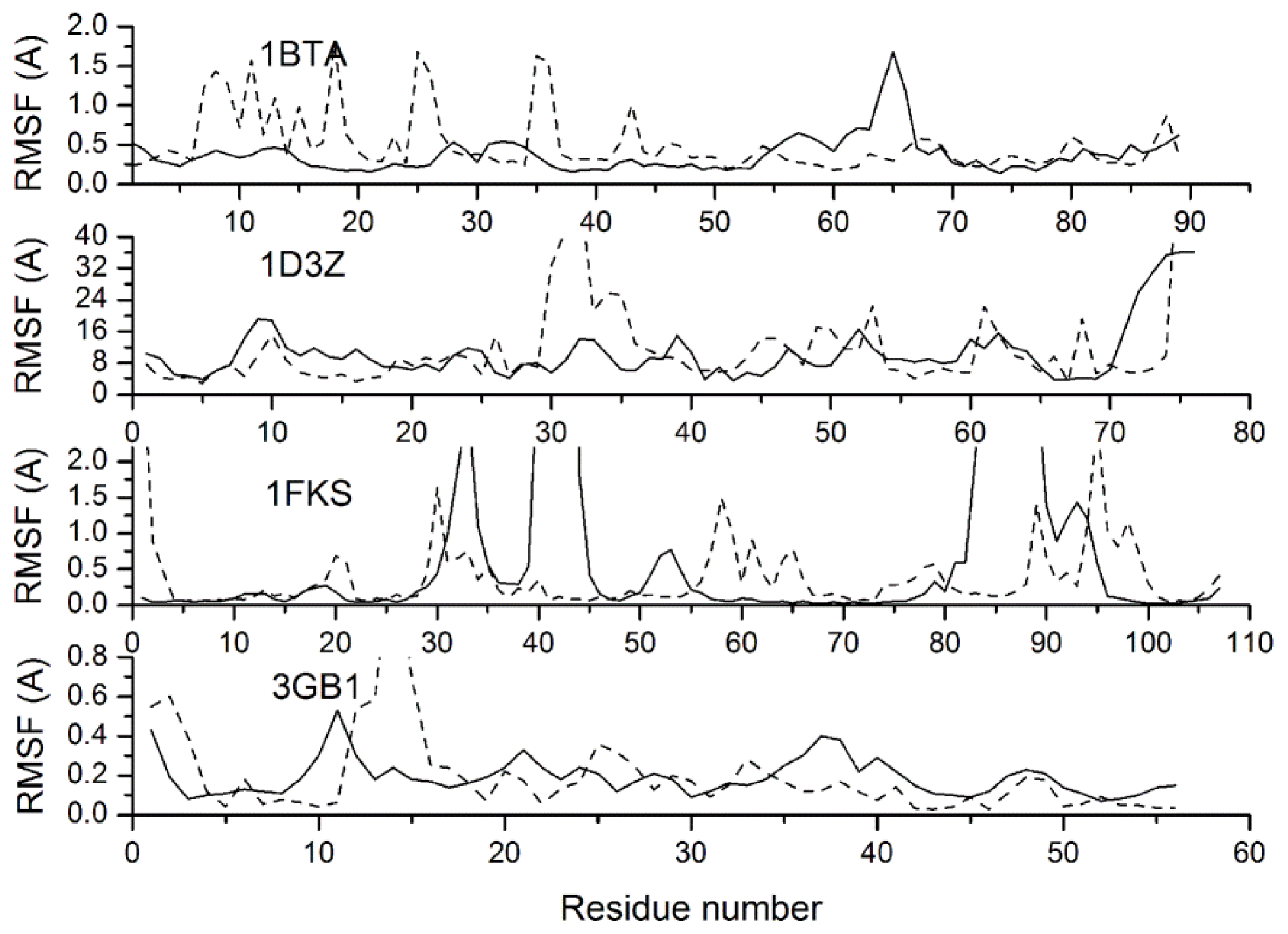
2.3. Verification of the Force Field
2.4. Efficiency of the Force Field
3. Materials and Methods
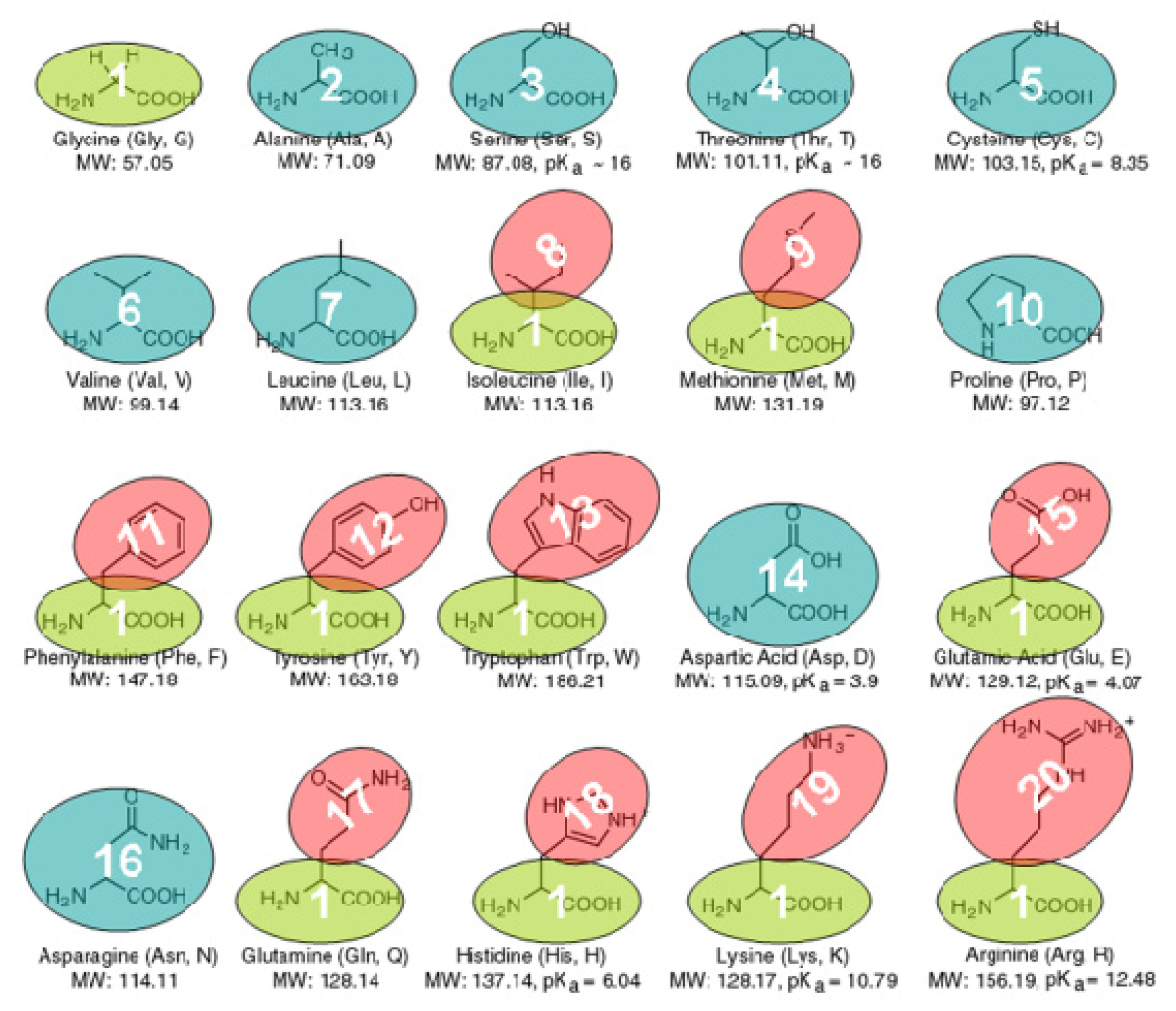
3.1. The Coarse-Grained Protein Models
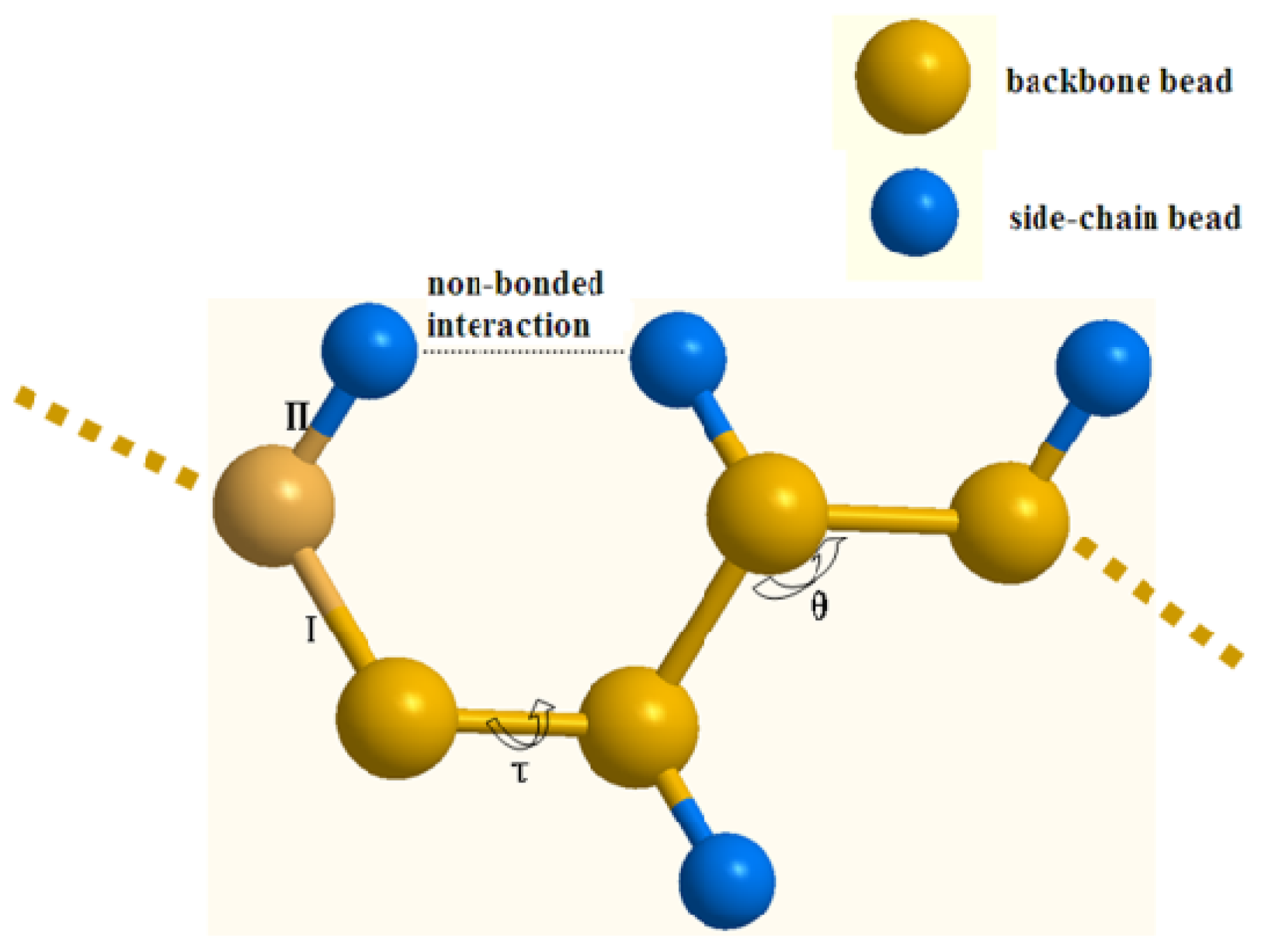
3.2. The Coarse-Grained Force Field
3.3. The Bonded Potential and Parameterization
3.4. The Non-Bonded Potential and Parameterization
3.5. Coarse-Grained Water Model and Parameterization
4. Conclusions
Supplementary Information
ijms-13-14451-s001.pdfAcknowledgments
References
- Gunsteren, W.F.; Dolenc, J. Biomolecular simulation: Historical picture and future perspectives. Biochem. Soc. Trans 2008, 36, 11–15. [Google Scholar]
- Adcock, S.A.; McCammon, J.A. Molecular dynamics: Survey of methods simulating the activity of proteins. Chem. Rev 2006, 106, 1589–1615. [Google Scholar]
- McCammon, J.A.; Gelin, B.R.; Kaplus, M. Dynamics of folded proteins. Nature 1977, 267, 585–590. [Google Scholar]
- Klepeis, J.L.; Lindorff-Larsen, K.; Dror, R.O.; Shaw, D.E. Long-timescale molecular dynamics simulations of protein structure and function. Curr. Opin. Struct. Biol 2009, 19, 120–127. [Google Scholar]
- Sanbonmatsun, K.Y.; Tung, C.-S. High performance computing in biology: Multimillion atom simulations of nanoscale systems. J. Struct. Biol 2007, 157, 470–480. [Google Scholar]
- Muller-Plathe, F. Coarse-graining in polymer simulation: From the atomistic to the mesoscopic scale and back. ChemPhysChem 2002, 3, 754–769. [Google Scholar]
- Kolinski, A.; Skolnick, J. Reduced models of proteins and their applications. Polymer 2004, 45, 511–524. [Google Scholar]
- Tozzini, V. Coarse-grained models for proteins. Curr. Opin. Struct. Biol 2005, 15, 144–150. [Google Scholar]
- Clementi, C. Coarse-grained models of protein folding: Toy models or predictive tools. Curr. Opin. Struct. Biol 2008, 18, 10–15. [Google Scholar]
- Lindahl, E.; Sansom, M.S. Membrane proteins: Molecular dynamics simulation. Curr. Opin. Struct. Biol 2008, 18, 425–431. [Google Scholar]
- Khalili-Araghi, F.; Gumbart, J.; Wen, P.; Sotomayor, M.; Tajkhorshid, E.; Schulten, K. Molecular dynamics simulations of membrane channels and transporters. Curr. Opin. Struct. Biol 2009, 19, 128–137. [Google Scholar]
- Monticelli, L.; Kandasamy, S.K.; Periole, X.; Larson, R.G.; Tieleman, D.P.; Marrink, S. The MARTINI coarse-grained force field: Extension to proteins. J. Chem. Theory Comput 2008, 4, 819–834. [Google Scholar]
- Ha-Duong, T. Protein backbone dynamics simulations using coarse-grained bonded potentials and simplified hydrogen bonds. J. Chem. Theory Comput 2010, 6, 761–773. [Google Scholar]
- Han, W.; Wan, C.K.; Jiang, F.; Wu, Y.D. PACE force field for protein simulations. 1. Full parameterization of version1 and verification. J. Chem. Theory Comput 2010, 6, 3373–3389. [Google Scholar]
- Han, W.; Wan, C.K.; Jiang, F.; Wu, Y.D. PACE force field for protein simulations. 2. Folding simulations of peptides. J. Chem. Theory Comput 2010, 6, 3390–3402. [Google Scholar]
- Basdevant, N.; Borgis, D.; Ha-Duong, T. A coarse-grained protein-protein potential derived from an all-atom force field. J. Phys. Chem. B 2007, 111, 9390–9399. [Google Scholar]
- Han, W.; Wu, Y.D. Coarse-grained protein model coupled with a coarse-grained water model: Molecular dynamics study of polyalanine-based peptides. J. Chem. Theory Comput 2007, 3, 2146–2161. [Google Scholar]
- Bereau, T.; Deserno, M. Generic coarse-grained model for protein folding and aggregation. J. Chem. Phys 2009, 130, 235106. [Google Scholar]
- Han, W.; Wan, C.K.; Wu, Y.D. Toward a coarse-grained protein model coupled with a coarse-grained solvent model: Solvation free energies of amino acid side chains. J. Chem. Theory Comput 2008, 4, 1891–1901. [Google Scholar]
- DeVane, R.; Shinoda, W.; Moore, P.B.; Klein, M.L. Transferable coarse grain nonbonded interaction model for amino acids. J. Chem. Theory Comput 2009, 5, 2115–2124. [Google Scholar]
- Shih, A.Y.; Arkhipov, A.; Freddolino, P.L.; Schulten, K. A coarse grained protein-lipid model with application to lipprotein particles. J. Phys. Chem. B 2006, 110, 3674–3684. [Google Scholar]
- Zhou, J.; Thorpe, L.F.; Izvekov, S.; Voth, G.A. Coarse-grained peptide modeling using a systematic multiscale approach. Biophys. J 2007, 92, 4289–4303. [Google Scholar]
- Korkut, A.; Hendrickson, W.A. A force field for virtual atom molecular mechanics of proteins. Proc. Natl. Acad. Sci. USA 2009, 106, 15667–15672. [Google Scholar]
- Tozzini, V.; Rocchia, W.; McCammon, J.A. Mapping all-atom models onto one-bead coarse grained models: General properties and applications to a minimal polypeptide model. J. Chem. Theory Comput 2006, 2, 667–673. [Google Scholar]
- Chang, C.A.; Trylska, J.; Tozzini, V.; McCammon, J.A. Binding pathways of ligands to HIV-1 protease: Coarse-grained and atomistic simulations. Chem. Biol. Drug Des 2007, 69, 5–13. [Google Scholar]
- Korkut, A.; Hendrickson, W.A. Computation of conformational transitions in proteins by virtual atom molecular mechanics as validated in application to adenylate kinase. Proc. Natl. Acad. Sci. USA 2009, 106, 15673–15678. [Google Scholar]
- Alemani, D.; Collu, F.; Cascella, M.; Peraro, M.D. A nonradial coarse-grained potential for proteins produces naturally stable secondary structure elements. J. Chem. Theory Comput 2010, 6, 315–324. [Google Scholar]
- Arkhipov, A.; Freddolino, P.L.; Schulten, K. Stability and dynamics of virus capsids described by coarse-grained modeling. Structure 2006, 14, 1767–1777. [Google Scholar]
- Arkhipov, A.; Yin, Y.; Schulten, K. Four-scale description of membrane sculpting by BAR domains. Biophys. J 2008, 95, 2806–2861. [Google Scholar]
- Arkhipov, A.; Freddolino, P.L.; Imada, K.; Namba, K.; Schulten, K. Coarse-grained molecular dynamics simulations of a rotating bacterial Flagellum. Biophys. J 2006, 91, 4589–4597. [Google Scholar]
- West, B.; Brown, F.L.H.; Schmid, B. Membrane-protein interactions in a generic coarse-grained model for lipid bilayers. Biophys. J 2009, 96, 101–115. [Google Scholar]
- Spijker, P.; Hoof, B.V.; Debertrand, M.; Markvoort, A.J.; Vaidehi, N.; Hilbers, P.A.J. Coarse grained molecular dynamics simulations of transmembrane protein-lipid systems. Int. J. Mol. Sci 2010, 11, 2393–2420. [Google Scholar]
- Treptow, W.; Marrink, S.; Tarek, M. Gating motions in voltage-gated potassium channels revealed by coarse-grained molecular dynamics simulations. J. Phys. Chem. B 2008, 112, 3277–3282. [Google Scholar]
- Guardiani, C.; Livi, R.; Cecconi, F. Coarse grained modeling and approaches to protein folding. Curr. Bioinforma 2010, 5, 217–240. [Google Scholar]
- Liwo, A.; Khalili, M.; Scheraga, H.A. Ab initio simulations of protein-folding pathways by molecular dynamics with the united-residue model of polypeptide chains. Proc. Natl. Acad. Sci. USA 2005, 102, 2362–2367. [Google Scholar]
- Hall, B.; Sansom, M.S.P. Coarse-grained MD simulations and protein-protein interactions: The cohesion-dockerin system. J. Chem. Theory Comput 2009, 5, 2465–2471. [Google Scholar]
- Feig, M. Is alanine dipeptide a good model for representing the torsional preferences of protein backbone? J. Chem. Theory Comput 2008, 4, 1555–1564. [Google Scholar]
- Hess, B.; Kutzner, C.; Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput 2008, 4, 435–447. [Google Scholar]
- Meyerguz, L.; Grasso, C.; Kleinberg, J.; Elber, R. Computational analysis of sequence selection mechanisms. Structure 2004, 12, 547–557. [Google Scholar]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protien data bank. Nucl. Acids Res 2000, 28, 235–242. [Google Scholar]
- Reith, D.; Putz, M.; Muller-plathe, F. Deriving effective mesoscale potentials from atomistic simulations. J. Comput. Chem 1997, 29, 292–308. [Google Scholar]
- Tozzini, V.; McCammon, J.A. A coarse grained model for the dynamics of flap opening in HIV-1 protease. Chem. Phys. Lett 2005, 413, 123–128. [Google Scholar]
- Torrie, G.M.; Valleau, J.P. Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J. Comput. Phys 1977, 23, 187–199. [Google Scholar]
- Kumar, S.; Bouzida, D.; Swendsen, R.H.; Kollman, P.A.; Rosenberg, J.M. The weighted histogram analysis method for free-energy calculations on biomolecular. I. The method. J. Comput. Chem 1992, 13, 1011–1021. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bond | Length (nm) | Bond | Length (nm) |
|---|---|---|---|
| B–SARG | 0.406 | B–SLYS | 0.344 |
| B–SGLN | 0.301 | B–SMET | 0.287 |
| B–SGLU | 0.295 | B–SPHE | 0.333 |
| B–SHIS | 0.307 | B–STRP | 0.381 |
| B–SILE | 0.226 | B–STYR | 0.371 |
| Interacting beads | Distance cij (nm) | Interacting beads | Distance cij (nm) |
|---|---|---|---|
| BALA–BALA | 0.50 | SARG–SARG | 0.60 |
| BASN–BASN | 0.60 | SGLN–SGLN | 0.45 |
| BASP–BASP | 0.55 | SGLU–SGLU | 0.45 |
| BCYS–BCYS | 0.50 | SHIS–SHIS | 0.45 |
| BGLY–BGLY | 0.40 | SILE–SILE | 0.50 |
| BLEU–BLEU | 0.55 | SLYS–SLYS | 0.45 |
| BPRO–BPRO | 0.65 | SMET–SMET | 0.45 |
| BSER–BSER | 0.50 | SPHE–SPHE | 0.45 |
| BTHR–BTHR | 0.50 | STRP–STRP | 0.65 |
| BVAL–BVAL | 0.50 | STYR–STYR | 0.55 |
| System | PDB ID | Number of residues | Number of CG waters | Number of CG beads |
|---|---|---|---|---|
| Barstar | 1BTA | 89 | 939 | 1069 |
| CheY | 1CYE | 129 | 1196 | 1375 |
| Ubiquitin | 1D3Z | 76 | 1013 | 1124 |
| FKBP12 | 1FKS | 107 | 1264 | 1417 |
| Barnase | 1FW7 | 110 | 1157 | 1312 |
| RNase H | 1RCH | 155 | 1982 | 2207 |
| RNase A | 2AAS | 124 | 1126 | 1296 |
| protein G | 3GB1 | 56 | 887 | 963 |
| PDB | CG-MD | AA-MD * | ||||
|---|---|---|---|---|---|---|
| Simulation length (ns) | Avg. Ca RMSD (nm) | Final Ca RMSD (nm) | Simulation length (ns) | Avg. Ca RMSD (nm) | Final Ca RMSD (nm) | |
| 1bta | 1000 | 0.393(0.010) | 0.396 | 142.9 | 0.134(0.016) | 0.121 |
| 1cye | 1000 | 0.389(0.036) | 0.422 | 124.7 | 0.143(0.020) | 0.170 |
| 1d3z | 1000 | 0.394(0.020) | 0.395 | 22.0 | 0.141(0.021) | 0.128 |
| 1fks | 1000 | 0.379(0.021) | 0.415 | 143.5 | 0.358(0.074) | 0.477 |
| 1fw7 | 1000 | 0.391(0.033) | 0.408 | 148.0 | 0.171(0.015) | 0.167 |
| 1rch | 1000 | 0.415(0.025) | 0.431 | 121.5 | 0.278(0.017) | 0.289 |
| 2aas | 1000 | 0.364(0.034) | 0.400 | 148.3 | 0.249(0.043) | 0.321 |
| 3gb1 | 1000 | 0.316(0.015) | 0.323 | 50.0 | 0.106(0.020) | 0.143 |
| PDB | The proposed CG-MD | MARTINI | AA-MD | ||||
|---|---|---|---|---|---|---|---|
| Simulation time (s) | Avg. Ca RMSD (nm) | Avg. Ca RMSD in vacuum (nm) | Simulation time (s) | Avg. Ca RMSD (nm) | Simulation time (s) | Avg. Ca RMSD (nm) | |
| 1bta | 3501 | 0.210 | 0.637 | 4002 | 0.341 | 313062 | 0.148 |
| 1cye | 4309 | 0.292 | 0.440 | 4972 | 0.503 | 398432 | 0.148 |
| 1d3z | 4032 | 0.283 | 0.416 | 4261 | 0.426 | 334203 | 0.185 |
| 1fks | 4484 | 0.324 | 0.505 | 5242 | 0.378 | 436792 | 0.220 |
| 1fw7 | 4391 | 0.247 | 0.574 | 4902 | 0.400 | 388712 | 0.171 |
| 1rch | 7330 | 0.337 | 0.681 | 7845 | 0.357 | 650507 | 0.234 |
| 2aas | 4424 | 0.284 | 0.689 | 4801 | 0.421 | 387623 | 0.259 |
| 3gb1 | 3432 | 0.275 | 0.501 | 3721 | 0.339 | 274854 | 0.128 |
| Side-chain bead | Analogous compound | Side-chain bead | Analogous compound |
|---|---|---|---|
| SARG | n-propylguanidine | SLYS | n-butylamine |
| SGLN | propionamide | SMET | methyl propyl sulfide |
| SGLU | propionic acid | SPHE | toluene |
| SHIS | 4-methylimidazole | STRP | 3-methylindole |
| SILE | n-butane | STYR | p-cresol |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gu, J.; Bai, F.; Li, H.; Wang, X. A Generic Force Field for Protein Coarse-Grained Molecular Dynamics Simulation. Int. J. Mol. Sci. 2012, 13, 14451-14469. https://doi.org/10.3390/ijms131114451
Gu J, Bai F, Li H, Wang X. A Generic Force Field for Protein Coarse-Grained Molecular Dynamics Simulation. International Journal of Molecular Sciences. 2012; 13(11):14451-14469. https://doi.org/10.3390/ijms131114451
Chicago/Turabian StyleGu, Junfeng, Fang Bai, Honglin Li, and Xicheng Wang. 2012. "A Generic Force Field for Protein Coarse-Grained Molecular Dynamics Simulation" International Journal of Molecular Sciences 13, no. 11: 14451-14469. https://doi.org/10.3390/ijms131114451