3.1. PECs of the BeF and Spectroscopic Parameters
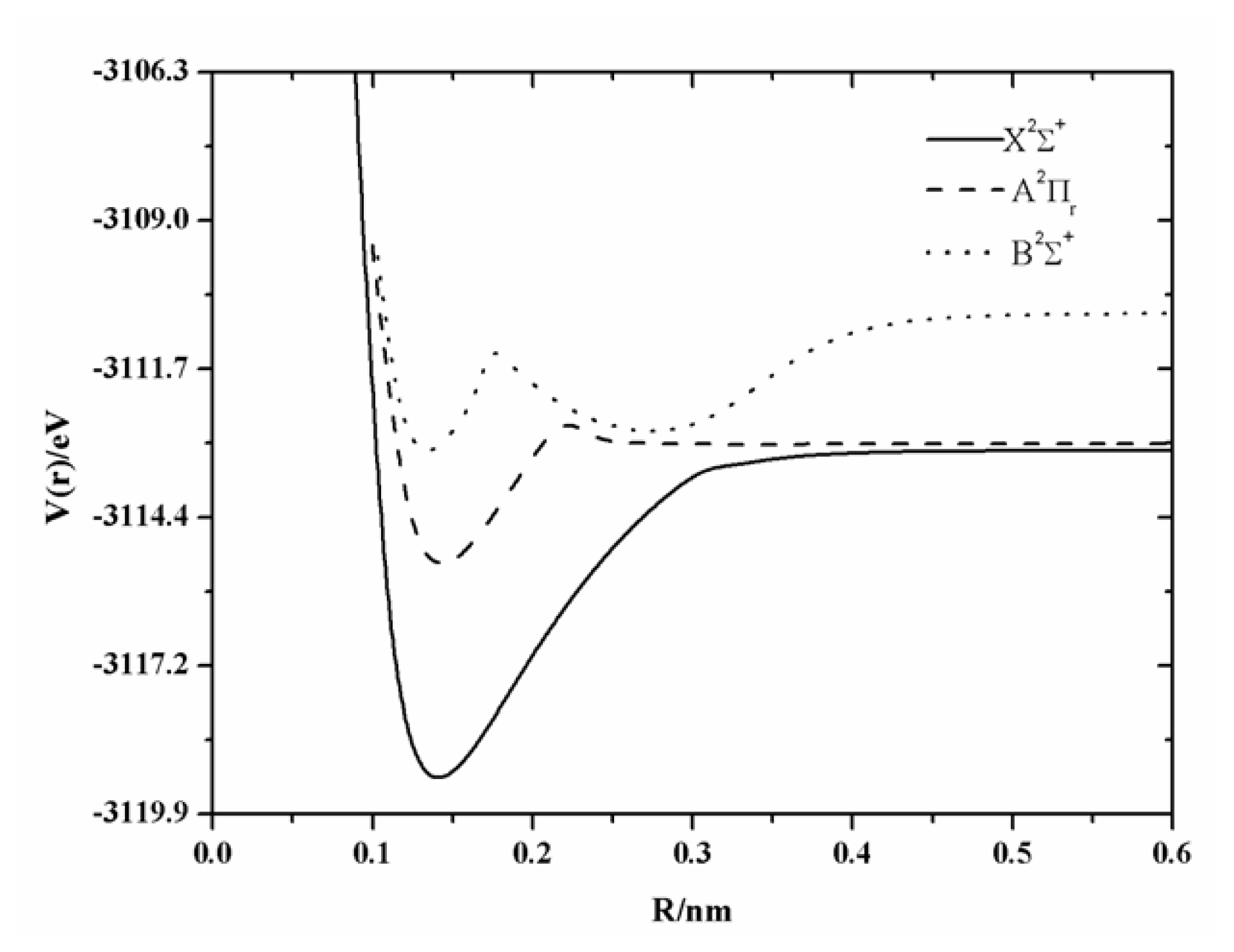
The PECs of BeF radical are shown in
Figure 1. As shown in the figure, the A
2Π
r curve and the B
2∑
+ curve are all marginally repulsive at long range, but they do not converge. The A
2Π
r state and the X
2∑
+ state have the same dissociation channel Be(
1S
g) +F(
2P
u), which is different from Be(
3P
u) +F(
2P
u) for the B
2∑
+ state. During the course of the PEC investigation of the X
2∑
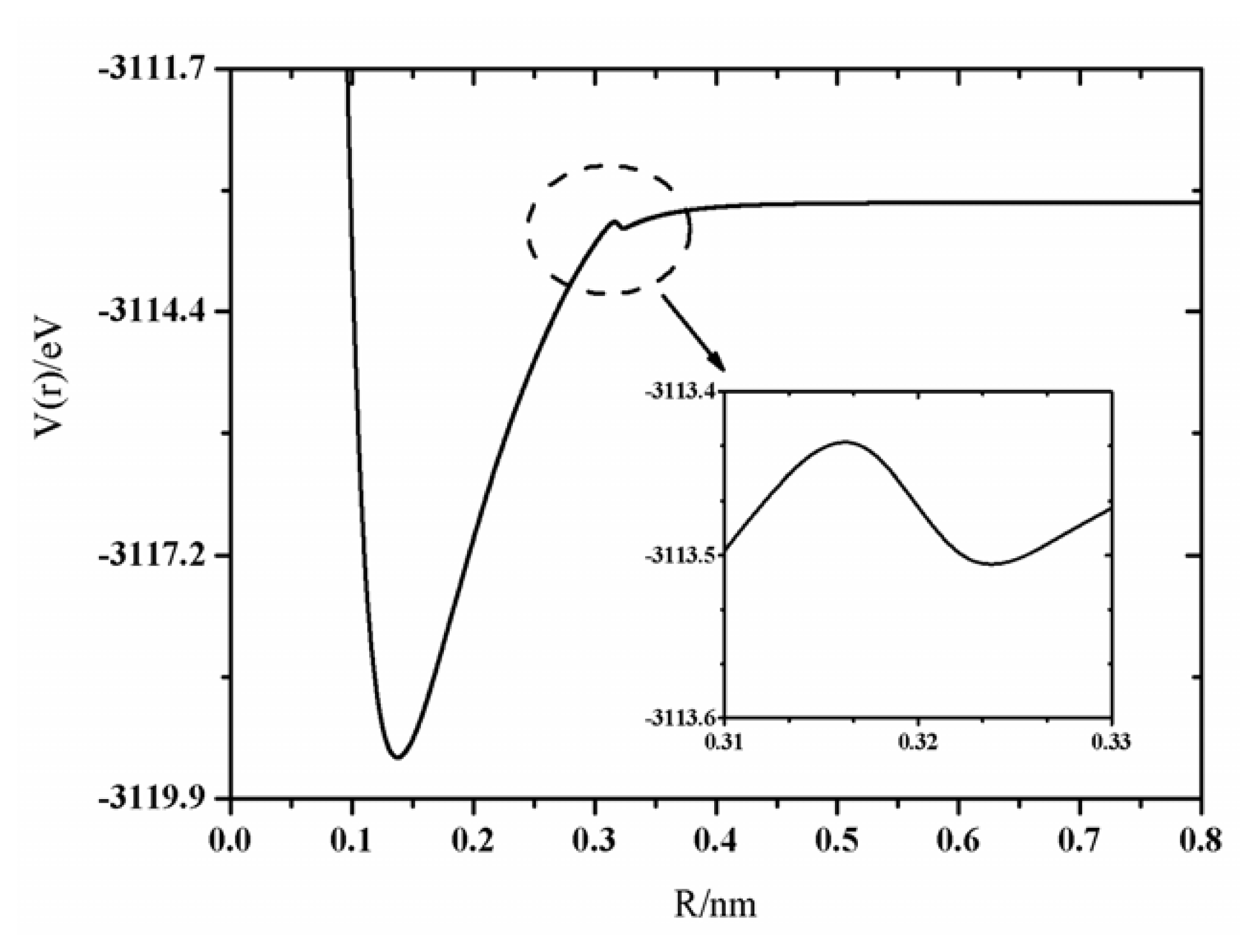
+ state, the existence of the barrier was a hot topic and should be stressed here, however, that it is not the main goal of the present work. To illustrate the existence of the barrier of the PEC of the X
2∑
+ state, a magnified image for the PEC of the X
2∑
+ state has been shown in
Figure 2. It has been found in our calculations that there is a small barrier in the curve of X
2∑
+ state which has been found at the internuclear separation, 0.3372 nm, and the barrier height is of 0.18 eV. A similar situation was also found by Roach [
12] and Machado [
17], but not by Marian [
14] and Ornellas
et al. [
18]. Ornellas
et al. [
18] did not observe the small hump since the interval used was too large when they calculated the PEC. Marian [
14] paid attention to calculating the spin-orbit coupling, and he considered 42 reference state functions to generate the CI wavefunction. In similarity with Reference [
18], the interval was also too large in his calculations [
14]. A wide barrier of 0.79 eV has been found in the PEC of the A
2Π
r state, similar to the value reported by Marian [
14] and Ornellas
et al. [
18], 0.81 eV and 0.79 eV, respectively. A similar feature has also been found for the B
2∑
+ curve of the BeF radical. Near 0.18nm, the B
2∑
+ state unfolds a sharp avoided crossing with the repulsive covalent state correlating with the dissociation channel Be(
3P
u) +F(
2P
u). So the avoided crossing and the ionic character are responsible for the unusual shape of these potential curves.
With the PECs determined, the spectroscopic parameters and molecular constants are evaluated with the VIBROT module in MOLCAS 7.4 program package. In order to conveniently compare the present results, we compiled the spectroscopic parameters together with the available experiments [
7–
11] and other theories [
12–
21] in
Table 1 for the BeF radical.
A number of theoretical investigations had been made on the spectroscopic parameters of the X
2∑
+ state of the BeF radical. Partridge
et al. [
13] in 1984 carried out the
Re,
De and
D0 calculations using Hartree-Fock (HF) method and some empirical formulas with Slater-type orbital (STO) basis set. Although their calculational results are close to the experiments, the existing experimental values and some empirical formulas were used and only two spectroscopic parameters were evaluated in their investigations. In 1985, Marian [
14] investigated the PEC using multireference doubles configuration interaction approach (MRDCI) method with the GTO DZP AO basis set. With the aid of PEC, they calculated several spectroscopic parameters. We can find that his
ωeχe is slightly smaller than the present one when compared with the corresponding experiments, though his
Re is in more agreement with the experiments than ours. Langhoff
et al. [
15] in 1986 calculated
Re and
ωe by two methods. We find that their most favorable results were obtained by the configuration interaction (CI) approach. As shown in
Table 1, it is believed that these results are the most accurate values so far, but only limited spectroscopic parameters are derived. Langhoff
et al. [
16] later evaluated the
Re and
ωe by three approaches. By comparison with the experiments, we find that their most favorable results were obtained with the singles and doubles configuration interaction (SDCI) approach. Also, the values are in more agreement with the experiments when compared with the present ones. However, their investigations were not concerned with other spectroscopic parameters.
Later, Machado and Ornellas [
17] in 1989 made the PEC calculations by multireference singles and doubles configuration interaction approach (MRSDCI) with the Gaussian sets (5
s, 3
p) for Be and (7
s, 4
p) for F. As can be seen in
Table 1, their
ωe and
ωeχe are too large when compared with the experiments. Three years later, Ornellas
et al. [
18] in 1992 made the PEC calculation for ground state. In the calculations, their approach is the MRSDCI and the basis sets are (14
s10
p3
d1
f)/[8
s6
p3
d1
f] for F and (11
s6
p1
d)/[6
s4
p1
d] for Be. By comparison with the present ones, it is not difficult to find that their
ωeχe and
ωe are slightly larger than the present experiments. Recently, Li and Hamilton [
19] in 2001 calculated the
Re using density functional theory (DFT) and MØller-Plesset (MP2) methods with three basis sets. Their most favorable results were obtained by DFT (BH and HLYP) approach with 6 − 311 + G* basis sets. However, they did not compute spectroscopic parameters apart from the
Re and
ωe. Recently, Pelegrini
et al. [
20] in 2005 performed some spectroscopic parameter calculations by the MRCI method with the aug-cc-pVQZ basis set. As tabulated in
Table 1, their
ωeχe is far from the measurements when compared with the present work. Furthermore, other important spectroscopic parameters (such as
Be and
αe) were not evaluated in their investigations.
For the A
2Π
r state, Walker and Richards [
21] performed the
Re and
ωe calculations using two methods in 1967. We find that their optimal results were obtained by the configuration interaction (CI) approach. As shown in
Table 1, their
ωe is slightly smaller than the experiment data and other important spectroscopic parameters were not evaluated in their investigations. In 1985, Marian [
14] investigated the PEC using MRDCI method with a GTO DZP AO basis set, with the aid of PEC, they calculated several spectroscopic parameters. We can find that his
ωeχe is too large and his
De is too small when compared with the experiments. Furthermore,
αe was not evaluated in his investigations. Ornellas
et al. [
18] in 1992 made the PEC calculation for lowest-lying state. In the calculations, their approach is the MRSDCI and the basis sets are (14
s10
p3
d1
f)/[8
s6
p3
d1
f] for F and (11
s6
p1
d)/[6
s4
p1
d] for Be. By comparison, it is not difficult to find that their
ωeχe and
ωe are slightly larger than the present experiments when compared with the present ones. Pelegrini
et al. [
20] also performed some spectroscopic parameter calculations for the A
2Π
r state of the BeF radical using the MRCI method with the aug-cc-pVQZ basis set. As tabulated in
Table 1, their
ωeχe and
ωe are far from the available measurements when compared with our work.
For the B
2∑
+ of BeF radical, few theoretical investigations have been made on the spectroscopic parameters. The earlier theoretical calculations were performed by Marian [
14]. He investigated the PEC of BeF(B
2∑
+) using MRDCI method with a GTO DZP AO basis set. We can find that his
ωe and
ωeχe are too large when compared with the experiments. Furthermore,
De and
αe were not evaluated in his investigations.
According to the above analysis and discussion, on the whole, the spectroscopic parameters obtained in the present work have improved when compared with previous theoretical results. For example, for the X
2∑
+ state, the spectroscopic parameters,
ωeχe,
αe, ωe,
Be and
Re, deviate from the experiments [
11] only by 0.11%, 0.57%, 0.90%, 1.60% and 0.81%, respectively. For the BeF(A
2Π
r), the spectroscopic parameters,
ωeχe,
αe, ωe,
Be and
Re, deviate from the experiments [
11] only by 0.00%, 2.86%, 1.69%, 0.51% and 0.25%, respectively.
As for the dissociation energy
De of BeF(X
2∑
+), it shows a wide variation. Roach and Kuntz [
12] in 1982 made valence-bond (VB) calculations on the BeF(X
2∑
+) radical, and they obtained the value to be 3.94 eV. But they claimed that their VB calculations are not accurate enough to deduce the accurate value of
De in Reference [
12]. Partridge
et al. [
13] calculated the
D0 with empirical formula and obtained the direct value of
D0 to be 5.86 eV, and also gave the estimate result of 5.91 eV. The precision of the method is slightly lower than this work. Marian [
14] investigated the PEC using MRDCI method with a GTO DZP AO basis set. They obtained
De of 5.5 eV, however, he thought that the value is a little small. Langhoff
et al. [
15] calculated the
De by the SCF method. As we know, the method is too simple so that the
De result they obtained is not very credible. Machado and Ornellas [
17] calculated the
De by MRSDCI approach with the Gaussian sets (5s,3p) for Be and (7s,4p) for F. Ornellas
et al. [
18] computed the
De by the MRSDCI method and the basis sets are (11s6p1d)/[6s4p1d] for Be and (14
s10
p3
d1
f)/[8
s6
p3
d1
f] for F. The basis sets they used are very small. Therefore, their values are less accurate. In the present work, the PEC of BeF(X
2∑
+) is computed using the highly accurate MRCI approach with the large basis sets, cc-pV5Z for Be and aug-cc-pV6Z for F. With the aid of PEC, the
De is determined to be 6.22 eV, which should be relatively close to the true value.
In this paper, we also calculate the
ΔTe of the A
2Π
r state is of 32,343.9 cm
−1, while the value obtained by Marian [
14], Ornellas
et al. [
18] and Pelegrini
et al. [
20] to be 34,814 cm
−1, 33,974 cm
−1 and 34,902 cm
−1, respectively. And the
ΔTe of the B
2∑
+ state is also calculated, and the value is of 48,877 cm
−1, the data reported by Marian [
14] to be 50,844 cm
−1.
It is widely recognized that the accuracy of the spectroscopic parameters calculations mainly depends on the scanned results for the PEC of the electronic state by using CASSCF AND MRCI approach. The scanned results of the electronic state are related to the choice of the active space for a CASSCF and of the basis sets. For BeF radical, the each electronic state possesses different bonding orbitals at various internuclear sparations [
14]. In order to obtain more accurate calculational results of PECS of BeF radical, eight molecular orbitals, including four
a1, two
b1 and two
b2 symmetry MOs, are put into the active space, and the rest of the electrons in the BeF radical are put into two
a1 symmetry closed-shell orbitals, which differ from Reference [
20]. In addition, the appropriate choices of the basis sets and the calculational interval in the CASSCF calculation also conduce to the accurate calculational results. So we have reasons to believe that the present results are reliable.
3.2. Vibrational Manifolds
Based on the reliable PECs of the X
2∑
+, A
2Π
r and B
2∑
+ states, we determine their vibrational levels, inertial rotation and centrifugal constants when
J = 0. And we also compute classical turning points for the ground state. Owing to the length limitation of the paper, we only tabulate some of these results for the vibrational states in
Tables 2–
7. To the best of our knowledge, no experimental data of molecular constants have been found in the literature, except several groups of theoretical results. But according to the remarkable agreement between the present spectroscopic parameters and the available experiments and the excellent accordance between the theoretical and the corresponding RKR data, we have reasons to believe that the results collected in
Tables 2–
7 are accurate.
As can be seen from
Table 2, the present results are in excellent agreement with the theoretical data reported in the literature. For example, the deviations from the theories [
17] are of only 0.25%, 0.12%, 0.02% and 0.23% when
υ = 1, 3, 5 and 7, respectively, and the deviations from the theories [
18] deviate only by 0.23%, 0.33%, 0.45% and 0.64%, respectively. Therefore, we can say that the present calculations are accurate. Furthermore we can conclude that the values of vibrational levels and classical turning points presented in
Table 3 must be reliable.
Similar to the vibrational level spacings, there are two groups of theoretical data [
17,
18] concerned with the inertial rotation constant
Bυ and centrifugal distortion constant
Dυ of BeF(X
2∑
+). For a convenient comparison with the present results, we also tabulate them in
Table 4. By simple calculations, it is not difficult to find that excellent agreement exists between the present results and the theoretical data. For example for the
Bυ, the deviations from the theory [
17] are only 0.14%, 0.47%, and 0.51% when
υ =0, 2 and 4, respectively. As to the centrifugal distortion constant
Dυ, good accord also exists between the present results and the available theoretical data [
17,
18]. Therefore, the present calculations are accurate. According to these, the calculations of the centrifugal distortion constants presented in
Table 5 should be reliable.
As can be seen from
Table 6, the present results are in excellent agreement with the experiments [
14]. For example, the deviations from the experiments [
14] are only 0.13%, 0.19%, 0.27% and 0.38% when
υ = 0, 2, 4 and 6, respectively. Therefore, we can say that the present calculations are accurate. For the inertial rotation constant
Bυ, the deviations of the present values from the experiments [
8] are of 0.50% and 0.45%, when
υ = 0 and 1, respectively.
To the best of our knowledge, no experimental and theoretical data of vibrational levels and molecular constants for BeF(B
2∑
+) has been found in the literature. However, according to the remarkable agreement between the present spectroscopic parameters and the available experiments [
8,
11], we have reasons to believe that the results collected in
Tables 5 are accurate.




{kind=link}
{kind=link}