Dynamic Control of Electron Transfers in Diflavin Reductases

Abstract
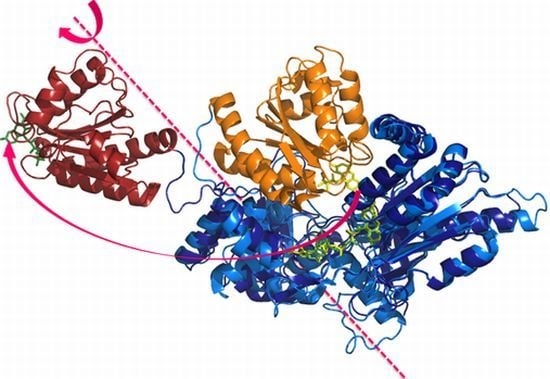
:
1. Introduction
1.1. Generalities
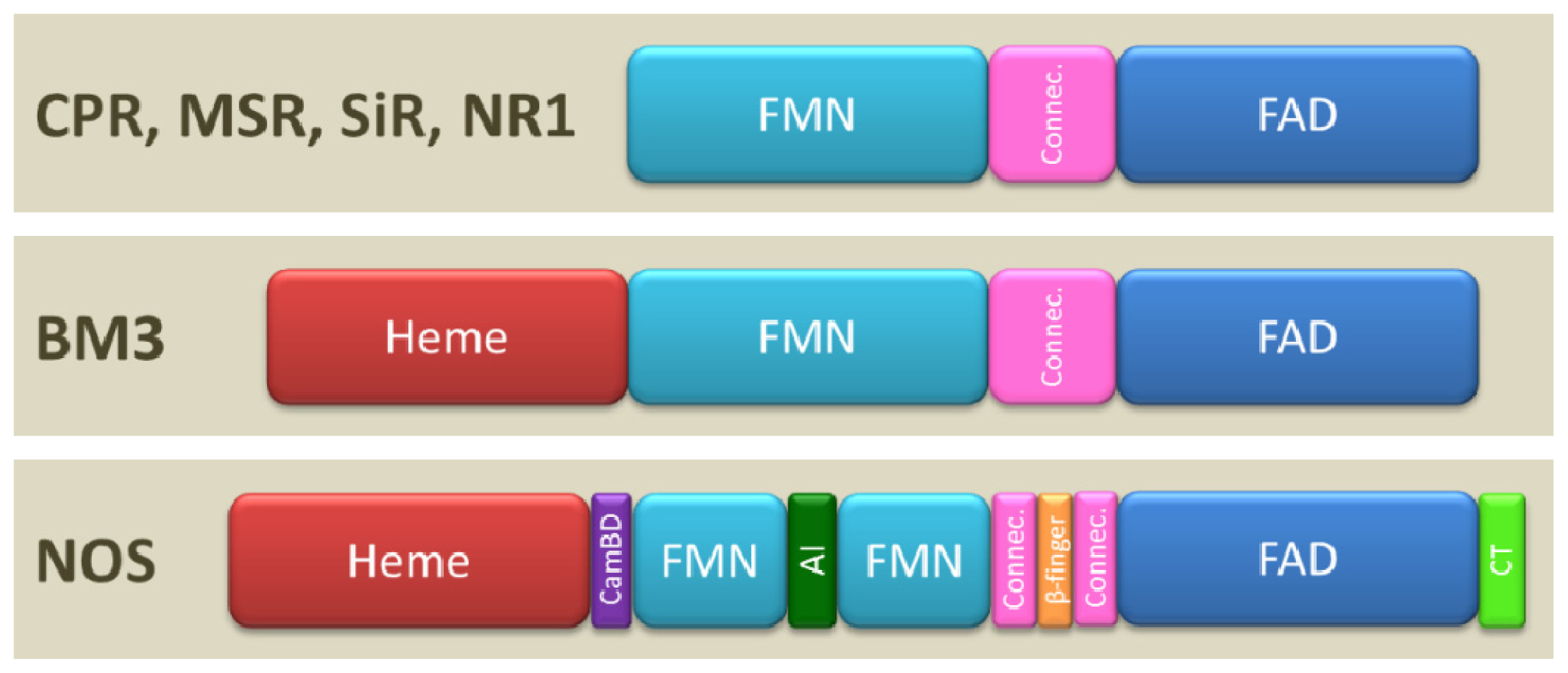
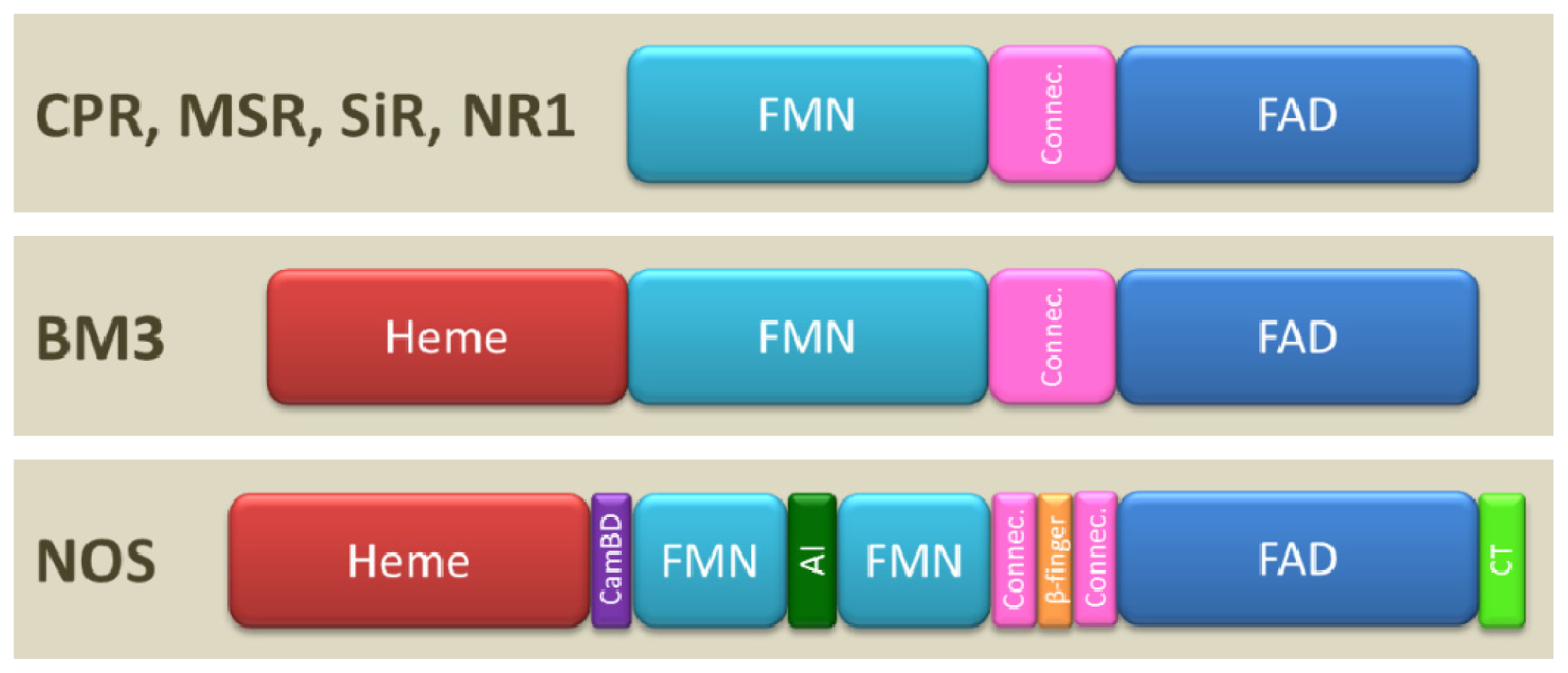
1.2. Domain Architecture
1.3. Oligomerization
1.4. Modular Assembly of Domains
2. First Evidences of Interdomain Dynamics
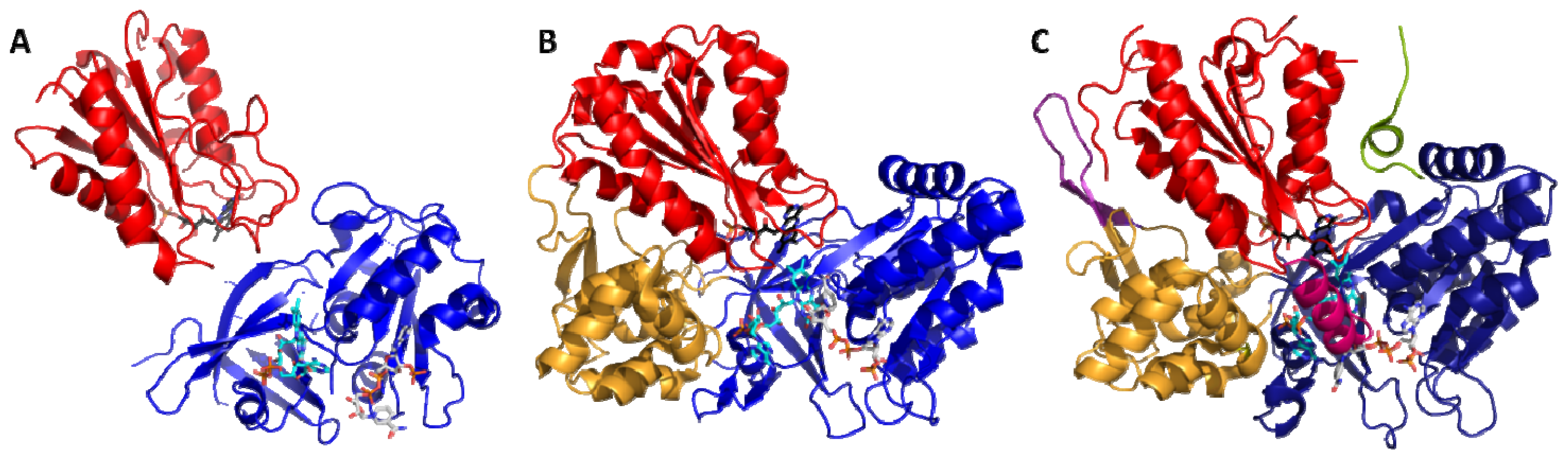
2.1. Crystallography
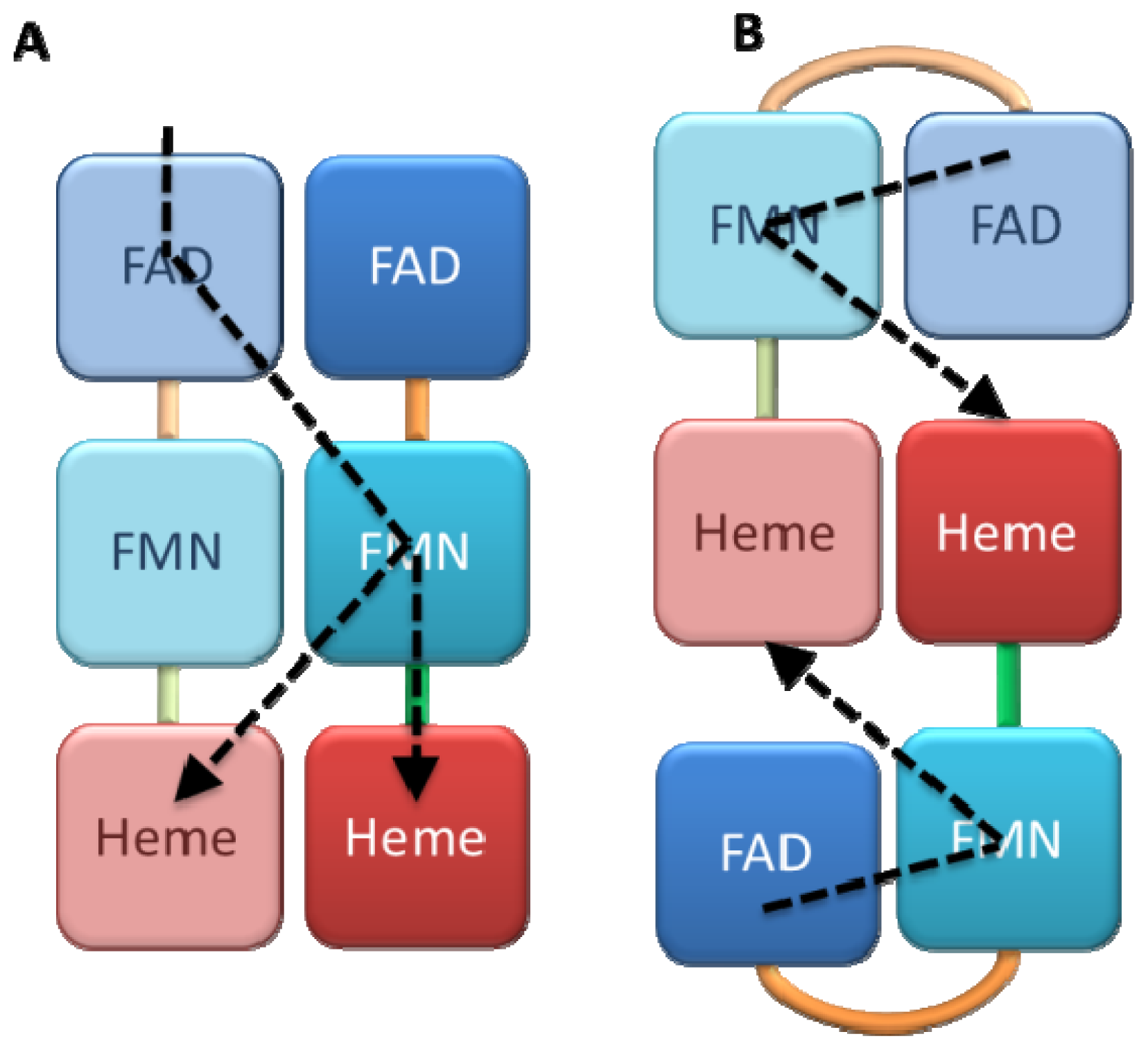
2.2. Alternate Hypothesis
3. Biochemical Characterization of Dynamical Behaviors
3.1. NOS
3.2. CPR
3.3. Chimeric Diflavin Reductases
4. Structural Characterization of Dynamical Behaviors
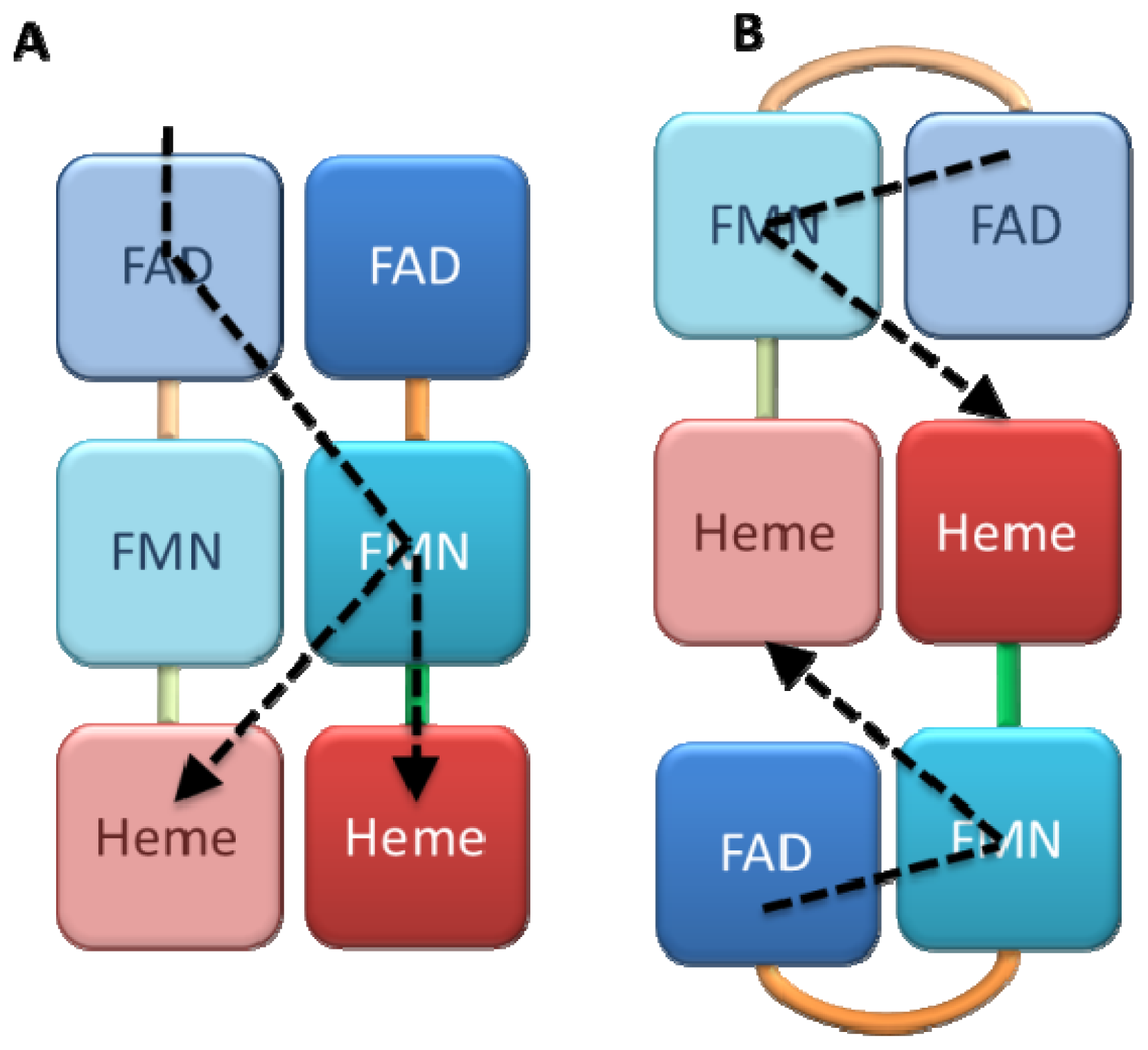
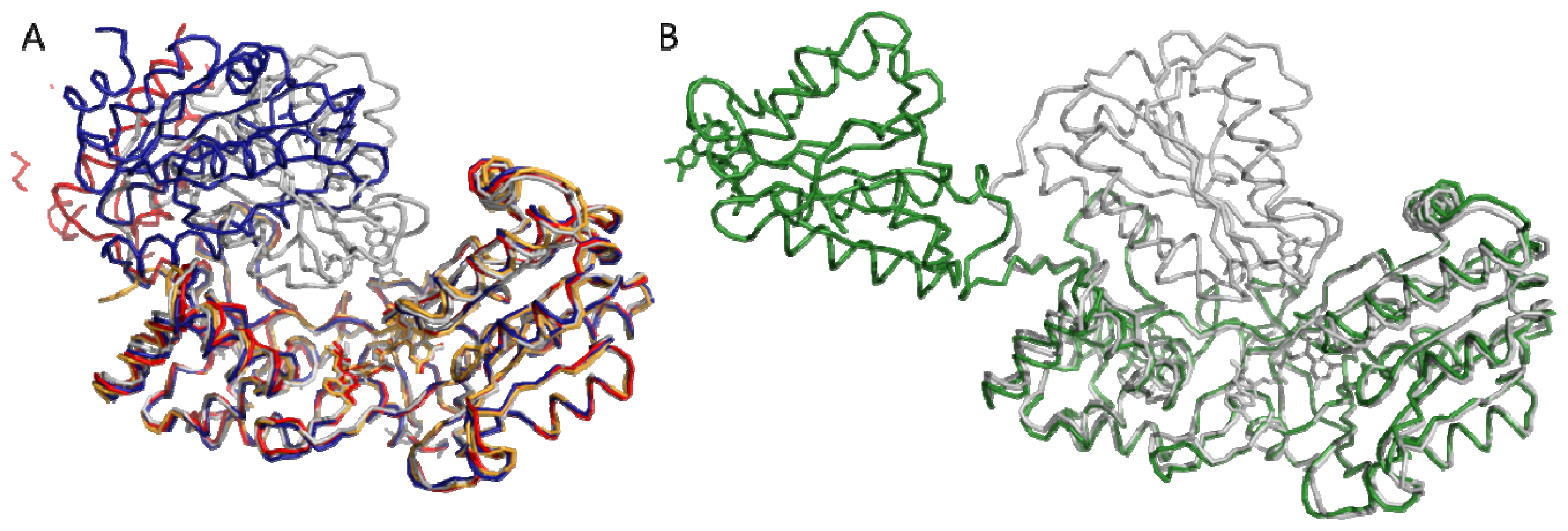
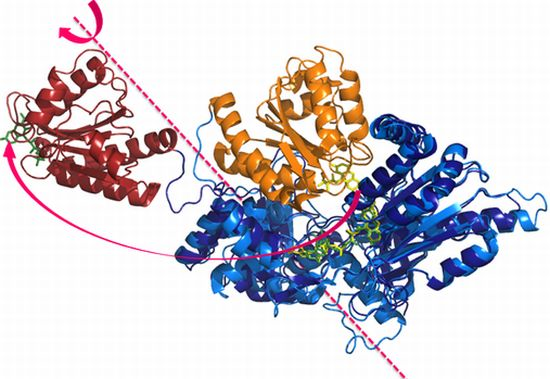
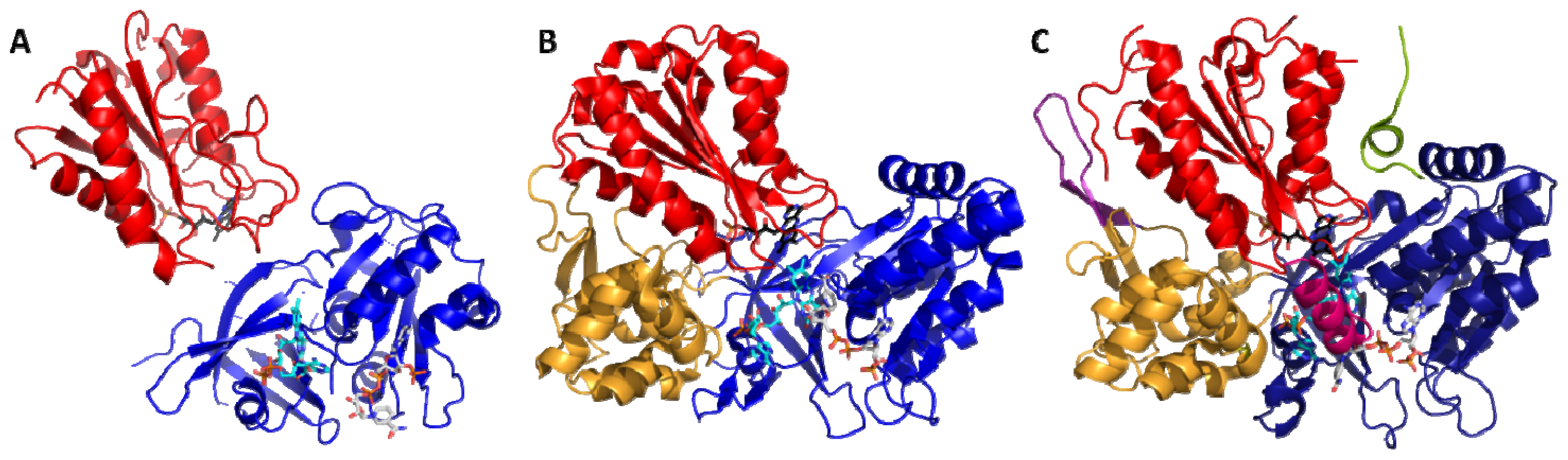
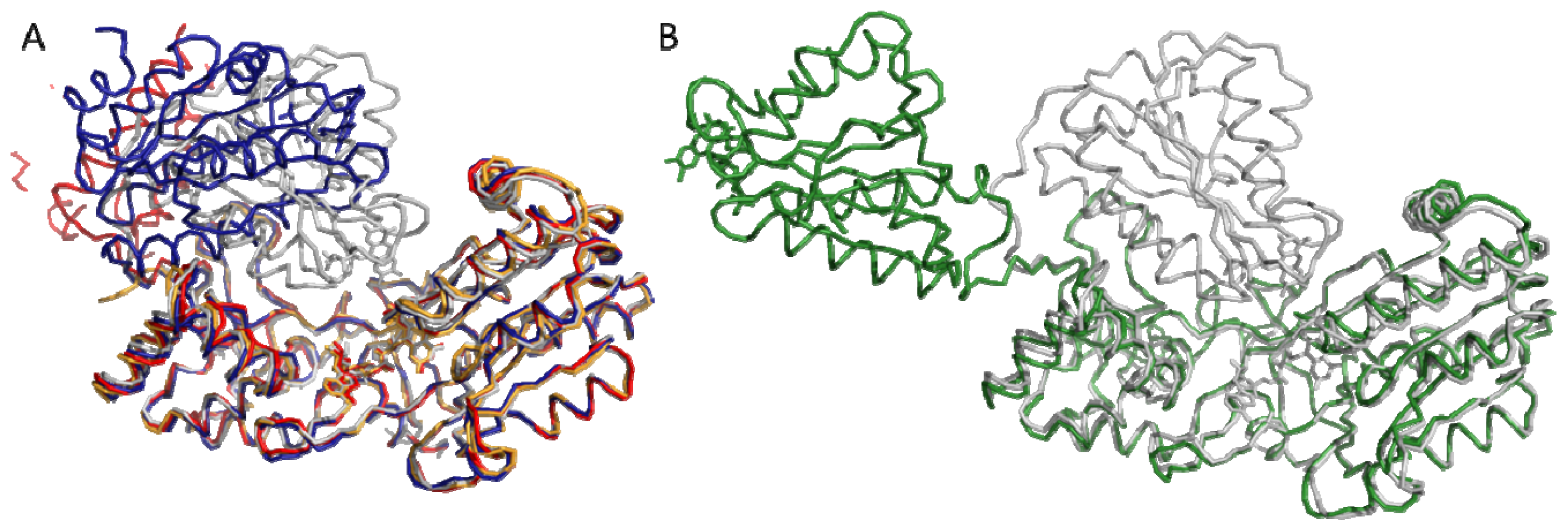
4.1. Crystallography
4.2. SAXS and NMR Experiments
4.3. ELDOR, Mass and Fluorescence Spectroscopy
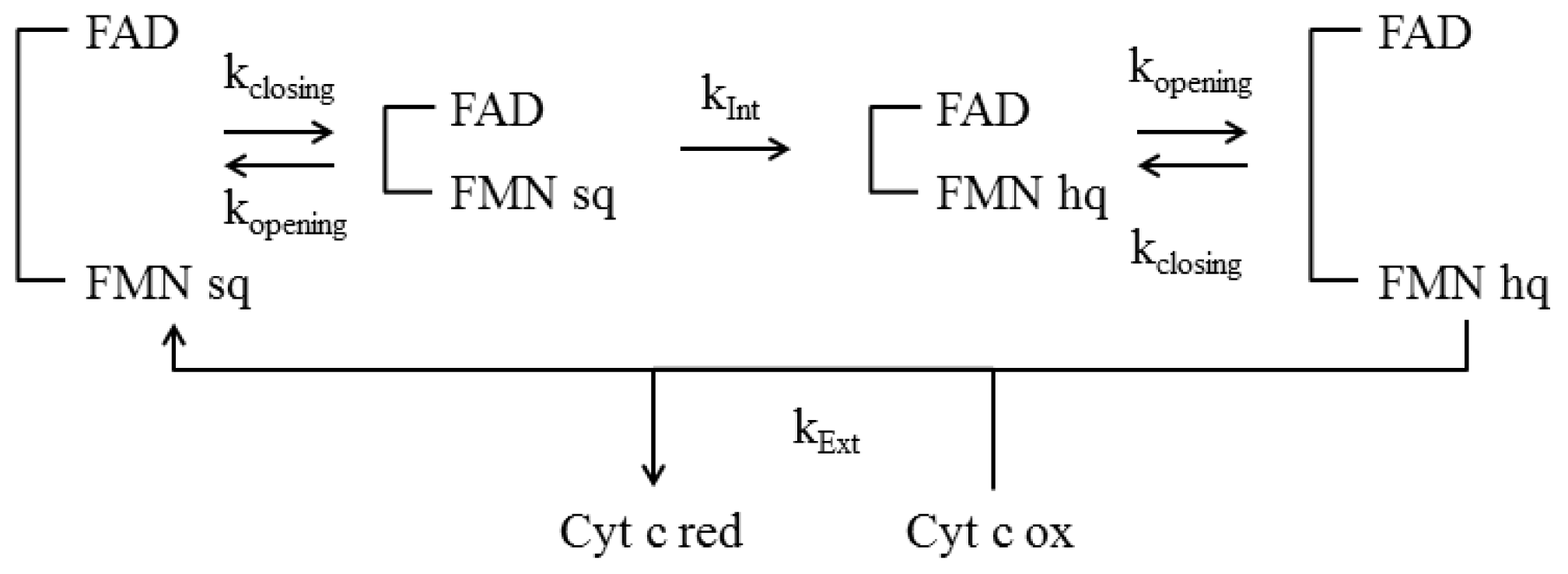
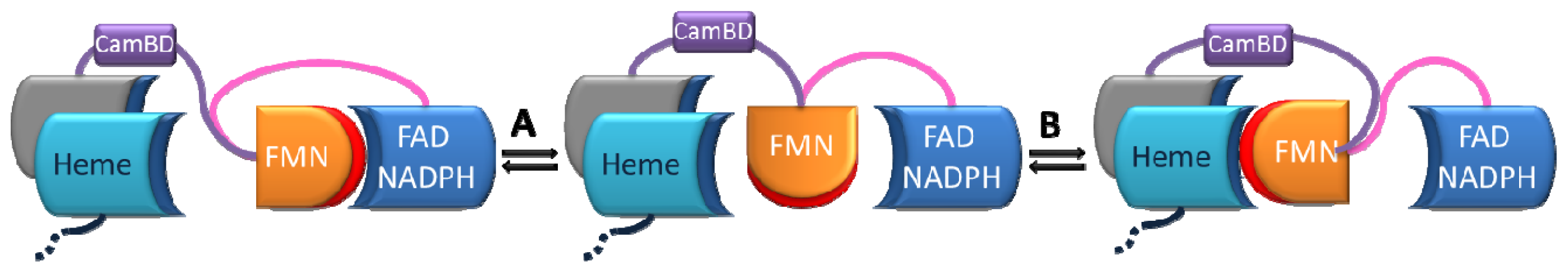
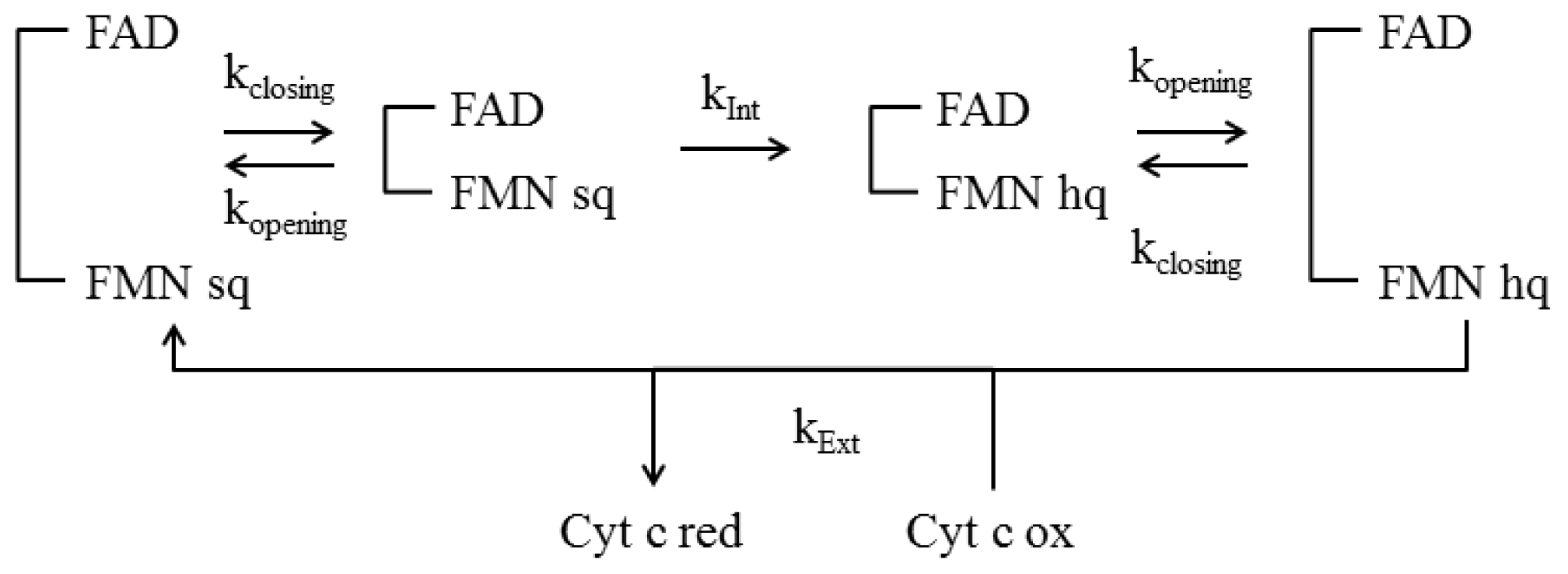
4.4 Kinetic Models
5. Conclusions and Future Perspectives
Acknowledgments
References
- De Colibus, L.; Mattevi, A. New frontiers in structural flavoenzymology. Curr. Opin. Struct. Biol 2006, 16, 722–728. [Google Scholar]
- Joosten, V.; van Berkel, W.J. Flavoenzymes. Curr. Opin. Chem. Biol 2007, 11, 195–202. [Google Scholar]
- Gomez-Moreno, C. Structural and mechanistic aspects of flavoproteins. FEBS J 2009, 276, 3929. [Google Scholar]
- Murataliev, M.B.; Feyereisen, R.; Walker, F.A. Electron transfer by diflavin reductases. Biochim. Biophys. Acta 2004, 1698, 1–26. [Google Scholar]
- Horecker, B.L. Triphosphopyridine nucleotide-cytochrome c reductase in liver. J. Biol. Chem 1950, 183, 12. [Google Scholar]
- Philips, A.H.; Langdon, R.G. Hepatic triphosphopyridine nucleotide cytochrome c reductase: Isolation, characteization, and kinetics studies. J. Biol. Chem 1962, 237, 2652–2660. [Google Scholar]
- Williams, C.H., Jr; Kamin, H. Microsomal triphosphopyridine nucleotide-cytochrome c reductase of liver. J. Biol. Chem. 1962, 237, 587–595. [Google Scholar]
- Kamin, H.; Masters, B.S.; Gibson, Q.H.; Williams, C.H., Jr. Microsomal TPNH-cytochrome c reductase. Fed. Proc. 1965, 24, 1164–1171. [Google Scholar]
- Masters, B.S.; Bilimoria, M.H.; Kamin, H.; Gibson, Q.H. The mechanism of 1- and 2-electron transfers catalyzed by reduced triphosphopyridine nucleotide-cytochrome c reductase. J. Biol. Chem 1965, 240, 4081–4088. [Google Scholar]
- Dignam, J.D.; Strobel, H.W. NADPH-cytochrome P-450 reductase from rat liver: Purification by affinity chromatography and characterization. Biochemistry 1977, 16, 1116–1123. [Google Scholar]
- Vermilion, J.L.; Coon, M.J. Purified liver microsomal NADPH-cytochrome P-450 reductase. Spectral characterization of oxidation-reduction states. J. Biol. Chem 1978, 253, 2694–2704. [Google Scholar]
- Welton, A.F.; Pederson, T.C.; Buege, J.A.; Aust, S.D. The molecular weight of NADPH-cytochrome C reductase isolated by immunoprecipitation from detergent-solubilized rat liver microsomes. Biochem. Biophys. Res. Commun 1973, 54, 161–167. [Google Scholar]
- Vermilion, J.L.; Coon, M.J. Identification of the high and low potential flavins of liver microsomal NADPH-cytochrome P-450 reductase. J. Biol. Chem 1978, 253, 8812–8819. [Google Scholar]
- Miura, Y.; Fulco, A.J. (Omega-2) hydroxylation of fatty acids by a soluble system from bacillus megaterium. J. Biol. Chem. 1974, 249, 1880–1888. [Google Scholar]
- Narhi, L.O.; Fulco, A.J. Characterization of a catalytically self-sufficient 119,000-dalton cytochrome P-450 monooxygenase induced by barbiturates in Bacillus megaterium. J. Biol. Chem 1986, 261, 7160–7169. [Google Scholar]
- Gulati, S.; Chen, Z.; Brody, L.C.; Rosenblatt, D.S.; Banerjee, R. Defects in auxiliary redox proteins lead to functional methionine synthase deficiency. J. Biol. Chem 1997, 272, 19171–19175. [Google Scholar]
- Leclerc, D.; Wilson, A.; Dumas, R.; Gafuik, C.; Song, D.; Watkins, D.; Heng, H.H.; Rommens, J.M.; Scherer, S.W.; Rosenblatt, D.S.; et al. Cloning and mapping of a cDNA for methionine synthase reductase, a flavoprotein defective in patients with homocystinuria. Proc. Natl. Acad. Sci. USA 1998, 95, 3059–3064. [Google Scholar]
- Olteanu, H.; Banerjee, R. Human methionine synthase reductase, a soluble P-450 reductase-like dual flavoprotein, is sufficient for NADPH-dependent methionine synthase activation. J. Biol. Chem 2001, 276, 35558–35563. [Google Scholar]
- Siegel, L.M.; Davis, P.S. Reduced nicotinamide adenine dinucleotide phosphate-sulfite reductase of enterobacteria. IV. The Escherichia coli hemoflavoprotein: Subunit structure and dissociation into hemoprotein and flavoprotein components. J. Biol. Chem 1974, 249, 1587–1598. [Google Scholar]
- Siegel, L.M.; Murphy, M.J.; Kamin, H. Reduced nicotinamide adenine dinucleotide phosphate-sulfite reductase of enterobacteria. I. The Escherichia coli hemoflavoprotein: Molecular parameters and prosthetic groups. J. Biol. Chem 1973, 248, 251–264. [Google Scholar]
- Eschenbrenner, M.; Coves, J.; Fontecave, M. NADPH-sulfite reductase flavoprotein from Escherichia coli: Contribution to the flavin content and subunit interaction. FEBS Lett 1995, 374, 82–84. [Google Scholar]
- Ostrowski, J.; Barber, M.J.; Rueger, D.C.; Miller, B.E.; Siegel, L.M.; Kredich, N.M. Characterization of the flavoprotein moieties of NADPH-sulfite reductase from Salmonella typhimurium and Escherichia coli. Physicochemical and catalytic properties, amino acid sequence deduced from DNA sequence of cysJ, and comparison with NADPH-cytochrome P-450 reductase. J. Biol. Chem 1989, 264, 15796–15808. [Google Scholar]
- Bredt, D.S.; Snyder, S.H. Isolation of nitric oxide synthetase, a calmodulin-requiring enzyme. Proc. Natl. Acad. Sci. USA 1990, 87, 682–685. [Google Scholar]
- Miller, R.T.; Hinck, A.P. Characterization of hydride transfer to flavin adenine dinucleotide in neuronal nitric oxide synthase reductase domain: Geometric relationship between the nicotinamide and isoalloxazine rings. Arch. Biochem. Biophys 2001, 395, 129–135. [Google Scholar]
- Ruan, J.; Xie, Q.; Hutchinson, N.; Cho, H.; Wolfe, G.C.; Nathan, C. Inducible nitric oxide synthase requires both the canonical calmodulin-binding domain and additional sequences in order to bind calmodulin and produce nitric oxide in the absence of free Ca2+. J. Biol. Chem 1996, 271, 22679–22686. [Google Scholar]
- Schmidt, H.H.; Smith, R.M.; Nakane, M.; Murad, F. Ca2+/calmodulin-dependent NO synthase type I: A biopteroflavoprotein with Ca2+/calmodulin-independent diaphorase and reductase activities. Biochemistry 1992, 31, 3243–3249. [Google Scholar]
- Stuehr, D.J. Mammalian nitric oxide synthases. Biochim. Biophys. Acta 1999, 1411, 217–230. [Google Scholar]
- Roman, L.J.; Martasek, P.; Masters, B.S. Intrinsic and extrinsic modulation of nitric oxide synthase activity. Chem. Rev 2002, 102, 1179–1190. [Google Scholar]
- Paine, M.J.; Garner, A.P.; Powell, D.; Sibbald, J.; Sales, M.; Pratt, N.; Smith, T.; Tew, D.G.; Wolf, C.R. Cloning and characterization of a novel human dual flavin reductase. J. Biol. Chem 2000, 275, 1471–1478. [Google Scholar]
- Varadarajan, J.; Guilleminot, J.; Saint-Jore-Dupas, C.; Piegu, B.; Chaboute, M.E.; Gomord, V.; Coolbaugh, R.C.; Devic, M.; Delorme, V. ATR3 encodes a diflavin reductase essential for Arabidopsis embryo development. New Phytol 2010, 187, 67–82. [Google Scholar]
- Olteanu, H.; Banerjee, R. Redundancy in the pathway for redox regulation of mammalian methionine synthase: Reductive activation by the dual flavoprotein, novel reductase 1. J. Biol. Chem 2003, 278, 38310–38314. [Google Scholar]
- Eschenbrenner, M.; Coves, J.; Fontecave, M. The flavin reductase activity of the flavoprotein component of sulfite reductase from Escherichia coli. A new model for the protein structure. J. Biol. Chem 1995, 270, 20550–20555. [Google Scholar]
- Iyanagi, T.; Mason, H.S. Some properties of hepatic reduced nicotinamide adenine dinucleotide phosphate-cytochrome c reductase. Biochemistry 1973, 12, 2297–2308. [Google Scholar]
- Porter, T.D.; Kasper, C.B. Coding nucleotide sequence of rat NADPH-cytochrome P-450 oxidoreductase cDNA and identification of flavin-binding domains. Proc. Natl. Acad. Sci. USA 1985, 82, 973–977. [Google Scholar]
- Porter, T.D.; Kasper, C.B. NADPH-cytochrome P-450 oxidoreductase: flavin mononucleotide and flavin adenine dinucleotide domains evolved from different flavoproteins. Biochemistry 1986, 25, 1682–1687. [Google Scholar]
- Smith, G.C.; Tew, D.G.; Wolf, C.R. Dissection of NADPH-cytochrome P450 oxidoreductase into distinct functional domains. Proc. Natl. Acad. Sci. USA 1994, 91, 8710–8714. [Google Scholar]
- Black, S.D. On the domain structure of cytochrome P450 102 (BM-3): Isolation and properties of a 45-kDa FAD/NADP domain. Biochem. Biophys. Res. Commun 1994, 203, 162–168. [Google Scholar]
- Champier, L.; Sibille, N.; Bersch, B.; Brutscher, B.; Blackledge, M.; Coves, J. Reactivity, secondary structure, and molecular topology of the Escherichia coli sulfite reductase flavodoxin-like domain. Biochemistry 2002, 41, 3770–3780. [Google Scholar]
- Coves, J.; Zeghouf, M.; Macherel, D.; Guigliarelli, B.; Asso, M.; Fontecave, M. Flavin mononucleotide-binding domain of the flavoprotein component of the sulfite reductase from Escherichia coli. Biochemistry 1997, 36, 5921–5928. [Google Scholar]
- Sevrioukova, I.; Truan, G.; Peterson, J.A. The flavoprotein domain of P450BM-3: Expression, purification, and properties of the flavin adenine dinucleotide- and flavin mononucleotide-binding subdomains. Biochemistry 1996, 35, 7528–7535. [Google Scholar]
- Vermilion, J.L.; Ballou, D.P.; Massey, V.; Coon, M.J. Separate roles for FMN and FAD in catalysis by liver microsomal NADPH-cytochrome P-450 reductase. J. Biol. Chem 1981, 256, 266–277. [Google Scholar]
- Boddupalli, S.S.; Oster, T.; Estabrook, R.W.; Peterson, J.A. Reconstitution of the fatty acid hydroxylation function of cytochrome P-450BM-3 utilizing its individual recombinant hemoand flavoprotein domains. J. Biol. Chem 1992, 267, 10375–10380. [Google Scholar]
- Higashimoto, Y.; Sato, H.; Sakamoto, H.; Takahashi, K.; Palmer, G.; Noguchi, M. The reactions of heme- and verdoheme-heme oxygenase-1 complexes with FMN-depleted NADPH-cytochrome P450 reductase. Electrons required for verdoheme oxidation can be transferred through a pathway not involving FMN. J. Biol. Chem 2006, 281, 31659–31667. [Google Scholar]
- Adak, S.; Ghosh, S.; Abu-Soud, H.M.; Stuehr, D.J. Role of reductase domain cluster 1 acidic residues in neuronal nitric-oxide synthase. Characterization of the FMN-FREE enzyme. J. Biol. Chem 1999, 274, 22313–22320. [Google Scholar]
- Gherasim, C.G.; Zaman, U.; Raza, A.; Banerjee, R. Impeded electron transfer from a pathogenic FMN domain mutant of methionine synthase reductase and its responsiveness to flavin supplementation. Biochemistry 2008, 47, 12515–12522. [Google Scholar]
- Klein, M.L.; Fulco, A.J. Critical residues involved in FMN binding and catalytic activity in cytochrome P450BM-3. J. Biol. Chem 1993, 268, 7553–7561. [Google Scholar]
- Neeli, R.; Girvan, H.M.; Lawrence, A.; Warren, M.J.; Leys, D.; Scrutton, N.S.; Munro, A.W. The dimeric form of flavocytochrome P450 BM3 is catalytically functional as a fatty acid hydroxylase. FEBS Lett 2005, 579, 5582–5588. [Google Scholar]
- Porter, T.D.; Beck, T.W.; Kasper, C.B. NADPH-cytochrome P-450 oxidoreductase gene organization correlates with structural domains of the protein. Biochemistry 1990, 29, 9814–9818. [Google Scholar]
- Wang, M.; Roberts, D.L.; Paschke, R.; Shea, T.M.; Masters, B.S.; Kim, J.J. Three-dimensional structure of NADPH-cytochrome P450 reductase: prototype for FMN- and FAD-containing enzymes. Proc. Natl. Acad. Sci. USA 1997, 94, 8411–8416. [Google Scholar]
- Gruez, A.; Pignol, D.; Zeghouf, M.; Coves, J.; Fontecave, M.; Ferrer, J.L.; Fontecilla-Camps, J.C. Four crystal structures of the 60 kDa flavoprotein monomer of the sulfite reductase indicate a disordered flavodoxin-like module. J. Mol. Biol 2000, 299, 199–212. [Google Scholar]
- Garcin, E.D.; Bruns, C.M.; Lloyd, S.J.; Hosfield, D.J.; Tiso, M.; Gachhui, R.; Stuehr, D.J.; Tainer, J.A.; Getzoff, E.D. Structural basis for isozyme-specific regulation of electron transfer in nitric-oxide synthase. J. Biol. Chem 2004, 279, 37918–37927. [Google Scholar]
- Black, S.D.; Martin, S.T. Evidence for conformational dynamics and molecular aggregation in cytochrome P450 102 (BM-3). Biochemistry 1994, 33, 12056–12062. [Google Scholar]
- Kitazume, T.; Haines, D.C.; Estabrook, R.W.; Chen, B.; Peterson, J.A. Obligatory intermolecular electron-transfer from FAD to FMN in dimeric P450BM-3. Biochemistry 2007, 46, 11892–11901. [Google Scholar]
- Pant, K.; Crane, B.R. Structure of a loose dimer: an intermediate in nitric oxide synthase assembly. J. Mol. Biol 2005, 352, 932–940. [Google Scholar]
- Stuehr, D.J.; Tejero, J.; Haque, M.M. Structural and mechanistic aspects of flavoproteins: Electron transfer through the nitric oxide synthase flavoprotein domain. FEBS J 2009, 276, 3959–3974. [Google Scholar]
- Feng, C. Mechanism of nitric oxide synthase regulation: Electron transfer and interdomain interactions. Coord. Chem. Rev 2012, 256, 393–411. [Google Scholar]
- Batey, S.; Randles, L.G.; Steward, A.; Clarke, J. Cooperative folding in a multi-domain protein. J. Mol. Biol 2005, 349, 1045–1059. [Google Scholar]
- Kuriyan, J.; Eisenberg, D. The origin of protein interactions and allostery in colocalization. Nature 2007, 450, 983–990. [Google Scholar]
- Ryabov, Y.E.; Fushman, D. A model of interdomain mobility in a multidomain protein. J. Am. Chem. Soc 2007, 129, 3315–3327. [Google Scholar]
- Batey, S.; Clarke, J. Apparent cooperativity in the folding of multidomain proteins depends on the relative rates of folding of the constituent domains. Proc. Natl. Acad. Sci. USA 2006, 103, 18113–18118. [Google Scholar]
- Chothia, C.; Gough, J. Genomic and structural aspects of protein evolution. Biochem. J 2009, 419, 15–28. [Google Scholar]
- Gokhale, R.S.; Khosla, C. Role of linkers in communication between protein modules. Curr. Opin. Chem. Biol 2000, 4, 22–27. [Google Scholar]
- Hawkins, A.R.; Lamb, H.K. The molecular biology of multidomain proteins. Selected examples. Eur. J. Biochem 1995, 232, 7–18. [Google Scholar]
- Ramachandran, R.; Surka, M.; Chappie, J.S.; Fowler, D.M.; Foss, T.R.; Song, B.D.; Schmid, S.L. The dynamin middle domain is critical for tetramerization and higher-order self-assembly. EMBO J 2007, 26, 559–566. [Google Scholar]
- Sauer, J.; Sigurskjold, B.W.; Christensen, U.; Frandsen, T.P.; Mirgorodskaya, E.; Harrison, M.; Roepstorff, P.; Svensson, B. Glucoamylase: Structure/function relationships, and protein engineering. Biochim. Biophys. Acta 2000, 1543, 275–293. [Google Scholar]
- Abu-Soud, H.M.; Yoho, L.L.; Stuehr, D.J. Calmodulin controls neuronal nitric-oxide synthase by a dual mechanism. Activation of intra- and interdomain electron transfer. J. Biol. Chem 1994, 269, 32047–32050. [Google Scholar]
- Gachhui, R.; Presta, A.; Bentley, D.F.; Abu-Soud, H.M.; McArthur, R.; Brudvig, G.; Ghosh, D.K.; Stuehr, D.J. Characterization of the reductase domain of rat neuronal nitric oxide synthase generated in the methylotrophic yeast Pichia pastoris. Calmodulin response is complete within the reductase domain itself. J. Biol. Chem 1996, 271, 20594–20602. [Google Scholar]
- Salerno, J.C.; Harris, D.E.; Irizarry, K.; Patel, B.; Morales, A.J.; Smith, S.M.; Martasek, P.; Roman, L.J.; Masters, B.S.; Jones, C.L.; et al. An autoinhibitory control element defines calcium-regulated isoforms of nitric oxide synthase. J. Biol. Chem 1997, 272, 29769–29777. [Google Scholar]
- Daff, S.; Sagami, I.; Shimizu, T. The 42-amino acid insert in the FMN domain of neuronal nitric-oxide synthase exerts control over Ca2+/calmodulin-dependent electron transfer. J. Biol. Chem 1999, 274, 30589–30595. [Google Scholar]
- Montgomery, H.J.; Romanov, V.; Guillemette, J.G. Removal of a putative inhibitory element reduces the calcium-dependent calmodulin activation of neuronal nitric-oxide synthase. J. Biol. Chem 2000, 275, 5052–5058. [Google Scholar]
- Roman, L.J.; Martasek, P.; Miller, R.T.; Harris, D.E.; de La Garza, M.A.; Shea, T.M.; Kim, J.J.; Masters, B.S. The C termini of constitutive nitric-oxide synthases control electron flow through the flavin and heme domains and affect modulation by calmodulin. J. Biol. Chem 2000, 275, 29225–29232. [Google Scholar]
- Roman, L.J.; Miller, R.T.; de La Garza, M.A.; Kim, J.J.; Siler Masters, B.S. The C terminus of mouse macrophage inducible nitric-oxide synthase attenuates electron flow through the flavin domain. J. Biol. Chem 2000, 275, 21914–21919. [Google Scholar]
- Sevrioukova, I.F.; Li, H.; Zhang, H.; Peterson, J.A.; Poulos, T.L. Structure of a cytochrome P450-redox partner electron-transfer complex. Proc. Natl. Acad. Sci. USA 1999, 96, 1863–1868. [Google Scholar]
- Joyce, M.G.; Ekanem, I.S.; Roitel, O.; Dunford, A.J.; Neeli, R.; Girvan, H.M.; Baker, G.J.; Curtis, R.A.; Munro, A.W.; Leys, D. The crystal structure of the FAD/NADPH-binding domain of flavocytochrome P450 BM3. FEBS J 2012, 279, 1694–1706. [Google Scholar]
- Zhao, Q.; Modi, S.; Smith, G.; Paine, M.; McDonagh, P.D.; Wolf, C.R.; Tew, D.; Lian, L.Y.; Roberts, G.C.; Driessen, H.P. Crystal structure of the FMN-binding domain of human cytochrome P450 reductase at 1.93 A resolution. Protein Sci 1999, 8, 298–306. [Google Scholar]
- Sibille, N.; Blackledge, M.; Brutscher, B.; Coves, J.; Bersch, B. Solution structure of the sulfite reductase flavodoxin-like domain from Escherichia coli. Biochemistry 2005, 44, 9086–9095. [Google Scholar]
- Wolthers, K.R.; Lou, X.; Toogood, H.S.; Leys, D.; Scrutton, N.S. Mechanism of coenzyme binding to human methionine synthase reductase revealed through the crystal structure of the FNR-like module and isothermal titration calorimetry. Biochemistry 2007, 46, 11833–11844. [Google Scholar]
- Zhang, J.; Martasek, P.; Paschke, R.; Shea, T.; Siler Masters, B.S.; Kim, J.J. Crystal structure of the FAD/NADPH-binding domain of rat neuronal nitric-oxide synthase. Comparisons with NADPH-cytochrome P450 oxidoreductase. J. Biol. Chem 2001, 276, 37506–37513. [Google Scholar]
- Xia, C.; Panda, S.P.; Marohnic, C.C.; Martasek, P.; Masters, B.S.; Kim, J.J. Structural basis for human NADPH-cytochrome P450 oxidoreductase deficiency. Proc. Natl. Acad. Sci. USA 2011, 108, 13486–13491. [Google Scholar]
- Lamb, D.C.; Kim, Y.; Yermalitskaya, L.V.; Yermalitsky, V.N.; Lepesheva, G.I.; Kelly, S.L.; Waterman, M.R.; Podust, L.M. A second FMN binding site in yeast NADPH-cytochrome P450 reductase suggests a mechanism of electron transfer by diflavin reductases. Structure 2006, 14, 51–61. [Google Scholar]
- Hubbard, P.A.; Shen, A.L.; Paschke, R.; Kasper, C.B.; Kim, J.J. NADPH-cytochrome P450 oxidoreductase. Structural basis for hydride and electron transfer. J. Biol. Chem 2001, 276, 29163–29170. [Google Scholar]
- Olteanu, H.; Wolthers, K.R.; Munro, A.W.; Scrutton, N.S.; Banerjee, R. Kinetic and thermodynamic characterization of the common polymorphic variants of human methionine synthase reductase. Biochemistry 2004, 43, 1988–1997. [Google Scholar]
- Ballou, D.P.; Entsch, B.; Cole, L.J. Dynamics involved in catalysis by single-component and two-component flavin-dependent aromatic hydroxylases. Biochem. Biophys. Res. Commun 2005, 338, 590–598. [Google Scholar]
- Ivanov, A.S.; Gnedenko, O.V.; Molnar, A.A.; Archakov, A.I.; Podust, L.M. FMN binding site of yeast NADPH-cytochrome P450 reductase exposed at the surface is highly specific. ACS Chem. Biol 2010, 5, 767–776. [Google Scholar]
- Louerat-Oriou, B.; Perret, A.; Pompon, D. Differential redox and electron-transfer properties of purified yeast, plant and human NADPH-cytochrome P-450 reductases highly modulate cytochrome P-450 activities. Eur. J. Biochem 1998, 258, 1040–1049. [Google Scholar]
- Warman, A.J.; Roitel, O.; Neeli, R.; Girvan, H.M.; Seward, H.E.; Murray, S.A.; McLean, K.J.; Joyce, M.G.; Toogood, H.; Holt, R.A.; et al. Flavocytochrome P450 BM3: an update on structure and mechanism of a biotechnologically important enzyme. Biochem. Soc. Trans 2005, 33, 747–753. [Google Scholar]
- Welland, A.; Daff, S. Conformation-dependent hydride transfer in neuronal nitric oxide synthase reductase domain. FEBS J 2010, 277, 3833–3843. [Google Scholar]
- Wolthers, K.R.; Scrutton, N.S. Electron transfer in human methionine synthase reductase studied by stopped-flow spectrophotometry. Biochemistry 2004, 43, 490–500. [Google Scholar]
- Neeli, R.; Sabri, M.; McLean, K.J.; Dunford, A.J.; Scrutton, N.S.; Leys, D.; Munro, A.W. Trp(359) regulates flavin thermodynamics and coenzyme selectivity in Mycobacterium tuberculosis FprA. Biochem. J 2008, 411, 563–570. [Google Scholar]
- Daff, S. An appraisal of multiple NADPH binding-site models proposed for cytochrome P450 reductase, NO synthase, and related diflavin reductase systems. Biochemistry 2004, 43, 3929–3932. [Google Scholar]
- Medina, M. Structural and mechanistic aspects of flavoproteins: photosynthetic electron transfer from photosystem I to NADP+. FEBS J 2009, 276, 3942–3958. [Google Scholar]
- Brunner, K.; Tortschanoff, A.; Hemmens, B.; Andrew, P.J.; Mayer, B.; Kungl, A.J. Sensitivity of flavin fluorescence dynamics in neuronal nitric oxide synthase to cofactor-induced conformational changes and dimerization. Biochemistry 1998, 37, 17545–17553. [Google Scholar]
- Noble, M.A.; Munro, A.W.; Rivers, S.L.; Robledo, L.; Daff, S.N.; Yellowlees, L.J.; Shimizu, T.; Sagami, I.; Guillemette, J.G.; Chapman, S.K. Potentiometric analysis of the flavin cofactors of neuronal nitric oxide synthase. Biochemistry 1999, 38, 16413–16418. [Google Scholar]
- Feng, C.; Tollin, G.; Holliday, M.A.; Thomas, C.; Salerno, J.C.; Enemark, J.H.; Ghosh, D.K. Intraprotein electron transfer in a two-domain construct of neuronal nitric oxide synthase: The output state in nitric oxide formation. Biochemistry 2006, 45, 6354–6362. [Google Scholar]
- Nishida, C.R.; Ortiz de Montellano, P.R. Electron transfer and catalytic activity of nitric oxide synthases. Chimeric constructs of the neuronal, inducible, and endothelial isoforms. J. Biol. Chem 1998, 273, 5566–5571. [Google Scholar]
- Ilagan, R.P.; Tejero, J.; Aulak, K.S.; Ray, S.S.; Hemann, C.; Wang, Z.Q.; Gangoda, M.; Zweier, J.L.; Stuehr, D.J. Regulation of FMN subdomain interactions and function in neuronal nitric oxide synthase. Biochemistry 2009, 48, 3864–3876. [Google Scholar]
- Ilagan, R.P.; Tiso, M.; Konas, D.W.; Hemann, C.; Durra, D.; Hille, R.; Stuehr, D.J. Differences in a conformational equilibrium distinguish catalysis by the endothelial and neuronal nitric-oxide synthase flavoproteins. J. Biol. Chem 2008, 283, 19603–19615. [Google Scholar]
- Tiso, M.; Tejero, J.; Panda, K.; Aulak, K.S.; Stuehr, D.J. Versatile regulation of neuronal nitric oxide synthase by specific regions of its C-terminal tail. Biochemistry 2007, 46, 14418–14428. [Google Scholar]
- Craig, D.H.; Chapman, S.K.; Daff, S. Calmodulin activates electron transfer through neuronal nitric-oxide synthase reductase domain by releasing an NADPH-dependent conformational lock. J. Biol. Chem 2002, 277, 33987–33994. [Google Scholar]
- Daff, S. Calmodulin-dependent regulation of mammalian nitric oxide synthase. Biochem. Soc. Trans 2003, 31, 502–505. [Google Scholar]
- Garnaud, P.E.; Koetsier, M.; Ost, T.W.; Daff, S. Redox properties of the isolated flavin mononucleotide- and flavin adenine dinucleotide-binding domains of neuronal nitric oxide synthase. Biochemistry 2004, 43, 11035–11044. [Google Scholar]
- Nishino, Y.; Yamamoto, K.; Kimura, S.; Kikuchi, A.; Shiro, Y.; Iyanagi, T. Mechanistic studies on the intramolecular one-electron transfer between the two flavins in the human endothelial NOS reductase domain. Arch. Biochem. Biophys 2007, 465, 254–265. [Google Scholar]
- Welland, A.; Garnaud, P.E.; Kitamura, M.; Miles, C.S.; Daff, S. Importance of the domain-domain interface to the catalytic action of the NO synthase reductase domain. Biochemistry 2008, 47, 9771–9780. [Google Scholar]
- Gutierrez, A.; Lian, L.Y.; Wolf, C.R.; Scrutton, N.S.; Roberts, G.C. Stopped-flow kinetic studies of flavin reduction in human cytochrome P450 reductase and its component domains. Biochemistry 2001, 40, 1964–1975. [Google Scholar]
- Murataliev, M.B.; Feyereisen, R. Functional interactions in cytochrome P450BM3. Fatty acid substrate binding alters electron-transfer properties of the flavoprotein domain. Biochemistry 1996, 35, 15029–15037. [Google Scholar]
- Shen, A.L.; Sem, D.S.; Kasper, C.B. Mechanistic studies on the reductive half-reaction of NADPH-cytochrome P450 oxidoreductase. J. Biol. Chem 1999, 274, 5391–5398. [Google Scholar]
- Li, H.; Das, A.; Sibhatu, H.; Jamal, J.; Sligar, S.G.; Poulos, T.L. Exploring the electron transfer properties of neuronal nitric-oxide synthase by reversal of the FMN redox potential. J. Biol. Chem 2008, 283, 34762–34772. [Google Scholar]
- Ray, S.S.; Sengupta, R.; Tiso, M.; Haque, M.M.; Sahoo, R.; Konas, D.W.; Aulak, K.; Regulski, M.; Tully, T.; Stuehr, D.J.; et al. Reductase domain of Drosophila melanogaster nitric-oxide synthase: Redox transformations, regulation, and similarity to mammalian homologues. Biochemistry 2007, 46, 11865–11873. [Google Scholar]
- Haque, M.M.; Fadlalla, M.A.; Aulak, K.S.; Ghosh, A.; Durra, D.; Stuehr, D.J. Control of electron transfer and catalysis in neuronal NOS by a hinge connecting its FMN and FAD-NADPH domains. J. Biol. Chem. 2012. [Google Scholar] [CrossRef]
- Haque, M.M.; Panda, K.; Tejero, J.; Aulak, K.S.; Fadlalla, M.A.; Mustovich, A.T.; Stuehr, D.J. A connecting hinge represses the activity of endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 2007, 104, 9254–9259. [Google Scholar]
- Feng, C.; Dupont, A.L.; Nahm, N.J.; Spratt, D.E.; Hazzard, J.T.; Weinberg, J.B.; Guillemette, J.G.; Tollin, G.; Ghosh, D.K. Intraprotein electron transfer in inducible nitric oxide synthase holoenzyme. J. Biol. Inorg. Chem 2009, 14, 133–142. [Google Scholar]
- Feng, C.; Tollin, G.; Hazzard, J.T.; Nahm, N.J.; Guillemette, J.G.; Salerno, J.C.; Ghosh, D.K. Direct measurement by laser flash photolysis of intraprotein electron transfer in a rat neuronal nitric oxide synthase. J. Am. Chem. Soc 2007, 129, 5621–5629. [Google Scholar]
- Shen, A.L.; Kasper, C.B. Role of acidic residues in the interaction of NADPH-cytochrome P450 oxidoreductase with cytochrome P450 and cytochrome c. J. Biol. Chem 1995, 270, 27475–27480. [Google Scholar]
- Estabrook, R.W.; Shet, M.S.; Fisher, C.W.; Jenkins, C.M.; Waterman, M.R. The interaction of NADPH-P450 reductase with P450: An electrochemical study of the role of the flavin mononucleotide-binding domain. Arch. Biochem. Biophys 1996, 333, 308–315. [Google Scholar]
- Higashimoto, Y.; Sugishima, M.; Sato, H.; Sakamoto, H.; Fukuyama, K.; Palmer, G.; Noguchi, M. Mass spectrometric identification of lysine residues of heme oxygenase-1 that are involved in its interaction with NADPH-cytochrome P450 reductase. Biochem. Biophys. Res. Commun 2008, 367, 852–858. [Google Scholar]
- Shen, A.L.; Kasper, C.B. Differential contributions of NADPH-cytochrome P450 oxidoreductase FAD binding site residues to flavin binding and catalysis. J. Biol. Chem 2000, 275, 41087–41091. [Google Scholar]
- Im, S.C.; Waskell, L. The interaction of microsomal cytochrome P450 2B4 with its redox partners, cytochrome P450 reductase and cytochrome b(5). Arch. Biochem. Biophys 2011, 507, 144–153. [Google Scholar]
- Xia, C.; Hamdane, D.; Shen, A.L.; Choi, V.; Kasper, C.B.; Pearl, N.M.; Zhang, H.; Im, S.C.; Waskell, L.; Kim, J.J. Conformational changes of NADPH-cytochrome P450 oxidoreductase are essential for catalysis and cofactor binding. J. Biol. Chem 2011, 286, 16246–16260. [Google Scholar]
- Gutierrez, A.; Grunau, A.; Paine, M.; Munro, A.W.; Wolf, C.R.; Roberts, G.C.; Scrutton, N.S. Electron transfer in human cytochrome P450 reductase. Biochem. Soc. Trans 2003, 31, 497–501. [Google Scholar]
- Gutierrez, A.; Munro, A.W.; Grunau, A.; Wolf, C.R.; Scrutton, N.S.; Roberts, G.C. Interflavin electron transfer in human cytochrome P450 reductase is enhanced by coenzyme binding. Relaxation kinetic studies with coenzyme analogues. Eur. J. Biochem 2003, 270, 2612–2621. [Google Scholar]
- Gutierrez, A.; Paine, M.; Wolf, C.R.; Scrutton, N.S.; Roberts, G.C. Relaxation kinetics of cytochrome P450 reductase: Internal electron transfer is limited by conformational change and regulated by coenzyme binding. Biochemistry 2002, 41, 4626–4637. [Google Scholar]
- Grunau, A.; Geraki, K.; Grossmann, J.G.; Gutierrez, A. Conformational dynamics and the energetics of protein-ligand interactions: Role of interdomain loop in human cytochrome P450 reductase. Biochemistry 2007, 46, 8244–8255. [Google Scholar]
- Daff, S.; Noble, M.A.; Craig, D.H.; Rivers, S.L.; Chapman, S.K.; Munro, A.W.; Fujiwara, S.; Rozhkova, E.; Sagami, I.; Shimizu, T. Control of electron transfer in neuronal NO synthase. Biochem. Soc. Trans 2001, 29, 147–152. [Google Scholar]
- Degregorio, D.; Sadeghi, S.J.; Di Nardo, G.; Gilardi, G.; Solinas, S.P. Understanding uncoupling in the multiredox centre P450 3A4-BMR model system. J. Biol. Inorg. Chem 2010, 16, 109–116. [Google Scholar]
- Dodhia, V.R.; Fantuzzi, A.; Gilardi, G. Engineering human cytochrome P450 enzymes into catalytically self-sufficient chimeras using molecular Lego. J. Biol. Inorg. Chem 2006, 11, 903–916. [Google Scholar]
- Hlavica, P. Assembly of non-natural electron transfer conduits in the cytochrome P450 system: A critical assessment and update of artificial redox constructs amenable to exploitation in biotechnological areas. Biotechnol. Adv 2009, 27, 103–121. [Google Scholar]
- Sakaki, T.; Kominami, S.; Takemori, S.; Ohkawa, H.; Akiyoshi-Shibata, M.; Yabusaki, Y. Kinetic studies on a genetically engineered fused enzyme between rat cytochrome P4501A1 and yeast NADPH-P450 reductase. Biochemistry 1994, 33, 4933–4939. [Google Scholar]
- Shiota, N.; Kodama, S.; Inui, H.; Ohkawa, H. Expression of human cytochromes P450 1A1 and P450 1A2 as fused enzymes with yeast NADPH-cytochrome P450 oxidoreductase in transgenic tobacco plants. Biosci. Biotechnol. Biochem 2000, 64, 2025–2033. [Google Scholar]
- Adak, S.; Aulak, K.S.; Stuehr, D.J. Chimeras of nitric-oxide synthase types I and III establish fundamental correlates between heme reduction, heme-NO complex formation, and catalytic activity. J. Biol. Chem 2001, 276, 23246–23252. [Google Scholar]
- Roman, L.J.; McLain, J.; Masters, B.S. Chimeric enzymes of cytochrome P450 oxidoreductase and neuronal nitric-oxide synthase reductase domain reveal structural and functional differences. J. Biol. Chem 2003, 278, 25700–25707. [Google Scholar]
- Porter, R.; Jachymova, M.; Martasek, P.; Kalyanaraman, B.; Vasquez-Vivar, J. Reductive activation of Cr(Vi) by nitric oxide synthase. Chem. Res. Toxicol 2005, 18, 834–843. [Google Scholar]
- Aigrain, L.; Pompon, D.; Truan, G. Role of the interface between the FMN and FAD domains in the control of redox potential and electronic transfer of NADPH-cytochrome P450 reductase. Biochem. J 2011, 435, 197–206. [Google Scholar]
- Jenkins, C.M.; Genzor, C.G.; Fillat, M.F.; Waterman, M.R.; Gomez-Moreno, C. Negatively charged anabaena flavodoxin residues (Asp144 and Glu145) are important for reconstitution of cytochrome P450 17alpha-hydroxylase activity. J. Biol. Chem 1997, 272, 22509–22513. [Google Scholar]
- Alfieri, A.; Malito, E.; Orru, R.; Fraaije, M.W.; Mattevi, A. Revealing the moonlighting role of NADP in the structure of a flavin-containing monooxygenase. Proc. Natl. Acad. Sci. USA 2008, 105, 6572–6577. [Google Scholar]
- Perret, A.; Pompon, D. Electron shuttle between membrane-bound cytochrome P450 3A4 and b5 rules uncoupling mechanisms. Biochemistry 1998, 37, 11412–11424. [Google Scholar]
- Aigrain, L.; Pompon, D.; Morera, S.; Truan, G. Structure of the open conformation of a functional chimeric NADPH cytochrome P450 reductase. EMBO Rep 2009, 10, 742–747. [Google Scholar]
- Hamdane, D.; Xia, C.; Im, S.C.; Zhang, H.; Kim, J.J.; Waskell, L. Structure and function of an NADPH-cytochrome P450 oxidoreductase in an open conformation capable of reducing cytochrome P450. J. Biol. Chem 2009, 284, 11374–11384. [Google Scholar]
- Hall, D.A.; Vander Kooi, C.W.; Stasik, C.N.; Stevens, S.Y.; Zuiderweg, E.R.; Matthews, R.G. Mapping the interactions between flavodoxin and its physiological partners flavodoxin reductase and cobalamin-dependent methionine synthase. Proc. Natl. Acad. Sci. USA 2001, 98, 9521–9526. [Google Scholar]
- Ellis, J.; Gutierrez, A.; Barsukov, I.L.; Huang, W.C.; Grossmann, J.G.; Roberts, G.C. Domain motion in cytochrome P450 reductase: Conformational equilibria revealed by NMR and small-angle X-ray scattering. J. Biol. Chem 2009, 284, 36628–36637. [Google Scholar]
- Vincent, B.; Morellet, N.; Fatemi, F.; Aigrain, L.; Truan, G.; Guittet, E.; Lescop, E. The closed and compact domain organization of the 70-kDa human cytochrome P450 reductase in its oxidized state as revealed by NMR. J. Mol. Biol 2012, 420, 296–309. [Google Scholar]
- Hay, S.; Brenner, S.; Khara, B.; Quinn, A.M.; Rigby, S.E.; Scrutton, N.S. Nature of the energy landscape for gated electron transfer in a dynamic redox protein. J. Am. Chem. Soc 2010, 132, 9738–9745. [Google Scholar]
- Pudney, C.R.; Khara, B.; Johannissen, L.O.; Scrutton, N.S. Coupled motions direct electrons along human microsomal P450 Chains. PLoS Biol 2011, 9, e1001222. [Google Scholar]
- Jenner, M.; Ellis, J.; Huang, W.C.; Raven, E.L.; Roberts, G.C.; Oldham, N.J. Detection of a protein conformational equilibrium by electrospray ionisation-ion mobility-mass spectrometry. Angew. Chem. Int. Ed. Engl 2011, 50, 8291–8294. [Google Scholar]
- Iyanagi, T. Structure and function of NADPH-cytochrome P450 reductase and nitric oxide synthase reductase domain. Biochem. Biophys. Res. Commun 2005, 338, 520–528. [Google Scholar]
- Haque, M.M.; Kenney, C.; Tejero, J.; Stuehr, D.J. A kinetic model linking protein conformational motions, interflavin electron transfer and electron flux through a dual-flavin enzyme-simulating the reductase activity of the endothelial and neuronal nitric oxide synthase flavoprotein domains. FEBS J 2011, 278, 4055–4069. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diflavin reductase | Domains | % NOS activity | References | |||
|---|---|---|---|---|---|---|
| Heme | CamBD | FMN | FAD | |||
| eNOS | 10 | |||||
| nNOS | 60 | |||||
| iNOS | 100 | |||||
| iNOS-nCamBD | 100 | [25] | ||||
| nNOS-iCamBD | 30 | [25] | ||||
| iOx-nRed | 100 | [25] | ||||
| nOx-iRed | 0 | [25] | ||||
| eOx-nRed | 60 | [129] | ||||
| nOx-eRed | 20 | [129] | ||||
| eNOS-nCamBD | 40 | [110] | ||||
| Diflavin reductase | Domains | % activity | |||||
|---|---|---|---|---|---|---|---|
| Heme | CamBD | FMN | FAD | NOS | cyt c | ferricyanide | |
| nNOS | 100 | 100 | 100 | ||||
| CPR | 75 | 20 | |||||
| Chimera 1 | >100 | 50 | 50 | ||||
| Chimera 2 | ≈0 | 15 | >100 | ||||
| Chimera3 | >100 | 50 | |||||
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Aigrain, L.; Fatemi, F.; Frances, O.; Lescop, E.; Truan, G. Dynamic Control of Electron Transfers in Diflavin Reductases. Int. J. Mol. Sci. 2012, 13, 15012-15041. https://doi.org/10.3390/ijms131115012
Aigrain L, Fatemi F, Frances O, Lescop E, Truan G. Dynamic Control of Electron Transfers in Diflavin Reductases. International Journal of Molecular Sciences. 2012; 13(11):15012-15041. https://doi.org/10.3390/ijms131115012
Chicago/Turabian StyleAigrain, Louise, Fataneh Fatemi, Oriane Frances, Ewen Lescop, and Gilles Truan. 2012. "Dynamic Control of Electron Transfers in Diflavin Reductases" International Journal of Molecular Sciences 13, no. 11: 15012-15041. https://doi.org/10.3390/ijms131115012