Single Molecule Fluorescence Detection and Tracking in Mammalian Cells: The State-of-the-Art and Future Perspectives

Abstract
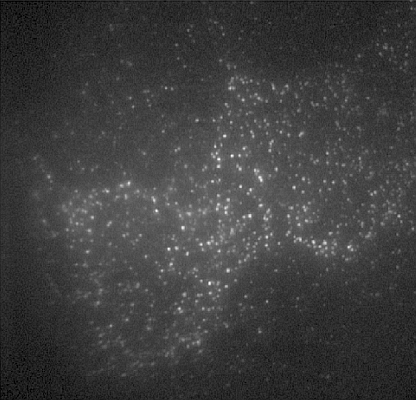
:
1. Introduction
2. Experimental Design
2.1. Choosing a Cell Line
2.2. Fluorescent Probes for Single Molecule Tracking
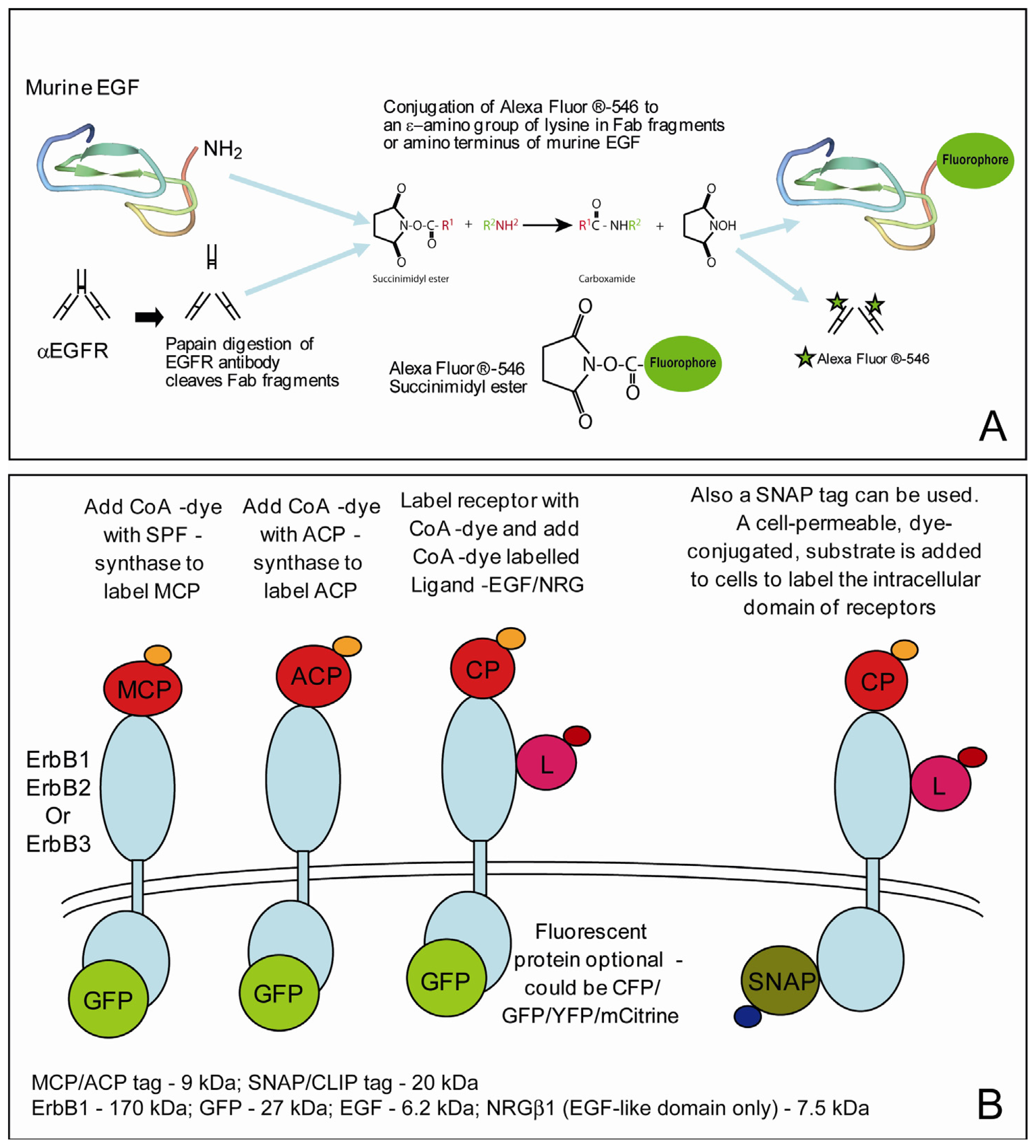
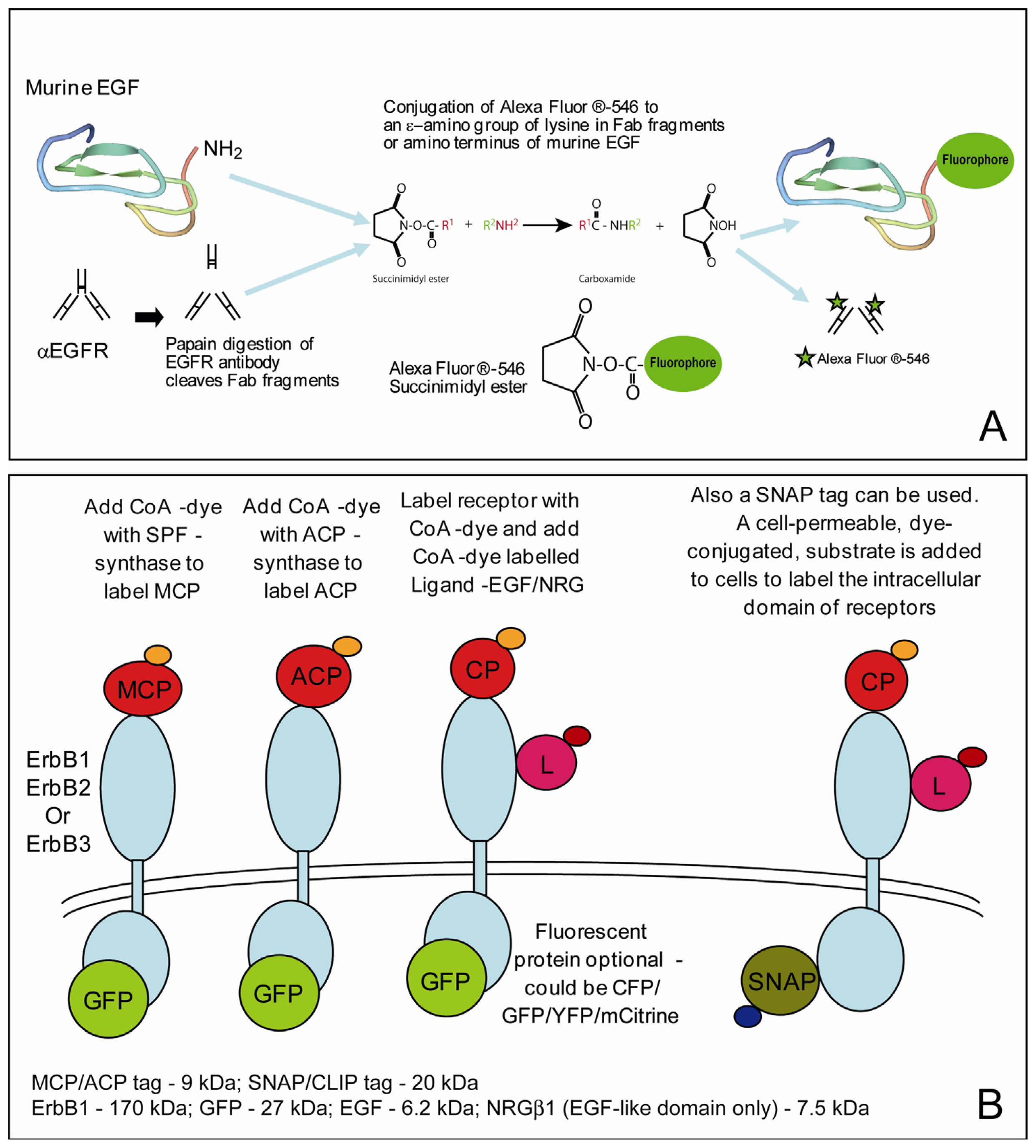
2.3. Labeling the Proteins of Interest
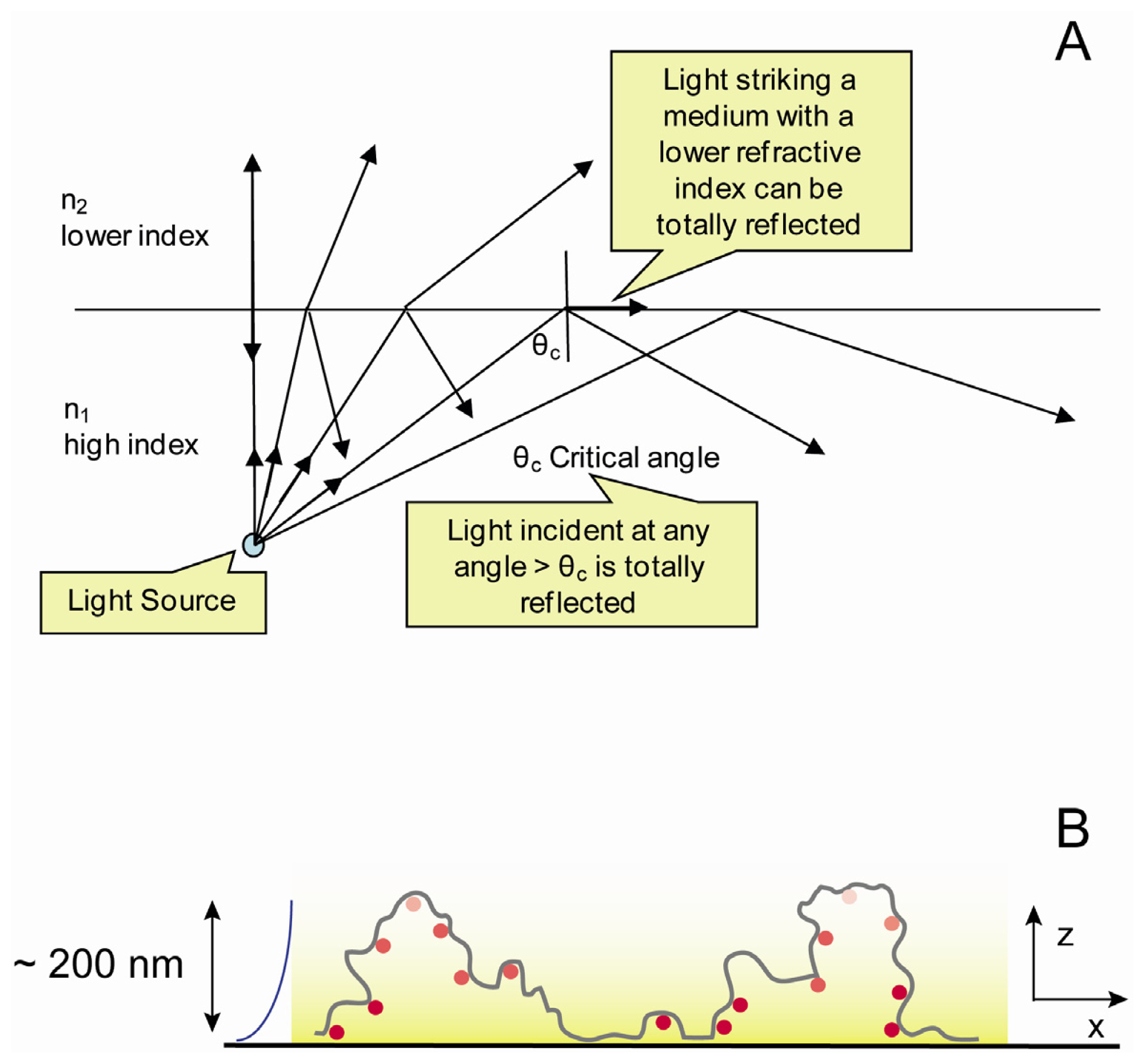
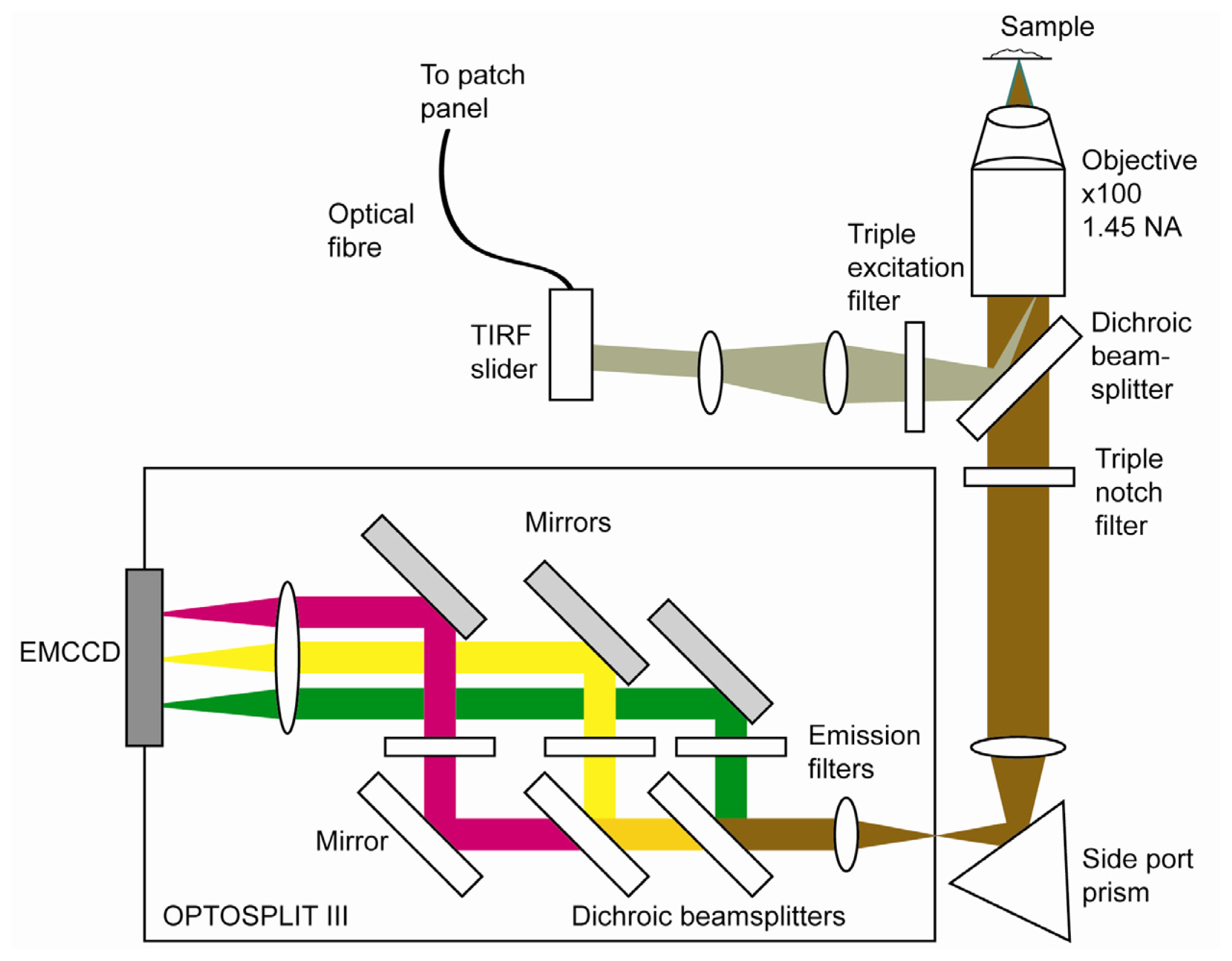
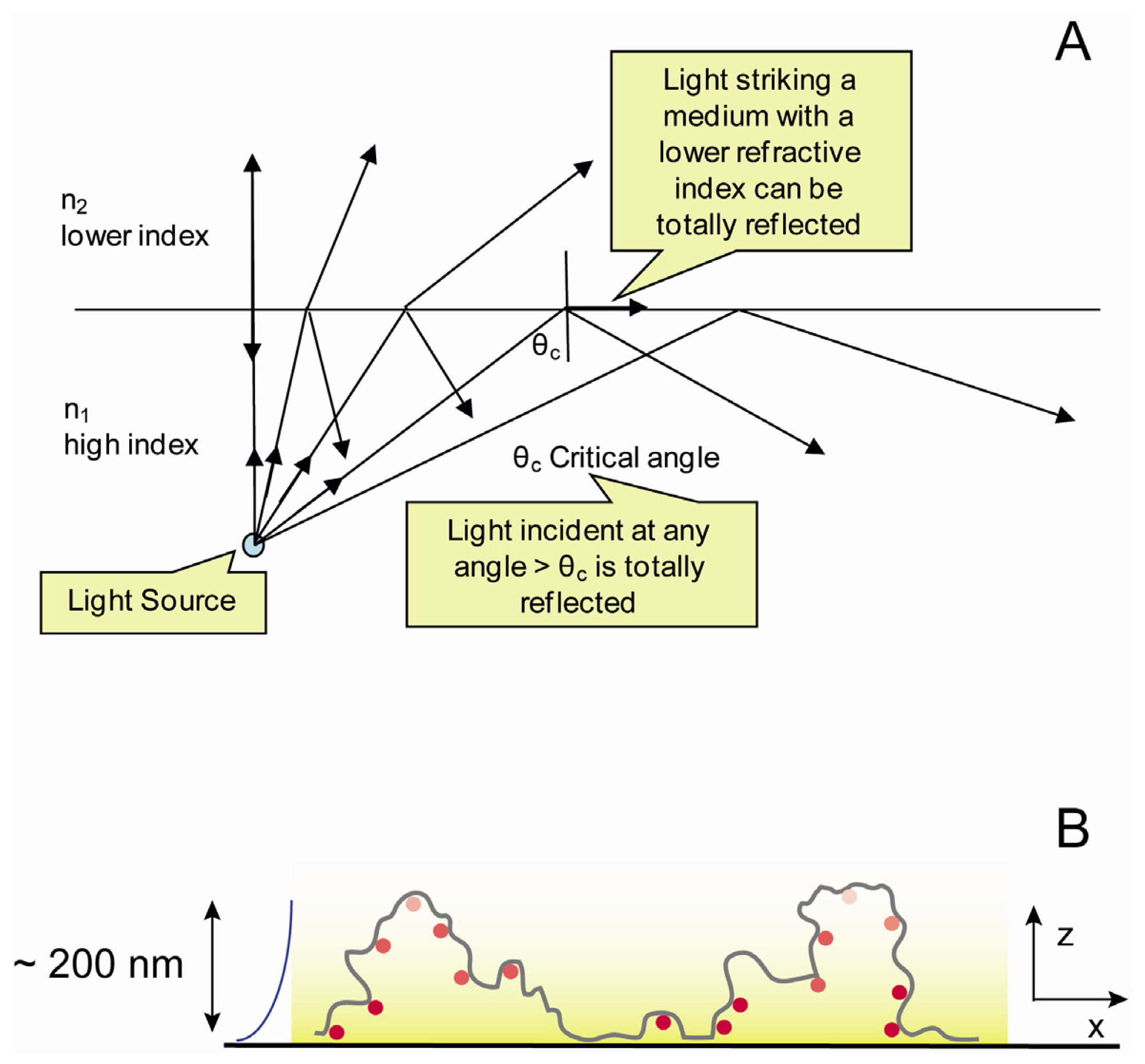
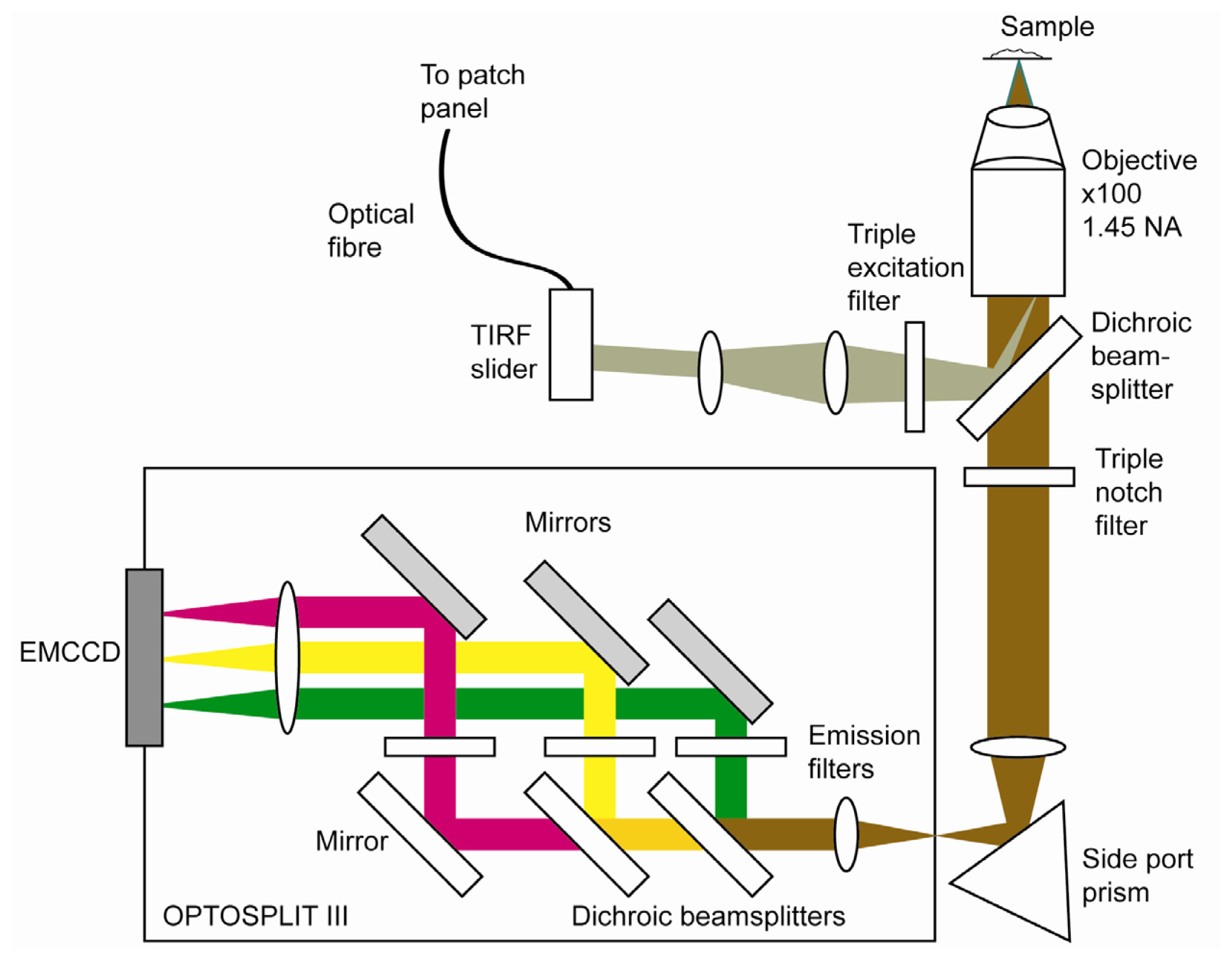
2.4. Optical Set up
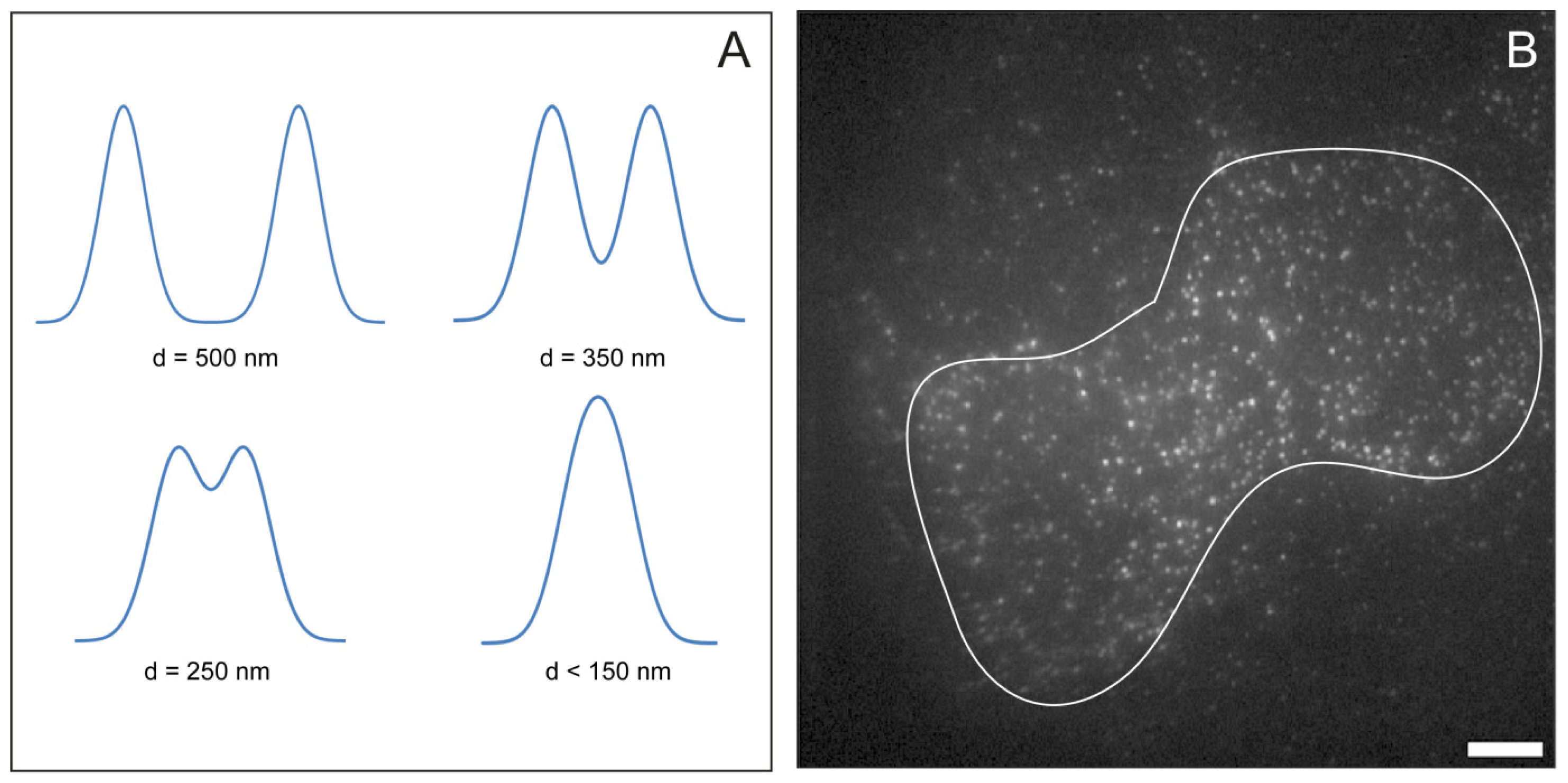
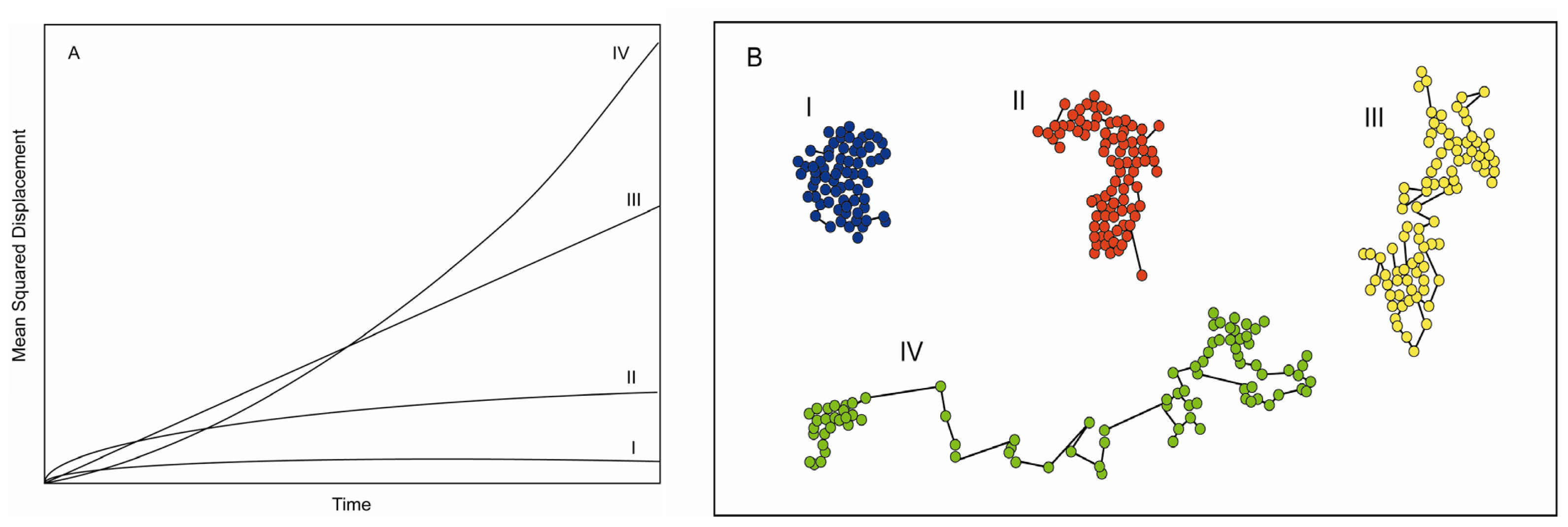
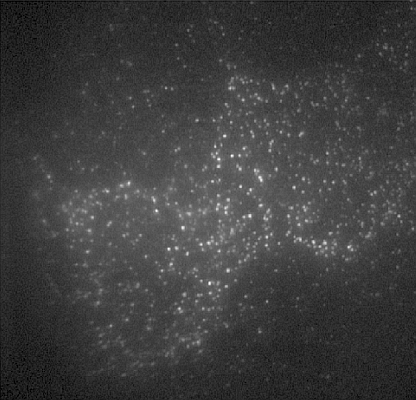
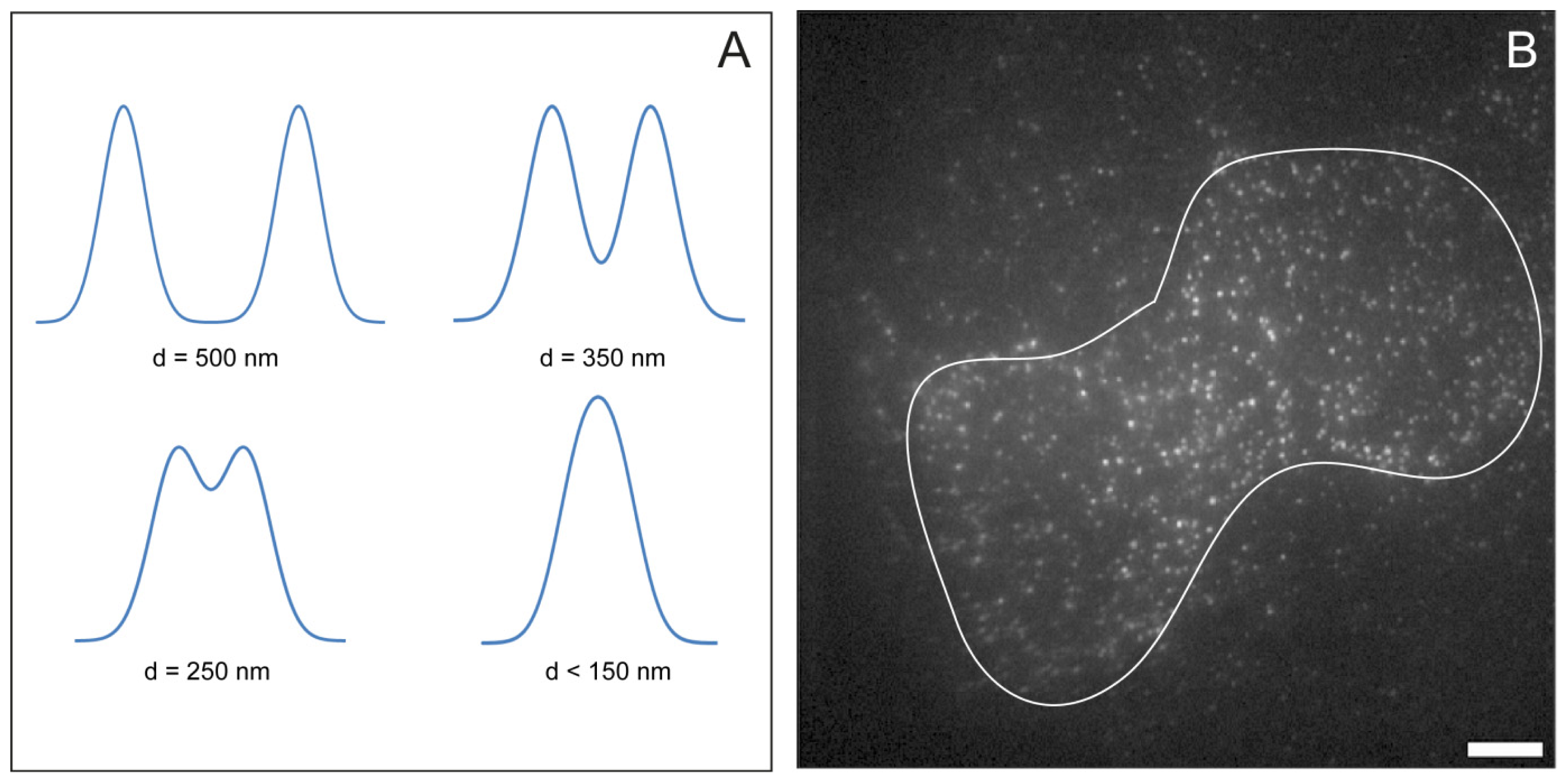
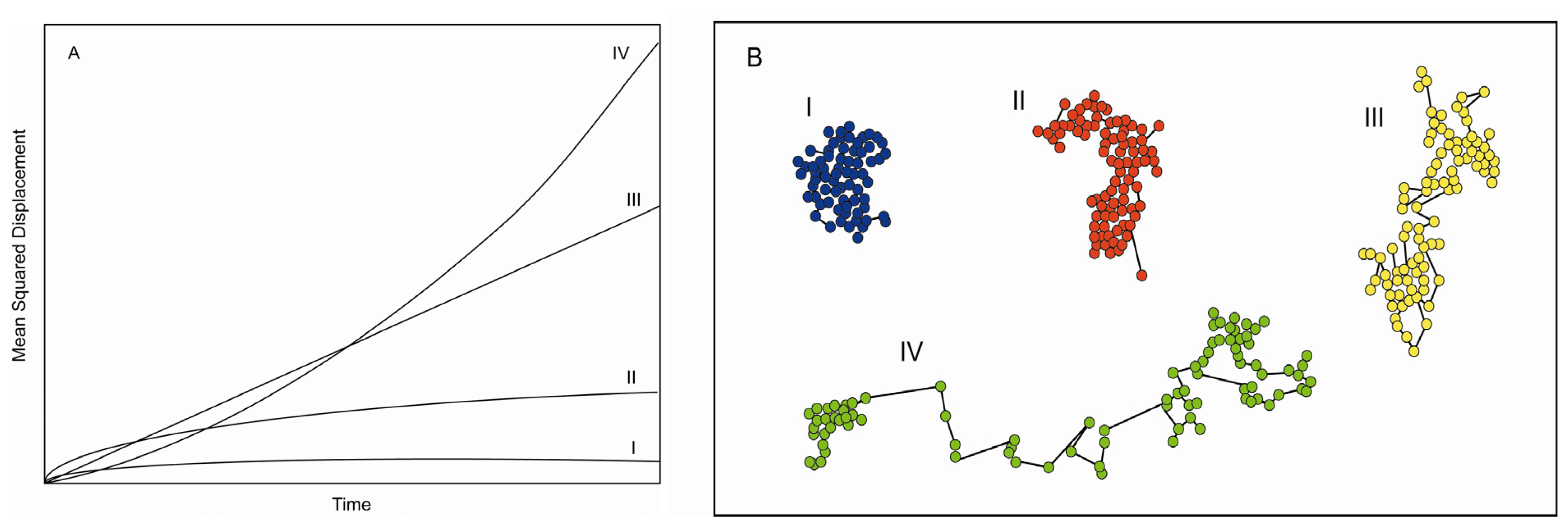
2.5. Feature Detection, Localisation and Analysis
3. Information Content
4. Examples of Applications
5. Conclusions and Future Perspectives
Acknowledgments
References
- Svoboda, K.; Schmidt, C.F.; Schnapp, B.J.; Block, S.M. Direct observation of kinesin stepping by optical trapping interferometry. Nature 1993, 365, 721–727. [Google Scholar]
- Shaklee, P.M.; Idema, T.; Bourel-Bonnet, L.; Dogterom, M.; Schmidt, T. Kinesin recycling in stationary membrane tubes. Biophys. J 2010, 99, 1835–1841. [Google Scholar]
- Yildiz, A.; Selvin, P.R. Fluorescence imaging with one nanometer accuracy: Application to molecular motors. Acc. Chem. Res 2005, 38, 574–582. [Google Scholar]
- Kapanidis, A.N.; Margeat, E.; Ho, S.O.; Kortkhonjia, E.; Weiss, S.; Ebright, R.H. Initial transcription by RNA polymerase proceeds through a DNA-scrunching mechanism. Science 2006, 314, 1144–1147. [Google Scholar]
- Roy, R.; Kozlov, A.G.; Lohman, T.M.; Ha, T. SSB protein diffusion on single-stranded DNA stimulates RecA filament formation. Nature 2009, 461, 1092–1097. [Google Scholar]
- Weiss, S. Measuring conformational dynamics of biomolecules by single molecule fluorescence spectroscopy. Nat. Struct. Biol 2000, 7, 724–729. [Google Scholar]
- Xie, X.S.; Choi, P.J.; Li, G.-W.; Lee, N.K.; Lia, G. Single-molecule approach to molecular biology in living bacterial cells. Annu. Rev. Biophys 2008, 37, 417–444. [Google Scholar]
- Biteen, J.S.; Shapiro, L.; Moerner, W.E. Exploring protein superstructures and dynamics in live bacterial cells using single-molecule and superresolution imaging. Methods Mol. Biol 2011, 783, 139–158. [Google Scholar]
- Kubitschek, H.E. Cell-growth and abrupt doubling of membrane-proteins in Escherichia-coli during the division cycle. J. Gen. Microbiol 1990, 136, 599–606. [Google Scholar]
- Sowa, Y.; Rowe, A.D.; Leake, M.C.; Yakushi, T.; Homma, M.; Ishijima, A.; Berry, R.M. Direct observation of steps in rotation of the bacterial flagellar motor. Nature 2005, 437, 916–919. [Google Scholar]
- Leake, M.C.; Chandler, J.H.; Wadhams, G.H.; Bai, F.; Berry, R.M.; Armitage, J.P. Stoichiometry and turnover in single, functioning membrane protein complexes. Nature 2006, 443, 355–358. [Google Scholar]
- Yu, J.; Xiao, J.; Ren, X.J.; Lao, K.Q.; Xie, X.S. Probing gene expression in live cells, one protein molecule at a time. Science 2006, 311, 1600–1603. [Google Scholar]
- Elf, J.; Li, G.-W.; Xie, X.S. Probing transcription factor dynamics at the single-molecule level in a living cell. Science 2007, 316, 1191–1194. [Google Scholar]
- Reyes-Lamothe, R.; Sherratt, D.J.; Leake, M.C. Stoichiometry and architecture of active DNA replication machinery in Escherichia coli. Science 2010, 328, 498–501. [Google Scholar]
- Li, G.-W.; Xie, X.S. Central dogma at the single-molecule level in living cells. Nature 2011, 475, 308–315. [Google Scholar]
- Monici, M. Cell and tissue autofluorescence research and diagnostic applications. Biotechnol. Annu. Rev 2005, 11, 227–256. [Google Scholar]
- Axelrod, D. Cell-substrate contacts illuminated by total internal reflection fluorescence. J. Cell Biol 1981, 89, 141–145. [Google Scholar]
- Kusumi, A.; Suzuki, K.G.N.; Kasai, R.S.; Ritchie, K.; Fujiwara, T.K. Hierarchical mesoscale domain organization of the plasma membrane. Trends Biochem. Sci 2011, 36, 604–615. [Google Scholar]
- Kusumi, A.; Nakada, C.; Ritchie, K.; Murase, K.; Suzuki, K.; Murakoshi, H.; Kasai, R.S.; Kondo, J.; Fujiwara, T. Paradigm shift of the plasma membrane concept from the two-dimensional continuum fluid to the partitioned fluid: High-speed single-molecule tracking of membrane molecules. Annu. Rev. Biophys. Biomol. Struct 2005, 34, 351–378. [Google Scholar]
- Hern, J.A.; Baig, A.H.; Mashanov, G.I.; Birdsall, B.; Corrie, J.E.T.; Lazareno, S.; Molloy, J.E.; Birdsall, N.J.M. Formation and dissociation of M(1) muscarinic receptor dimers seen by total internal reflection fluorescence imaging of single molecules. Proc. Natl. Acad. Sci. USA 2010, 107, 2693–2698. [Google Scholar]
- Kasai, R.S.; Suzuki, K.G.N.; Prossnitz, E.R.; Koyama-Honda, I.; Nakada, C.; Fujiwara, T.K.; Kusumi, A. Full characterization of GPCR monomer-dimer dynamic equilibrium by single molecule imaging. J. Cell Biol 2011, 192, 463–480. [Google Scholar]
- Ullrich, A.; Coussens, L.; Hayflick, J.S.; Dull, T.J.; Gray, A.; Tam, A.W.; Lee, J.; Yarden, Y.; Libermann, T.A.; Schlessinger, J.; et al. Human epidermal growth factor receptor cDNA sequence and aberrant expression of the amplified gene in A431 epidermoid carcinoma cells. Nature 1984, 309, 418–425. [Google Scholar]
- Fabricant, R.N.; De Larco, J.E.; Todaro, G.J. Nerve growth factor receptors on human melanoma cells in culture. Proc. Natl. Acad. Sci. USA 1977, 74, 565–569. [Google Scholar]
- Defize, L.H.; Boonstra, J.; Meisenhelder, J.; Kruijer, W.; Tertoolen, L.G.; Tilly, B.C.; Hunter, T.; van Bergen en Henegouwen, P.M.; Moolenaar, W.H.; de Laat, S.W. Signal transduction by epidermal growth factor occurs through the subclass of high affinity receptors. J. Cell Biol 1989, 109, 2495–2507. [Google Scholar]
- Sako, Y.; Minoghchi, S.; Yanagida, T. Single-molecule imaging of EGFR signalling on the surface of living cells. Nat. Cell Biol 2000, 2, 168–172. [Google Scholar]
- Yu, C.; Hale, J.; Ritchie, K.; Prasad, N.K.; Irudayaraj, J. Receptor overexpression or inhibition alters cell surface dynamics of EGF-EGFR interaction: New insights from real-time single molecule analysis. Biochem. Biophys. Res. Commun 2009, 378, 376–382. [Google Scholar]
- Manley, S.; Gillette, J.M.; Patterson, G.H.; Shroff, H.; Hess, H.F.; Betzig, E.; Lippincott-Schwartz, J. High-density mapping of single-molecule trajectories with photoactivated localization microscopy. Nat. Methods 2008, 5, 155–157. [Google Scholar]
- Hynes, N.E.; Lane, H.A. ERBB receptors and cancer: The complexity of targeted inhibitors. Nat. Rev. Cancer 2005, 5, 341–354. [Google Scholar]
- Cohen, B.D.; Kiener, P.A.; Green, J.M.; Foy, L.; Fell, H.P.; Zhang, K. The relationship between human epidermal growth-like factor receptor expression and cellular transformation in NIH3T3 cells. J. Biol. Chem 1996, 271, 30897–30903. [Google Scholar]
- Bunn, P.A.; Helfrich, B.; Soriano, A.F.; Franklin, W.A.; Varella-Garcia, M.; Hirsch, F.R.; Baron, A.; Zeng, C.; Chan, D.C. Expression of Her-2/neu in human lung cancer cell lines by immunohistochemistry and fluorescence in situ hybridization and its relationship to in vitro cytotoxicity by trastuzumab and chemotherapeutic agents. Clin. Cancer Res 2001, 7, 3239–3250. [Google Scholar]
- Wu, W.K.K.; Tse, T.T.M.; Sung, J.J.Y.; Li, Z.J.; Yu, L.; Ch, C.H. Expression of ErbB receptors and their cognate ligands in gastric and colon cancer cell lines. Anticancer Res 2009, 29, 229–234. [Google Scholar]
- Zhang, W.; Jiang, Y.; Wang, Q.; Ma, X.; Xiao, Z.; Zuo, W.; Fang, X.; Chen, Y.G. Single-molecule imaging reveals transforming growth factor-beta-induced type II receptor dimerization. Proc. Natl. Acad. Sci. USA 2009, 106, 15679–15683. [Google Scholar]
- Bauer, M.; Kristensen, B.W.; Meyer, M.; Gasser, T.; Widmer, H.R.; Zimmer, J.; Ueffing, M. Toxic effects of lipid-mediated gene transfer in ventral mesencephalic explant cultures. Basic Clin. Pharmacol. Toxicol 2006, 98, 395–400. [Google Scholar]
- Breunig, M.; Lungwitz, U.; Liebl, R.; Goepferich, A. Breaking up the correlation between efficacy and toxicity for nonviral gene delivery. Proc. Natl. Acad. Sci. USA 2007, 104, 14454–14459. [Google Scholar]
- Karra, D.; Dahm, R. Transfection techniques for neuronal cells. J. Neurosci 2010, 30, 6171–6177. [Google Scholar]
- Macdonald, J.; Taylor, L.; Sherman, A.; Kawakami, K.; Takahashi, Y.; Sang, H.M.; McGrew, M.J. Efficient genetic modification and germ-line transmission of primordial germ cells using piggyBac and Tol2 transposons. Proc. Natl. Acad. Sci. USA 2012, 109, E1466–E1472. [Google Scholar]
- Macdonald, J.L.; Pike, L.J. Heterogeneity in EGF-binding affinities arises from negative cooperativity in an aggregating system. Proc. Natl. Acad. Sci. USA 2008, 105, 112–117. [Google Scholar]
- Ha, T.; Tinnefeld, P. Photophysics of fluorescent probes for single-molecule biophysics and super-resolution imaging. Annu. Rev. Phys. Chem 2012, 63, 595–617. [Google Scholar]
- Tynan, C.J.; Clarke, D.T.; Coles, B.C.; Rolfe, D.J.; Martin-Fernandez, M.L.; Webb, S.E.D. Multicolour single molecule imaging in cells with near infra-red dyes. PLoS One 2012, 7, e36265. [Google Scholar]
- Berlier, J.E.; Rothe, A.; Buller, G.; Bradford, J.; Gray, D.R.; Filanoski, B.J.; Telford, W.G.; Yue, S.; Liu, J.; Cheung, C.Y.; et al. Quantitative comparison of long-wavelength Alexa Fluor dyes to Cy dyes: Fluorescence of the dyes and their bioconjugates. J. Histochem. Cytochem 2003, 51, 1699–1712. [Google Scholar]
- Kuimova, M.K.; Botchway, S.W.; Parker, A.W.; Balaz, M.; Collins, H.A.; Anderson, H.L.; Suhling, K.; Ogilby, P.R. Imaging intracellular viscosity of a single cell during photoinduced cell death. Nat. Chem 2009, 1, 69–73. [Google Scholar]
- Martin-Fernandez, M.L.; Clarke, D.T.; Tobin, M.J.; Jones, G.R. Real-time studies of the interactions between epidermal growth factor and its receptor during endocytic trafficking. Cell. Mol. Biol 2000, 46, 1103–1112. [Google Scholar]
- Forkey, J.N.; Quinlan, M.E.; Shaw, M.A.; Corrie, J.E.; Goldman, Y.E. Three-dimensional structural dynamics of myosin V by single-molecule fluorescence polarization. Nature 2003, 422, 399–404. [Google Scholar]
- Webb, S.E.; Rolfe, D.J.; Needham, S.R.; Roberts, S.K.; Clarke, D.T.; McLachlan, C.I.; Hobson, M.P.; Martin-Fernandez, M.L. Simultaneous widefield single molecule orientation and FRET microscopy in cells. Opt. Express 2008, 16, 20258–20265. [Google Scholar]
- Martin-Fernandez, M.; Clarke, D.T.; Tobin, M.J.; Jones, S.V.; Jones, G.R. Preformed oligomeric epidermal growth factor receptors undergo an ectodomain structure change during signaling. Biophys. J 2002, 82, 2415–2427. [Google Scholar]
- Zanetti-Domingues, L.C.; Martin-Fernandez, M.L.; Needham, S.R.; Rolfe, D.J.; Clarke, D.T. A systematic investigation of differential effects of cell culture substrates on the extent of artifacts in single-molecule tracking. PLoS One 2011, 7, e45655. [Google Scholar]
- Day, R.N.; Davidson, M.W. The fluorescent protein palette: Tools for cellular imaging. Chem. Soc. Rev 2009, 38, 2887–2921. [Google Scholar]
- Tsien, R.Y. The green fluorescent protein. Annu. Rev. Biochem 1998, 67, 509–544. [Google Scholar]
- Tavare, J.M.; Fletcher, L.M.; Welsh, G.I. Review—Using green fluorescent protein to study intracellular signalling. J. Endocrinol 2001, 170, 297–306. [Google Scholar]
- Felgner, P.L.; Gadek, T.R.; Holm, M.; Roman, R.; Chan, H.W.; Wenz, M.; Northrop, J.P.; Ringold, G.M.; Danielsen, M. Lipofection: A highly efficient, lipid-mediated DNA-transfection procedure. Proc. Natl. Acad. Sci. USA 1987, 84, 7413–7417. [Google Scholar]
- Chalfie, M.; Kain, S. Green Fluorescent Protein: Properties, Applications and Protocols, 2nd ed; Wiley: Chichester, UK, 2006. [Google Scholar]
- Colosimo, A.; Goncz, K.K.; Holmes, A.R.; Kunzelmann, K.; Novelli, G.; Malone, R.W.; Bennett, M.J.; Gruenert, D.C. Transfer and expression of foreign genes in mammalian cells. Biotechniques 2000, 29. [Google Scholar]
- Neumann, E.; Schaefer-Ridder, M.; Wang, Y.; Hofschneider, P.H. Gene transfer into mouse lyoma cells by electroporation in high electric fields. EMBO J 1982, 1, 841–845. [Google Scholar]
- Dunne, P.D.; Fernandes, R.A.; McColl, J.; Yoon, J.W.; James, J.R.; Davis, S.J.; Klenerman, D. DySCo: Quantitating associations of membrane proteins using two-color single-molecule tracking. Biophys. J 2009, 97, L5–L7. [Google Scholar]
- Wu, B.; Piatkevich, K.D.; Lionnet, T.; Singer, R.H.; Verkhusha, V.V. Modern fluorescent proteins and imaging technologies to study gene expression, nuclear localization, and dynamics. Curr. Opin. Cell Biol 2011, 23, 310–317. [Google Scholar]
- Baker, M. Bright light, better labels. Nature 2011, 478, 137–142. [Google Scholar]
- Moerner, W.E.; Fromm, D.P. Methods of single-molecule fluorescence spectroscopy and microscopy. Rev. Sci. Instrum 2003, 74, 3597–3619. [Google Scholar]
- Zhang, H.; Shu, D.; Browne, M.; Guo, P. Approaches for stoichiometry and distance determination of nanometer bio-complex by dual-channel single molecule imaging. IEEE NIH Life Sci. Syst. Appl. Workshop 2009, 2009, 124–127. [Google Scholar]
- Dickson, R.M.; Cubitt, A.B.; Tsien, R.Y.; Moerner, W.E. On/off blinking and switching behaviour of single molecules of green fluorescent protein. Nature 1997, 388, 355–358. [Google Scholar]
- Vogelsang, J.; Kasper, R.; Steinhauer, C.; Person, B.; Heilemann, M.; Sauer, M.; Tinnefeld, P. A reducing and oxidizing system minimizes photobleaching and blinking of fluorescent dyes. Angew. Chem. Int. Ed 2008, 47, 5465–5469. [Google Scholar]
- Steinhauer, C.; Forthmann, C.; Vogelsang, J.; Tinnefeld, P. Superresolution microscopy on the basis of engineered dark states. J. Am. Chem. Soc 2008, 130, 16840–16841. [Google Scholar]
- Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.S.; Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging intracellular fluorescent proteins at nanometer resolution. Science 2006, 313, 1642–1645. [Google Scholar]
- Fernandez-Suarez, M.; Ting, A.Y. Fluorescent probes for super-resolution imaging in living cells. Nat. Rev. Mol. Cell Biol 2008, 9, 929–943. [Google Scholar]
- Huang, B.; Bates, M.; Zhuang, X. Super-resolution fluorescence microscopy. Annu. Rev. Biochem 2009, 78, 993–1016. [Google Scholar]
- Barroso, M.M. Quantum dots in cell biology. J. Histochem. Cytochem 2011, 59, 237–251. [Google Scholar]
- Michalet, X.; Pinaud, F.F.; Bentolila, L.A.; Tsay, J.M.; Doose, S.; Li, J.J.; Sundaresan, G.; Wu, A.M.; Gambhir, S.S.; Weiss, S. Quantum dots for live cells, in vivo imaging, and diagnostics. Science 2005, 307, 538–544. [Google Scholar]
- Patel, G.S.; Kiuchi, T.; Lawler, K.; Ofo, E.; Fruhwirth, G.O.; Kelleher, M.; Shamil, E.; Zhang, R.; Selvin, P.R.; Santis, G.; et al. The challenges of integrating molecular imaging into the optimization of cancer therapy. Integr. Biol 2011, 3, 603–631. [Google Scholar]
- Lidke, D.S.; Nagy, P.; Barisas, B.G.; Heintzmann, R.; Post, J.N.; Lidke, K.A.; Clayton, A.H.; Arndt-Jovin, D.J.; Jovin, T.M. Imaging molecular interactions in cells by dynamic and static fluorescence anisotropy (rFLIM and emFRET). Biochem. Soc. Trans 2003, 31, 1020–1027. [Google Scholar]
- Low-Nam, S.T.; Lidke, K.A.; Cutler, P.J.; Roovers, R.C.; van Bergen en Henegouwen, P.M.; Wilson, B.S.; Lidke, D.S. ErbB1 dimerization is promoted by domain co-confinement and stabilized by ligand binding. Nat. Struct. Mol. Biol 2011, 18, 1244–1249. [Google Scholar]
- Leatherdale, C.A.; Woo, W.K.; Mikulec, F.V.; Bawendi, M.G. On the absorption cross section of CdSe nanocrystal quantum dots. J. Phys. Chem. B 2002, 106, 7619–7622. [Google Scholar]
- Lidke, D.S.; Nagy, P.; Heintzmann, R.; Arndt-Jovin, D.J.; Post, J.N.; Grecco, H.E.; Jares-Erijman, E.A.; Jovin, T.M. Quantum dot ligands provide new insights into erbB/HER receptor-mediated signal transduction. Nat. Biotechnol 2004, 22, 198–203. [Google Scholar]
- Chung, I.; Akita, R.; Vandlen, R.; Toomre, D.; Schlessinger, J.; Mellman, I. Spatial control of EGF receptor activation by reversible dimerization on living cells. Nature 2010, 464, 783–787. [Google Scholar]
- Howarth, M.; Takao, K.; Hayashi, Y.; Ting, A.Y. Targeting quantum dots to surface proteins in living cells with biotin ligase. Proc. Natl. Acad. Sci. USA 2005, 102, 7583–7588. [Google Scholar]
- Duchesne, L.; Octeau, V.; Bearon, R.N.; Beckett, A.; Prior, I.A.; Lounis, B.; Fernig, D.G. Transport of fibroblast growth factor 2 in the pericellular matrix is controlled by the spatial distribution of its binding sites in heparan sulfate. PLoS Biol 2012, 10, e1001361. [Google Scholar]
- Lundberg, E.; Hoiden-Guthenberg, I.; Larsson, B.; Uhlen, M.; Graslund, T. Site-specifically conjugated anti-HER2 Affibody (R) molecules as one-step reagents for target expression analyses on cells and xenograft samples. J. Immunol. Methods 2007, 319, 53–63. [Google Scholar]
- Stumpp, M.T.; Binz, H.K.; Amstutz, P. DARPins: A new generation of protein therapeutics. Drug Discovery Today 2008, 13, 695–701. [Google Scholar]
- Keppler, A.; Gendreizig, S.; Gronemeyer, T.; Pick, H.; Vogel, H.; Johnsson, K. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat. Biotechnol 2003, 21, 86–89. [Google Scholar]
- Gautier, A.; Juillerat, A.; Heinis, C.; Correa, I.R., Jr; Kindermann, M.; Beaufils, F.; Johnsson, K. An engineered protein tag for multiprotein labeling in living cells. Chem. Biol. 2008, 15, 128–136. [Google Scholar]
- Yin, J.; Straight, P.D.; McLoughlin, S.M.; Zhou, Z.; Lin, A.J.; Golan, D.E.; Kelleher, N.L.; Kolter, R.; Walsh, C.T. Genetically encoded short peptide tag for versatile protein labeling by Sfp phosphopantetheinyl transferase. Proc. Natl. Acad. Sci. USA 2005, 102, 15815–15820. [Google Scholar]
- Martin, B.R.; Giepmans, B.N.; Adams, S.R.; Tsien, R.Y. Mammalian cell-based optimization of the biarsenical-binding tetracysteine motif for improved fluorescence and affinity. Nat. Biotechnol 2005, 23, 1308–1314. [Google Scholar]
- Uttamapinant, C.; White, K.A.; Baruah, H.; Thompson, S.; Fernandez-Suarez, M.; Puthenveetil, S.; Ting, A.Y. A fluorophore ligase for site-specific protein labeling inside living cells. Proc. Natl. Acad. Sci. USA 2010, 107, 10914–10919. [Google Scholar]
- Slavoff, S.A.; Liu, D.S.; Cohen, J.D.; Ting, A.Y. Imaging protein-protein interactions inside living cells via interaction-dependent fluorophore ligation. J. Am. Chem. Soc 2011, 133, 19769–19776. [Google Scholar]
- Axelrod, D. Total internal reflection fluorescence microscopy. Methods Cell Biol 1989, 30, 245–270. [Google Scholar]
- Axelrod, D. Total internal reflection fluorescence microscopy in cell biology. Biophotonics B 2003, 361, 1–33. [Google Scholar]
- Stout, A.L.; Axelrod, D. Evanescent field excitation of fluorescence by epi-illumination microscopy. Appl. Opt 1989, 28, 5237–5242. [Google Scholar]
- Clarke, D.T.; Botchway, S.W.; Coles, B.C.; Needham, S.R.; Roberts, S.K.; Rolfe, D.J.; Tynan, C.J.; Ward, A.D.; Webb, S.E.D.; Yadav, R.; et al. Optics clustered to output unique solutions: A multi-laser facility for combined single molecule and ensemble microscopy. Rev. Sci. Instrum. 2011, 82, 093705:1–093705:11. [Google Scholar]
- Webb, S.E.D.; Zanetti-Domingues, L.; Coles, B.C.; Rolfe, D.J.; Wareham, R.J.; Martin-Fernandez, M.L. Multicolour single molecule imaging on cells using a supercontinuum source. Biomed. Opt. Express 2012, 3, 400–406. [Google Scholar]
- Webb, S.E.; Needham, S.R.; Roberts, S.K.; Martin-Fernandez, M.L. Multidimensional single-molecule imaging in live cells using total-internal-reflection fluorescence microscopy. Opt. Lett 2006, 31, 2157–2159. [Google Scholar]
- Denvir, D.J.; Coates, C.G. Electron Multiplying CCD Technology: Application to Ultrasensitive Detection of Biomolecules. In Biomedical Nanotechnology Architectures and Applications; Bornhop, D.J., Dunn, D.A., Mariella, R.P., Murphy, C.J., Nicolau, D.V., Nie, S., Palmer, M., Raghavachari, R., Eds.; Society of Photo Optical: Bellingham, WA, USA, 2002; Volume 4626, pp. 502–512. [Google Scholar]
- Mashanov, G.I.; Molloy, J.E. Automatic detection of single fluorophores in live cells. Biophys. J 2007, 92, 2199–2211. [Google Scholar]
- Askari, J.A.; Tynan, C.J.; Webb, S.E.D.; Martin-Fernandez, M.L.; Ballestrem, C.; Humphries, M.J. Focal adhesions are sites of integrin extension. J. Cell Biol 2010, 188, 891–903. [Google Scholar]
- Rolfe, D.J.; McLachlan, C.I.; Hirsch, M.; Needham, S.R.; Tynan, C.J.; Webb, S.E.D.; Martin-Fernandez, M.L.; Hobson, M.P. Automated multidimensional single molecule fluorescence microscopy feature detection and tracking. Eur. Biophys. J 2011, 40, 1167–1186. [Google Scholar]
- Ismail, T.M.; Zhang, S.; Fernig, D.G.; Gross, S.; Martin-Fernandez, M.L.; See, V.; Tozawa, K.; Tynan, C.J.; Wang, G.; Wilkinson, M.C.; et al. Self-association of calcium-binding protein S100A4 and metastasis. J. Biol. Chem 2010, 285, 914–922. [Google Scholar]
- Leung, B.O.; Chou, K.C. Review of super-resolution fluorescence microscopy for biology. Appl. Spectrosc 2011, 65, 967–980. [Google Scholar]
- Jaqaman, K.; Loerke, D.; Mettlen, M.; Kuwata, H.; Grinstein, S.; Schmid, S.L.; Danuser, G. Robust single-particle tracking in live-cell time-lapse sequences. Nat. Methods 2008, 5, 695–702. [Google Scholar]
- Yoon, J.W.; Bruckbauer, A.; Fitzgerald, W.J.; Klenerman, D. Bayesian inference for improved single molecule fluorescence tracking. Biophys. J 2008, 94, 4932–4947. [Google Scholar]
- Holden, S.J.; Uphoff, S.; Kapanidis, A.N. DAOSTORM: an algorithm for high-density super-resolution microscopy. Nat. Methods 2011, 8, 279–280. [Google Scholar]
- Churchman, L.S.; Okten, Z.; Rock, R.S.; Dawson, J.F.; Spudich, J.A. Single molecule high-resolution colocalization of Cy3 and Cy5 attached to macromolecules measures intramolecular distances through time. Proc. Natl. Acad. Sci. USA 2005, 102, 1419–1423. [Google Scholar]
- Semrau, S.; Holtzer, L.; Gonzalez-Gaitan, M.; Schmidt, T. Quantification of biological interactions with particle image cross-correlation spectroscopy (PICCS). Biophys. J 2011, 100, 1810–1818. [Google Scholar]
- Saxton, M.J. Single-particle tracking: connecting the dots. Nat. Methods 2008, 5, 671–672. [Google Scholar]
- Serge, A.; Bertaux, N.; Rigneault, H.; Marguet, D. Dynamic multiple-target tracing to probe spatiotemporal cartography of cell membranes. Nat. Methods 2008, 5, 687–694. [Google Scholar]
- Ji, W.; Xu, P.Y.; Li, Z.Z.; Lu, J.Z.; Liu, L.; Zhan, Y.; Chen, Y.; Hille, B.; Xu, T.; Chen, L.Y. Functional stoichiometry of the unitary calcium-release-activated calcium channel. Proc. Natl. Acad. Sci. USA 2008, 105, 13668–13673. [Google Scholar]
- Blunck, R.; McGuire, H.; Hyde, H.C.; Bezanilla, F. Fluorescence detection of the movement of single KcsA subunits reveals cooperativity. Proc. Natl. Acad. Sci. USA 2008, 105, 20263–20268. [Google Scholar]
- Saffman, P.G.; Delbruck, M. Brownian-motion in biological-membranes. Proc. Natl. Acad. Sci. USA 1975, 72, 3111–3113. [Google Scholar]
- Walters, P. An introduction to ergodic theory. Bull. Am. Math. Soc 1983, 9, 259–265. [Google Scholar]
- Axelrod, D.; Koppel, D.E.; Schlessinger, J.; Elson, E.; Webb, W.W. Mobility measurement by analysis of fluorescence photobleaching recovery kinetics. Biophys. J 1976, 16, 1055–1069. [Google Scholar]
- Schutz, G.J.; Schindler, H.; Schmidt, T. Single-molecule microscopy on model membranes reveals anomalous diffusion. Biophys. J 1997, 73, 1073–1080. [Google Scholar]
- Kusumi, A.; Sako, Y.; Yamamoto, M. Confined lateral diffusion of membrane-receptors as studied by single-particle tracking (nanovid microscopy)—Effects of calcium-induced differentiation in cultured epithelial-cells. Biophys. J 1993, 65, 2021–2040. [Google Scholar]
- Kusumi, A.; Ike, H.; Nakada, C.; Murase, K.; Fujiwara, T. Single-molecule tracking of membrane molecules: Plasma membrane compartmentalization and dynamic assembly of raft-philic signaling molecules. Semin. Immunol 2005, 17, 3–21. [Google Scholar]
- Fujiwara, T.; Ritchie, K.; Murakoshi, H.; Jacobson, K.; Kusumi, A. Phospholipids undergo hop diffusion in compartmentalized cell membrane. J. Cell Biol 2002, 157, 1071–1081. [Google Scholar]
- Pike, L.J.; Han, X.; Gross, R.W. Epidermal growth factor receptors are localized to lipid rafts that contain a balance of inner and outer leaflet lipids: A shotgun lipidomics study. J. Biol. Chem 2005, 280, 26796–26804. [Google Scholar]
- Pike, L.J.; Casey, L. Cholesterol levels modulate EGF receptor-mediated signaling by altering receptor function and trafficking. Biochemistry 2002, 41, 10315–10322. [Google Scholar]
- Weigel, A.V.; Simon, B.; Tamkun, M.M.; Krapf, D. Ergodic and nonergodic processes coexist in the plasma membrane as observed by single-molecule tracking. Proc. Natl. Acad. Sci. USA 2011, 108, 6438–6443. [Google Scholar]
- Kahana, A.; Kenan, G.; Feingold, M.; Elbaum, M.; Granek, R. Active transport on disordered microtubule networks: The generalized random velocity model. Phys. Rev. E Stat. Nonlin. Soft Matter Phys 2008, 78, 051912:1–051912:15. [Google Scholar]
- Bacher, C.P.; Reichenzeller, M.; Athale, C.; Herrmann, H.; Eils, R. 4-D single particle tracking of synthetic and proteinaceous microspheres reveals preferential movement of nuclear particles along chromatin—Poor tracks. BMC Cell Biol. 2004, 5. [Google Scholar] [CrossRef] [Green Version]
- Saxton, M.J. Two-dimensional continuum percolation threshold for diffusing particles of nonzero radius. Biophys. J 2010, 99, 1490–1499. [Google Scholar]
- Saxton, M.J. A biological interpretation of transient anomalous subdiffusion. I. Qualitative model. Biophys. J 2007, 92, 1178–1191. [Google Scholar]
- Teramura, Y.; Ichinose, J.; Takagi, H.; Nishida, K.; Yanagida, T.; Sako, Y. Single-molecule analysis of epidermal growth factor binding on the surface of living cells. EMBO J 2006, 25, 4215–4222. [Google Scholar]
- Webb, S.E.; Roberts, S.K.; Needham, S.R.; Tynan, C.J.; Rolfe, D.J.; Winn, M.D.; Clarke, D.T.; Barraclough, R.; Martin-Fernandez, M.L. Single-molecule imaging and fluorescence lifetime imaging microscopy show different structures for high- and low-affinity epidermal growth factor receptors in A431 cells. Biophys. J 2008, 94, 803–819. [Google Scholar]
- Cai, D.W.; McEwen, D.P.; Martens, J.R.; Meyhofer, E.; Verhey, K.J. Single molecule imaging reveals differences in microtubule track selection between kinesin motors. PLoS Biol 2009, 7, e1000216. [Google Scholar]
- Levey, A.I.; Kitt, C.A.; Simonds, W.F.; Price, D.L.; Brann, M.R. Identification and localization of muscarinic acetylcholine-receptor proteins in brain with subtype-specific antibodies. J. Neurosci 1991, 11, 3218–3226. [Google Scholar]
- Bruckbauer, A.; Dunne, P.D.; James, P.; Howes, E.; Zhou, D.J.; Jones, R.; Klenerman, D. Selective diffusion barriers separate membrane compartments. Biophys. J 2010, 99, L1–L3. [Google Scholar]
- Schaaf, M.J.; Koopmans, W.J.; Meckel, T.; van Noort, J.; Snaar-Jagalska, B.E.; Schmidt, T.S.; Spaink, H.P. Single-molecule microscopy reveals membrane microdomain organization of cells in a living vertebrate. Biophys. J 2009, 97, 1206–1214. [Google Scholar]
- Ritter, J.G.; Veith, R.; Veenendaal, A.; Siebrasse, J.P.; Kubitscheck, U. Light sheet microscopy for single molecule tracking in living tissue. PLoS One 2010, 5, e11639. [Google Scholar]
- Martiniere, A.; Lavagi, I.; Nageswaran, G.; Rolfe, D.J.; Maneta-Peyret, L.; Luu, D.T.; Botchway, S.W.; Webb, S.E.; Mongrand, S.; Maurel, C.; et al. Cell wall constrains lateral diffusion of plant plasma-membrane proteins. Proc. Natl. Acad. Sci. USA 2012, 109, 12805–12810. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Resistance to photo-bleaching | Brightness | Level of blinking | Emission range (nm) | Suitable for polarization | Narrow fluorescence bands | |
|---|---|---|---|---|---|---|
| Quantum dots | √√√ | √√√ | √ | 400–800 | NO | √ (excit.) √√√ (emiss.) |
| Organic dyes | √√ | √√ | √ | 400–800 | YES | √√√ |
| Fluorescent proteins | √ | √ | √ | 450–650 | YES | √√ (excit.) √√√ (emiss.) |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Martin-Fernandez, M.L.; Clarke, D.T. Single Molecule Fluorescence Detection and Tracking in Mammalian Cells: The State-of-the-Art and Future Perspectives. Int. J. Mol. Sci. 2012, 13, 14742-14765. https://doi.org/10.3390/ijms131114742
Martin-Fernandez ML, Clarke DT. Single Molecule Fluorescence Detection and Tracking in Mammalian Cells: The State-of-the-Art and Future Perspectives. International Journal of Molecular Sciences. 2012; 13(11):14742-14765. https://doi.org/10.3390/ijms131114742
Chicago/Turabian StyleMartin-Fernandez, Marisa L., and David T. Clarke. 2012. "Single Molecule Fluorescence Detection and Tracking in Mammalian Cells: The State-of-the-Art and Future Perspectives" International Journal of Molecular Sciences 13, no. 11: 14742-14765. https://doi.org/10.3390/ijms131114742