Increased Activity of Cell Surface Peptidases in HeLa Cells Undergoing UV-Induced Apoptosis Is Not Mediated by Caspase 3


Abstract
:1. Introduction
2. Results
2.1. Cellular Protein Content
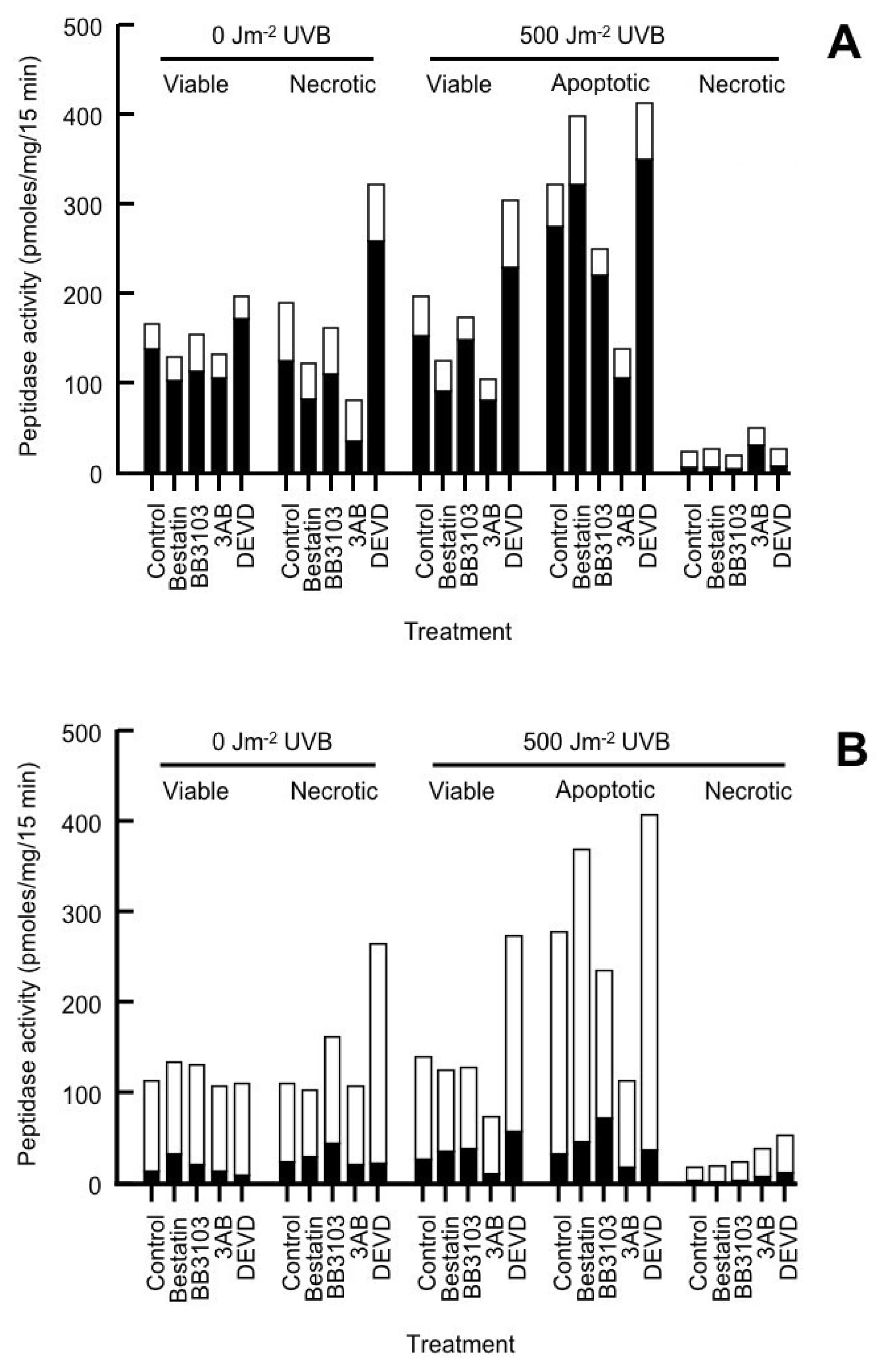
2.2. Cell Surface Peptidase (CSP) Activity
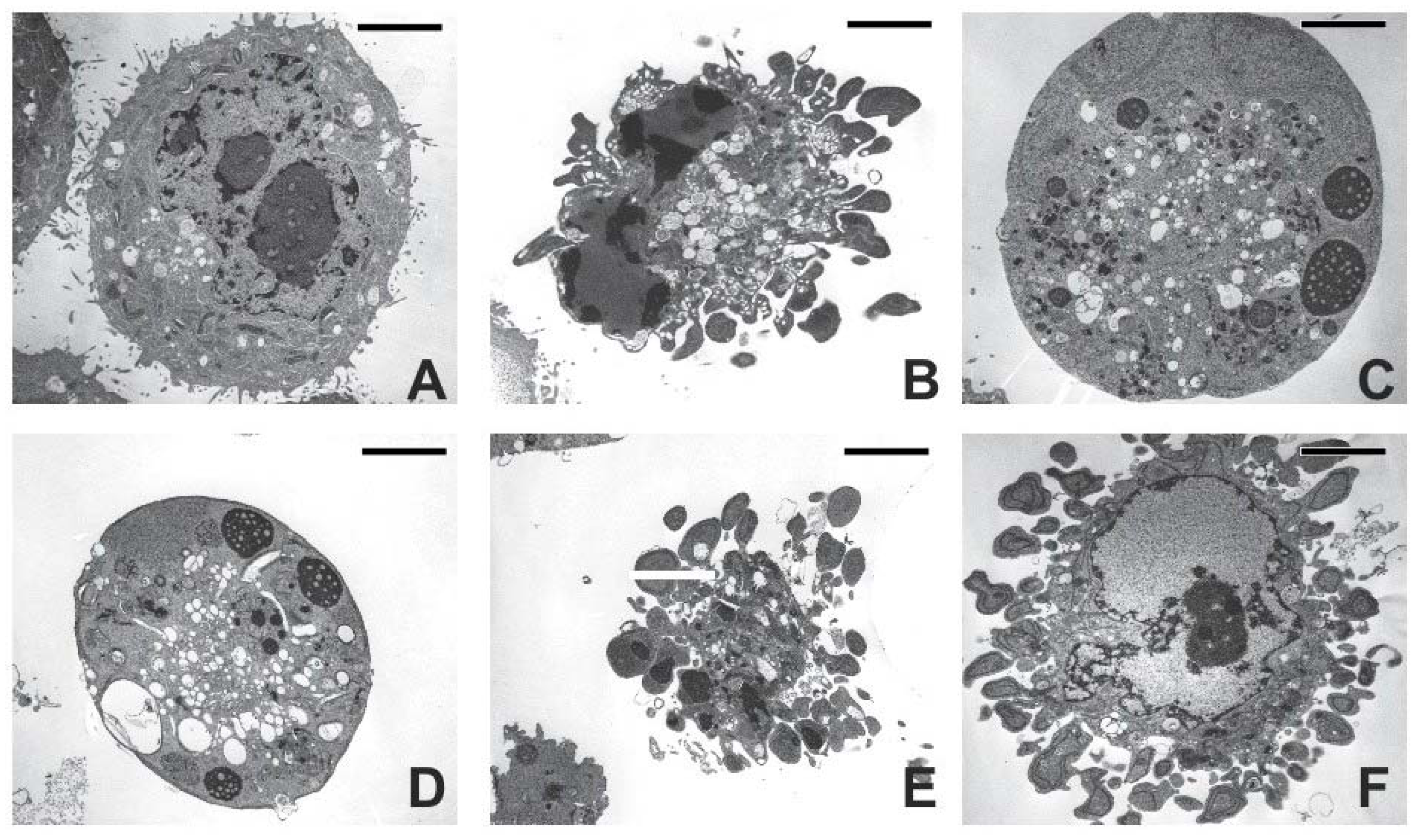
2.3. Cell Morphology
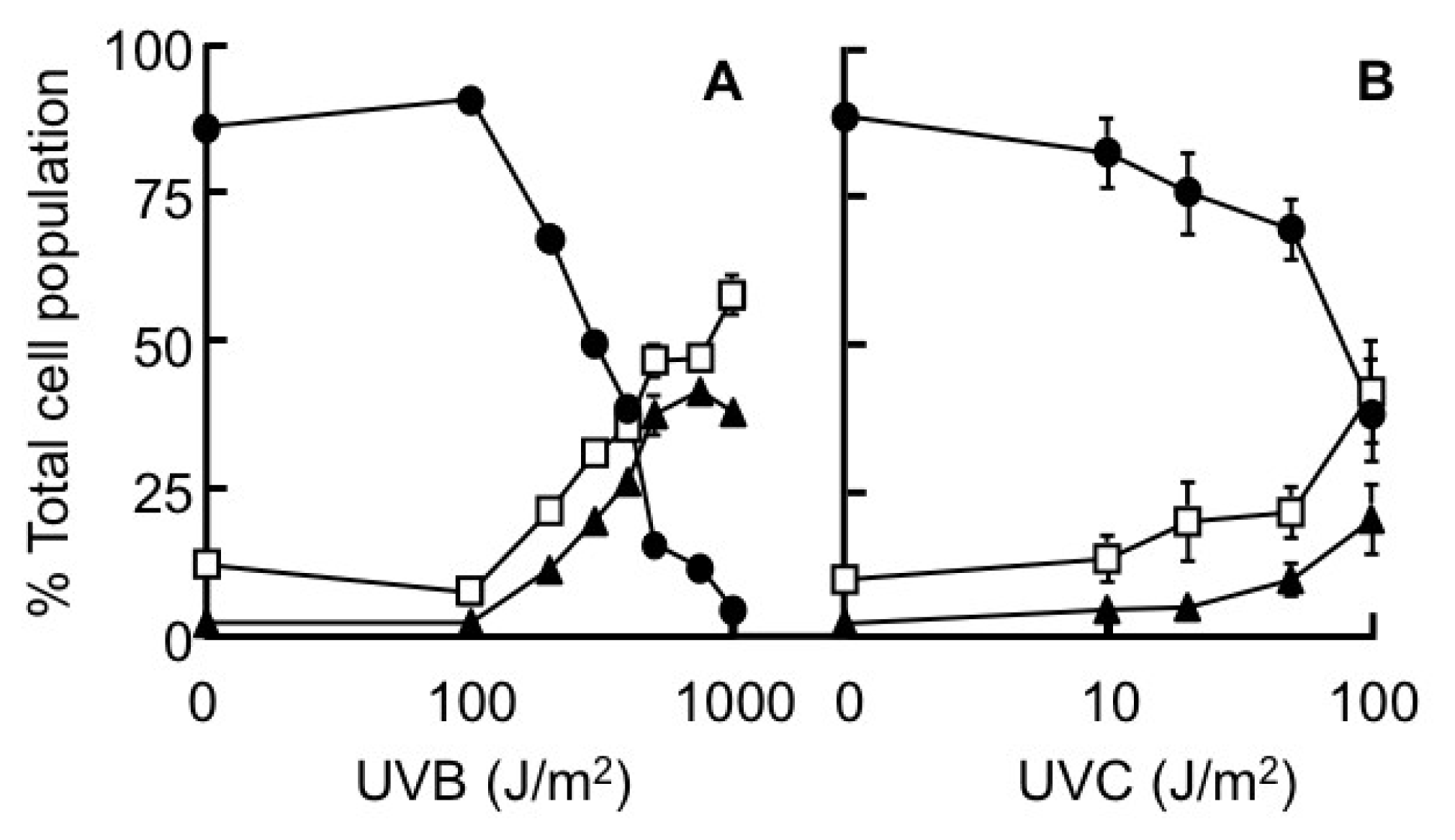
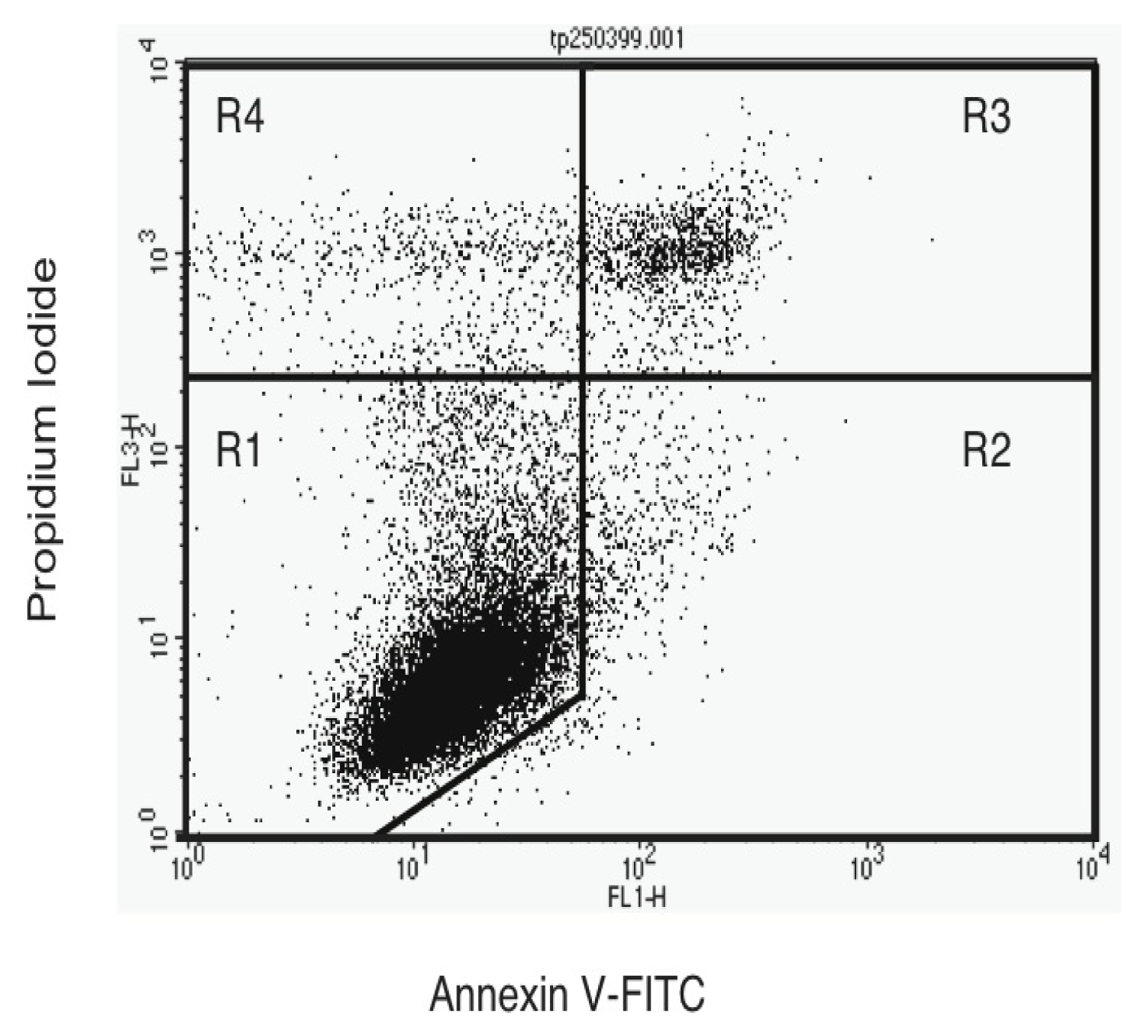
2.4. Annexin V-Staining Profiles
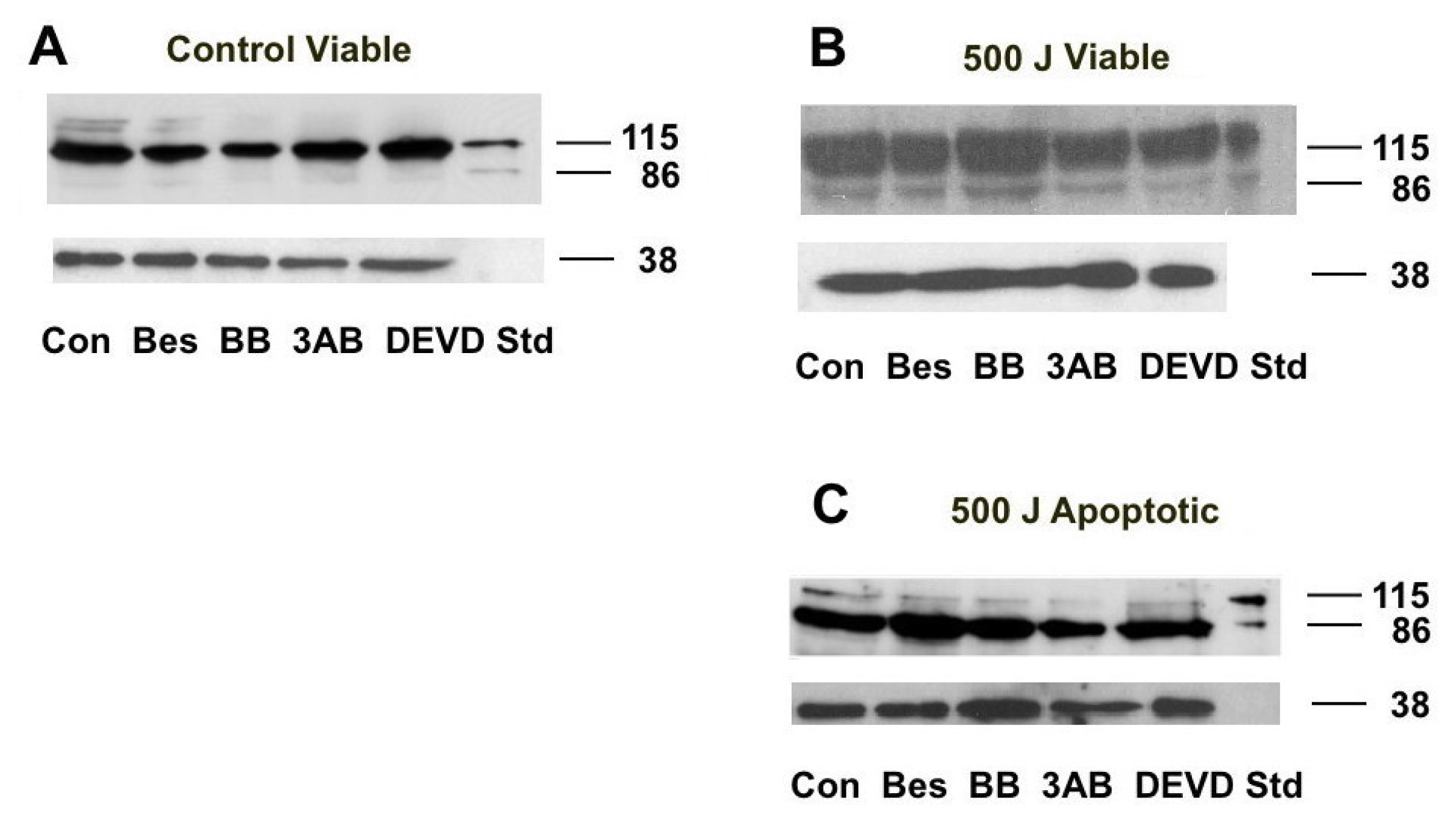
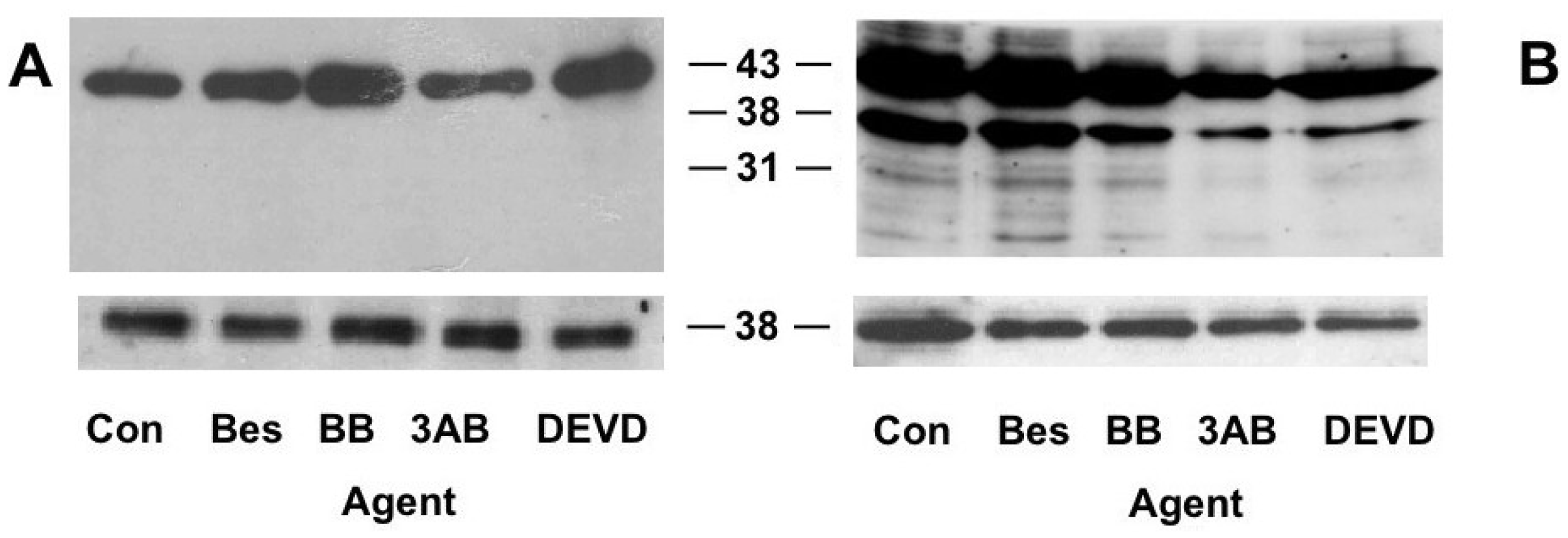
2.5. PARP Cleavage
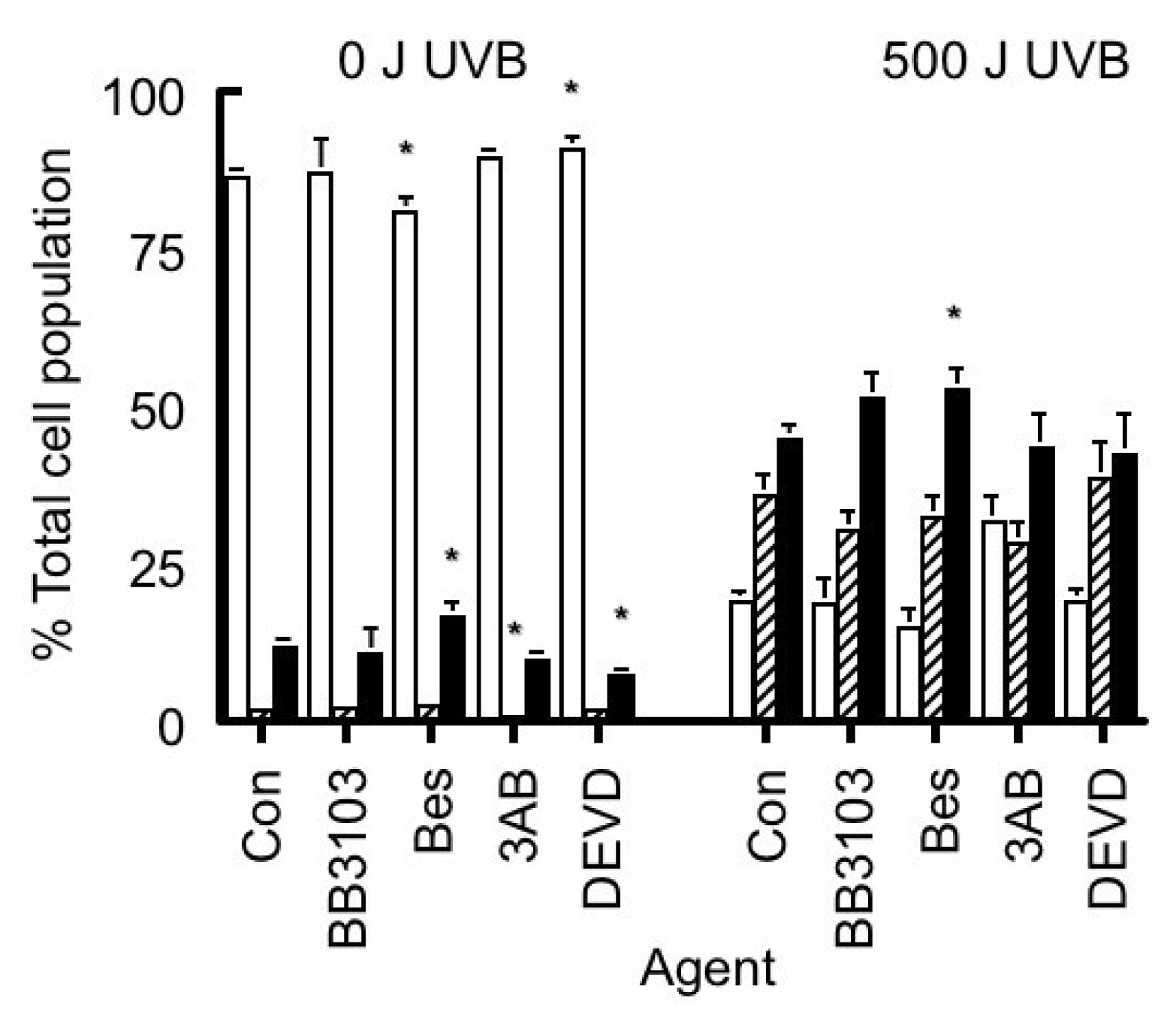
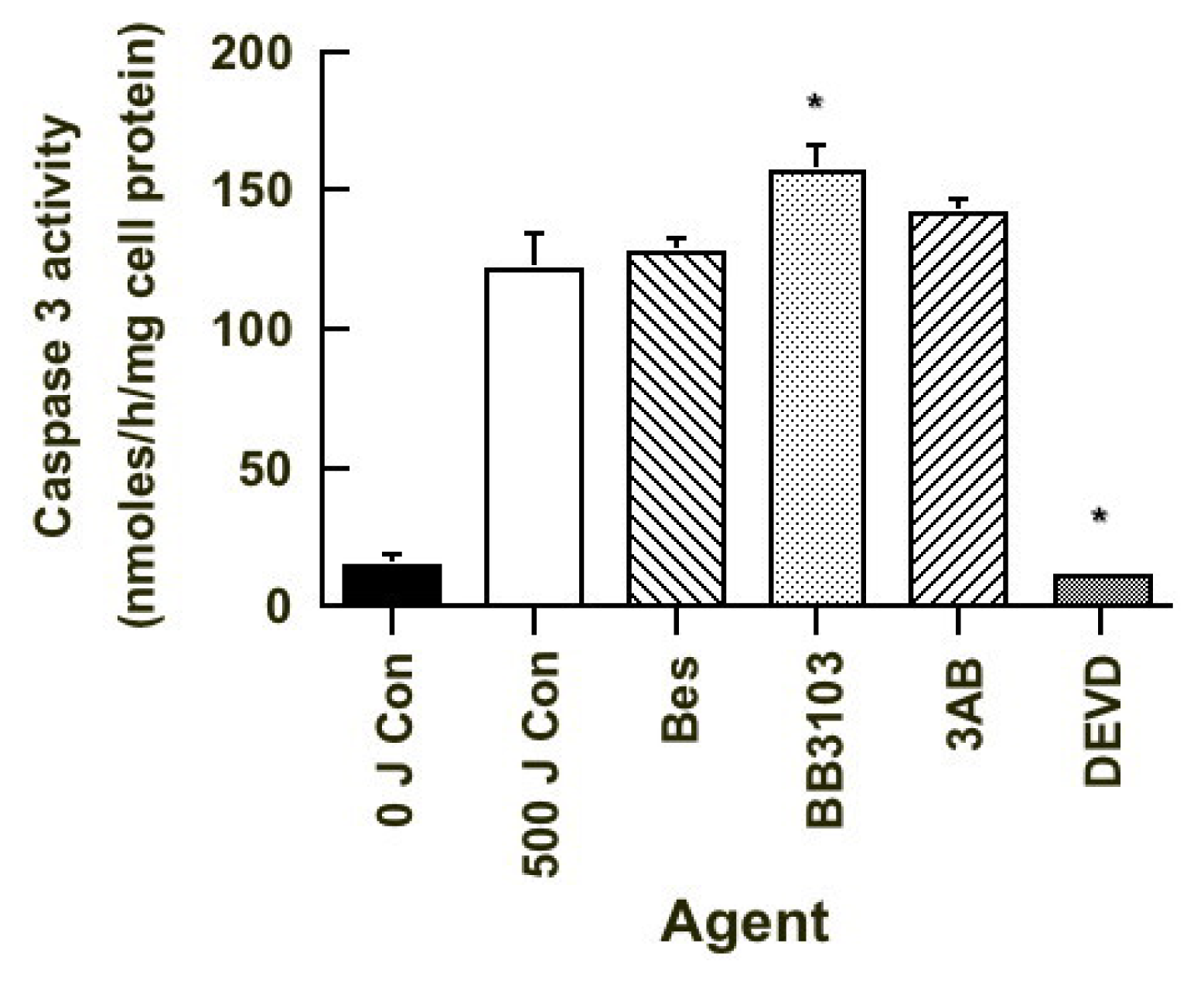
2.6. Caspase 3 Activity
2.7. Actin Cleavage
3. Discussion
4. Experimental Section
4.1. Materials
4.2. HeLa Cell Culture
4.3. Preparation of Cells for Sorting by Flow Cytometry
4.4. Flow Cytometry
4.5. Electron Microscopy
4.6. Annexin V Staining
4.7. Western Blot Assay
4.8. Peptidase Activity
4.9. Caspase 3 Activity Studies
4.10. Calculations
5. Conclusions
Acknowledgements
References
- Assefa, Z.; van Laethem, A.; Garmyn, M.; Agostinis, P. Ultraviolet radiation-induced apoptosis in keratinocytes: On the role of cytosolic factors. Biochim. Biophys. Acta 2005, 1755, 90–106. [Google Scholar]
- Kulms, D.; Schwarz, T. Independent contribution of three different pathways to ultraviolet-B-induced apoptosis. Biochem. Pharmacol 2002, 64, 837–841. [Google Scholar]
- van Laethem, A.; Garmyn, M.; Agostinis, P. Starting and propagating apoptotic signals in UVB irradiated keratinocytes. Photochem. Photobiol. Sci 2009, 8, 299–308. [Google Scholar]
- Kerr, J.F.R.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar]
- Ivanova, S.; Gregorc, U.; Vidergar, N.; Javier, R.; Bredt, D.S.; Vandenabeele, P.; Pardo, J.; Simon, M.M.; Turk, V.; Banks, L.; et al. MAGUKs, scaffolding proteins at cell junctions, are substrates of different proteases during apoptosis. Cell Death Dis 2011, 2, e116. [Google Scholar]
- Luo, H.; Wang, A. Induction of apoptosis in K562 cells by jolkinolide B. Can. J. Physiol. Pharmacol 2006, 84, 959–965. [Google Scholar]
- Krysko, D.V.; Berghe, T.V.; D’Herde, K.; Vandenabeele, P. Apoptosis and necrosis: Detection, discrimination and phagocytosis. Methods 2008, 44, 205–221. [Google Scholar]
- Balasubramanian, K.; Mirnikjoo, B.; Schroit, A.J. Regulated externalization of phosphatidylserine at the cell surface: Implications for apoptosis. J. Biol. Chem 2007, 282, 18357–18364. [Google Scholar]
- Fadok, V.A.; Voelker, D.R.; Campbell, P.A.; Cohen, J.J.; Bratton, D.L.; Henson, P.M. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J. Immunol 1992, 148, 2207–2216. [Google Scholar]
- Fischer, U.; Janicke, R.U.; Schulze-Osthoff, K. Many cuts to ruin: A comprehensive update of caspase substrates. Cell Death Differ 2003, 10, 76–100. [Google Scholar]
- Timmer, J.C.; Salvesen, G.S. Caspase substrates. Cell Death Differ 2007, 14, 66–72. [Google Scholar]
- O’Connell, A.R.; Stenson-Cox, C. A more serine way to die: Defining the characteristics of serine protease-mediated cell death cascades. Biochim. Biophys. Acta 2007, 1773, 1491–1499. [Google Scholar]
- Hakulinen, J.; Keski-Oja, J. ADAM10-mediated release of complement membrane cofactor protein during apoptosis of epithelial cells. J. Biol. Chem 2006, 281, 21369–21376. [Google Scholar]
- Steinhusen, U.; Weiske, J.; Badock, V.; Tauber, R.; Bommert, K.; Huber, O. Cleavage and shedding of E-cadherin after induction of apoptosis. J. Biol. Chem 2001, 276, 4972–4980. [Google Scholar]
- Wang, Y.; Robertson, J.D.; Walcheck, B. Different signaling pathways stimulate a disintegrin and metalloprotease-17 (ADAM17) in neutrophils during apoptosis and activation. J. Biol. Chem 2011, 286, 38980–38988. [Google Scholar]
- Wang, Y.; Zhang, A.C.; Ni, Z.; Herrera, A.; Walcheck, B. ADAM17 activity and other mechanisms of soluble L-selectin production during death receptor-induced leukocyte apoptosis. J. Immunol 2010, 184, 4447–4454. [Google Scholar]
- Piva, T.J.; Davern, C.M.; Francis, K.G.; Chojnowski, G.M.; Hall, P.M.; Ellem, K.A. Increased ecto-metallopeptidase activity in cells undergoing apoptosis. J. Cell. Biochem 2000, 76, 625–638. [Google Scholar]
- Inoue, S.; Browne, G.; Melino, G.; Cohen, G.M. Ordering of caspases in cells undergoing apoptosis by the intrinsic pathway. Cell Death Differ 2009, 16, 1053–1061. [Google Scholar]
- Krishnakumar, R.; Kraus, W.L. The PARP side of the nucleus: Molecular actions, physiological outcomes, and clinical targets. Mol. Cell 2010, 39, 8–24. [Google Scholar]
- Megnin-Chanet, F.; Bollet, M.A.; Hall, J. Targeting poly(ADP-ribose) polymerase activity for cancer therapy. Cell. Mol. Life Sci 2010, 67, 3649–3662. [Google Scholar]
- Soldani, C.; Scovassi, A.I. Poly(ADP-ribose) polymerase-1 cleavage during apoptosis: An update. Apoptosis 2002, 7, 321–328. [Google Scholar]
- Piva, T.J.; Krause, D.R.; Ellem, K.O. UVC activation of the HeLa cell membrane “TGFαase,” a metalloenzyme. J. Cell. Biochem 1997, 64, 353–368. [Google Scholar]
- Takasawa, R.; Nakamura, H.; Mori, T.; Tanuma, S. Differential apoptotic pathways in human keratinocyte HaCaT cells exposed to UVB and UVC. Apoptosis 2005, 10, 1121–1130. [Google Scholar]
- Suda, H.; Aoyagi, T.; Takeuchi, T.; Umezawa, H. Inhibition of aminopeptidase B and leucine aminopeptidase by bestatin and its stereoisomer. Arch. Biochem. Biophys 1976, 177, 196–200. [Google Scholar]
- Malorni, W.; Rivabene, R.; Straface, E.; Rainaldi, G.; Monti, D.; Salvioli, S.; Cossarizza, A.; Franceschi, C. 3-Aminobenzamide protects cells from UV-B-induced apoptosis by acting on cytoskeleton and substrate adhesion. Biochem. Biophys. Res. Commun 1995, 207, 715–724. [Google Scholar]
- Tiozzo, R.; Monti, D.; Straface, E.; Capri, M.; Croce, M.A.; Rainaldi, G.; Franceschi, C.; Malorni, W. Antiproliferative activity of 3-aminobenzamide in A431 carcinoma cells is associated with a target effect on cytoskeleton. Biochem. Biophys. Res. Commun 1996, 225, 826–832. [Google Scholar]
- Wu, Y.; Wang, D.; Wang, X.; Wang, Y.; Ren, F.; Chang, D.; Chang, Z.; Jia, B. Caspase 3 is activated through caspase 8 instead of caspase 9 during H2O2-induced apoptosis in HeLa cells. Cell. Physiol. Biochem 2011, 27, 539–546. [Google Scholar]
- Darzynkiewicz, Z.; Bruno, S.; del Bino, G.; Gorczyca, W.; Hotz, M.A.; Lassota, P.; Traganos, F. Features of apoptotic cells measured by flow cytometry. Cytometry 1992, 13, 795–808. [Google Scholar]
- Brown, S.B.; Krause, D.; Townsend, E.; Ellem, K.A.O. Development of a sensitive peptidase assay: In search of cell associated proteases responsible for the cleavage of preproTGFα. J. Cell. Biochem 1992, 48, 411–423. [Google Scholar]
- Piva, T.J.; Francis, K.G.; Krause, D.R.; Chojnowski, G.M.; Ellem, K.A. Effect of UV irradiation on cell surface protease activity and amino acid uptake. Mutat. Res 1998, 422, 55–67. [Google Scholar]
- Li, X.; Perez, L.; Pan, Z.; Fan, H. The transmembrane domain of TACE regulates protein ectodomain shedding. Cell Res 2007, 17, 985–998. [Google Scholar]
- Merchant, N.B.; Voskresensky, I.; Rogers, C.M.; Lafleur, B.; Dempsey, P.J.; Graves-Deal, R.; Revetta, F.; Foutch, A.C.; Rothenberg, M.L.; Washington, M.K.; et al. TACE/ADAM-17: A component of the epidermal growth factor receptor axis and a promising therapeutic target in colorectal cancer. Clin. Cancer Res 2008, 14, 1182–1191. [Google Scholar]
- Juanes, P.P.; Ferreira, L.; Montero, J.C.; Arribas, J.; Pandiella, A. N-terminal cleavage of proTGFα occurs at the cell surface by a TACE-independent activity. Biochem. J 2005, 389, 161–172. [Google Scholar]
- Brown, S.B.; Kluck, R.M.; Ellem, K.A. Loss and shedding of surface markers from the leukemic myeloid monocytic line THP-1 induced to undergo apoptosis. J. Cell. Biochem 1996, 60, 246–259. [Google Scholar]
- Mucha, A.; Drag, M.; Dalton, J.P.; Kafarski, P. Metallo-aminopeptidase inhibitors. Biochimie 2010, 92, 1509–1529. [Google Scholar]
- Hart, S.P.; Ross, J.A.; Ross, K.; Haslett, C.; Dransfield, I. Molecular characterization of the surface of apoptotic neutrophils: Implications for functional downregulation and recognition by phagocytes. Cell Death Differ 2000, 7, 493–503. [Google Scholar]
- Yang, B.; Gao, J.; Rao, Z.; Zhang, B.; Ouyang, W.; Yang, C. Antisense oligonucleotide Targeting matrix metalloproteinase-7 (MMP-7) changes the ultrastructure of human A549 lung adenocarcinoma cells. Ultrastruct. Pathol 2011, 35, 256–259. [Google Scholar]
- van Engeland, M.; Ramaekers, F.C.; Schutte, B.; Reutelingsperger, C.P. A novel assay to measure loss of plasma membrane asymmetry during apoptosis of adherent cells in culture. Cytometry 1996, 24, 131–139. [Google Scholar]
- Mammone, T.; Gan, D.; Collins, D.; Lockshin, R.A.; Marenus, K.; Maes, D. Successful separation of apoptosis and necrosis pathways in HaCaT keratinocyte cells induced by UVB irradiation. Cell Biol. Toxicol 2000, 16, 293–302. [Google Scholar]
- Maverakis, E.; Miyamura, Y.; Bowen, M.P.; Correa, G.; Ono, Y.; Goodarzi, H. Light, including ultraviolet. J. Autoimmun 2010, 34, J247–J257. [Google Scholar]
- Muthusamy, V.; Piva, T.J. The UV response of the skin: A review of the MAPK, NFκB and TNFα signal transduction pathways. Arch. Dermatol. Res 2010, 302, 5–17. [Google Scholar]
- Bohnke, A.; Westphal, F.; Schmidt, A.; El-Awady, R.A.; Dahm-Daphi, J. Role of p53 mutations, protein function and DNA damage for the radiosensitivity of human tumour cells. Int. J. Radiat. Biol 2004, 80, 53–63. [Google Scholar]
- Aliouat-Denis, C.M.; Dendouga, N.; van den Wyngaert, I.; Goehlmann, H.; Steller, U.; van de Weyer, I.; van Slycken, N.; Andries, L.; Kass, S.; Luyten, W.; et al. p53-Independent regulation of p21Waf1/Cip1 expression and senescence by Chk2. Mol. Cancer Res 2005, 3, 627–634. [Google Scholar]
- Rodust, P.M.; Stockfleth, E.; Ulrich, C.; Leverkus, M.; Eberle, J. UV-induced squamous cell carcinoma—A role for antiapoptotic signalling pathways. Br. J. Dermatol 2009, 161, S107–S115. [Google Scholar]
- Lei, X.; Liu, B.; Han, W.; Ming, M.; He, Y.Y. UVB-Induced p21 degradation promotes apoptosis of human keratinocytes. Photochem. Photobiol. Sci 2010, 9, 1640–1648. [Google Scholar]
- Ziegler, A.; Jonason, A.S.; Leffell, D.J.; Simon, J.A.; Sharma, H.W.; Kimmelman, J.; Remington, L.; Jacks, T.; Brash, D.E. Sunburn and p53 in the onset of skin cancer. Nature 1994, 372, 773–776. [Google Scholar]
- Hazar-Rethinam, M.; Endo-Munoz, L.; Gannon, O.; Saunders, N. The role of the E2F transcription factor family in UV-induced apoptosis. Int. J. Mol. Sci 2011, 12, 8947–8960. [Google Scholar]
- Ravi, R.; Jain, A.J.; Schulick, R.D.; Pham, V.; Prouser, T.S.; Allen, H.; Mayer, E.G.; Yu, H.; Pardoll, D.M.; Ashkenazi, A.; et al. Elimination of hepatic metastases of colon cancer cells via p53-independent cross-talk between irinotecan and Apo2 ligand/TRAIL. Cancer Res 2004, 64, 9105–9114. [Google Scholar]
- Ansorge, S.; Bank, U.; Heimburg, A.; Helmuth, M.; Koch, G.; Tadje, J.; Lendeckel, U.; Wolke, C.; Neubert, K.; Faust, J.; et al. Recent insights into the role of dipeptidyl aminopeptidase IV (DPIV) and aminopeptidase N (APN) families in immune functions. Clin. Chem. Lab. Med 2009, 47, 253–261. [Google Scholar]
- Plakidou-Dymock, S.; Tanner, M.J.; McGivan, J.D. A role for aminopeptidase N in Na+-dependent amino acid transport in bovine renal brush-border membranes. Biochem. J 1993, 290, 59–65. [Google Scholar]
- Rowland, A.F.; Fazakerley, D.J.; James, D.E. Mapping insulin/GLUT4 circuitry. Traffic 2011, 12, 672–681. [Google Scholar]
- Krysko, D.V.; Vandenabeele, P. From regulation of dying cell engulfment to development of anti-cancer therapy. Cell Death Differ 2008, 15, 29–38. [Google Scholar]
- Chalaris, A.; Rabe, B.; Paliga, K.; Lange, H.; Laskay, T.; Fielding, C.A.; Jones, S.A.; Rose-John, S.; Scheller, J. Apoptosis is a natural stimulus of IL6R shedding and contributes to the proinflammatory trans-signaling function of neutrophils. Blood 2007, 110, 1748–1755. [Google Scholar]
- van Laethem, A.; Claerhout, S.; Garmyn, M.; Agostinis, P. The sunburn cell: Regulation of death and survival of the keratinocyte. Int. J. Biochem. Cell Biol 2005, 37, 1547–1553. [Google Scholar]
- Moali, C.; Hulmes, D.J. Extracellular and cell surface proteases in wound healing: New players are still emerging. Eur. J. Dermatol 2009, 19, 552–564. [Google Scholar]
- Ayyash, M.; Tamimi, H.; Ashhab, Y. Developing a powerful in silico tool for the discovery of novel caspase-3 substrates: A preliminary screening of the human proteome. BMC Bioinforma 2012, 13, 14. [Google Scholar]
- Chauvier, D.; Ankri, S.; Charriaut-Marlangue, C.; Casimir, R.; Jacotot, E. Broad-spectrum caspase inhibitors: From myth to reality? Cell Death Differ 2007, 14, 387–391. [Google Scholar]
- Nicolescu, A.C.; Holt, A.; Kandasamy, A.D.; Pacher, P.; Schulz, R. Inhibition of matrix metalloproteinase-2 by PARP inhibitors. Biochem. Biophys. Res. Commun 2009, 387, 646–650. [Google Scholar]
- Seidah, N.G. The proprotein convertases, 20 years later. Methods Mol. Biol 2011, 768, 23–57. [Google Scholar]
- Platonova, A.; Koltsova, S.V.; Hamet, P.; Grygorczyk, R.; Orlov, S.N. Swelling rather than shrinkage precedes apoptosis in serum-deprived vascular smooth muscle cells. Apoptosis 2012, in press. [Google Scholar]
- Ferguson, H.A.; Marietta, P.M.; van den Berg, C.L. UV-induced apoptosis is mediated independent of caspase-9 in MCF-7 cells: A model for cytochrome c resistance. J. Biol. Chem 2003, 278, 45793–45800. [Google Scholar]
- Wlodkowic, D.; Telford, W.; Skommer, J.; Darzynkiewicz, Z. Apoptosis and beyond: Cytometry in studies of programmed cell death. Methods Cell Biol 2011, 103, 55–98. [Google Scholar]







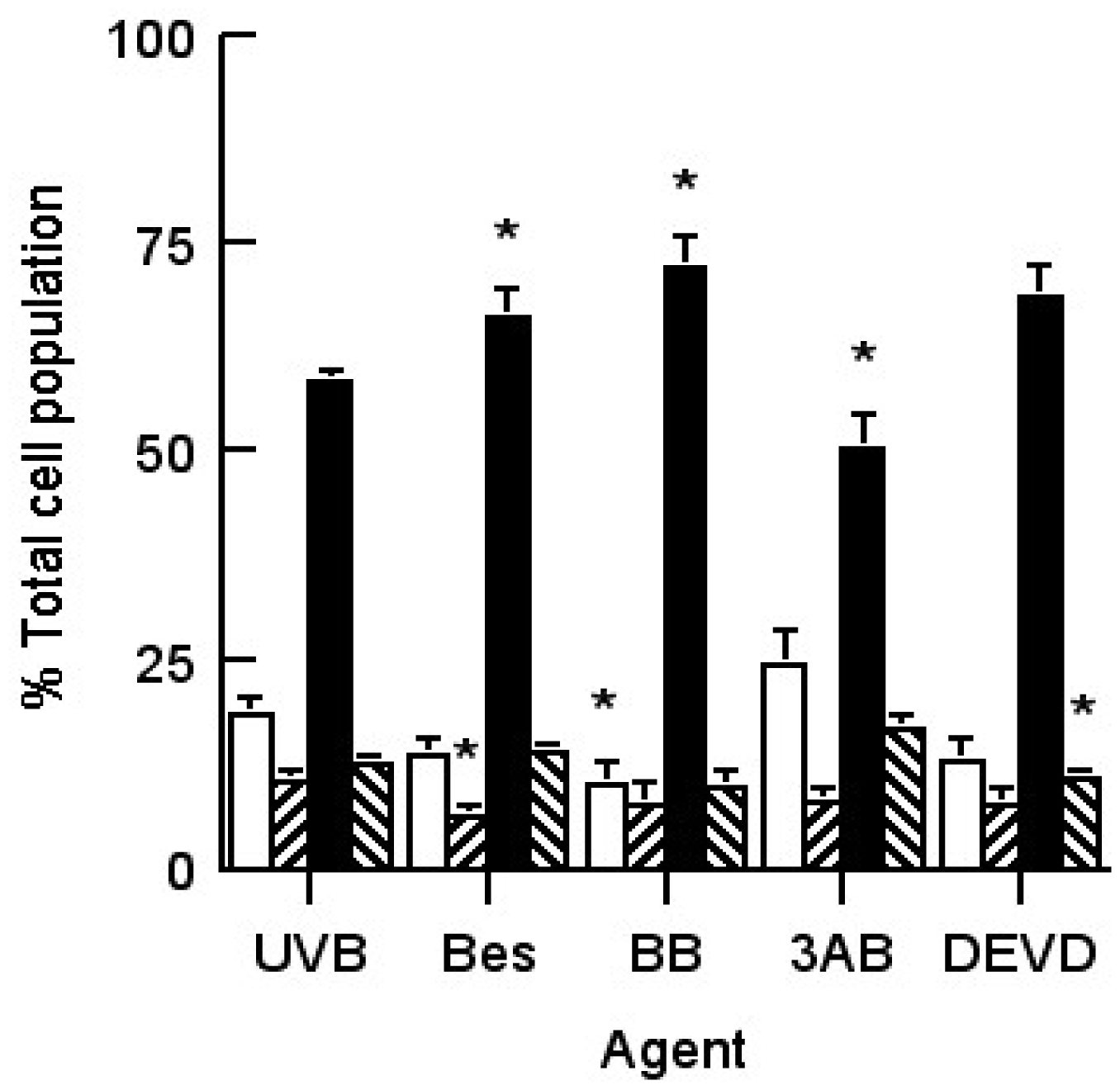
 ), 10 mM 3AB (▨) and 1 μM DEVD (
), 10 mM 3AB (▨) and 1 μM DEVD (
 ). Results expressed are the mean ± SEM of 3 separate experiments. The statistical significance of the difference between caspase 3 activity in the control population and in each treated group is represented as * p < 0.05.
), 10 mM 3AB (▨) and 1 μM DEVD (
). Results expressed are the mean ± SEM of 3 separate experiments. The statistical significance of the difference between caspase 3 activity in the control population and in each treated group is represented as * p < 0.05.
). Results expressed are the mean ± SEM of 3 separate experiments. The statistical significance of the difference between caspase 3 activity in the control population and in each treated group is represented as * p < 0.05.
), 10 mM 3AB (▨) and 1 μM DEVD (
). Results expressed are the mean ± SEM of 3 separate experiments. The statistical significance of the difference between caspase 3 activity in the control population and in each treated group is represented as * p < 0.05.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Content (mg/107 cells) of Sorted Cell Subpopulations | |||||
|---|---|---|---|---|---|
| Agent | Viable (V) | Apoptotic (A) | Ratio (A/V) | Necrotic (N) | Ratio (V/N) |
| 0 J UVB-irradiated cells | |||||
| Control | 5.8 ± 0.1 (26) | 2.1 ± 0.1 z (18) | 0.36 | ||
| Bestatin (10 μM) | 6.0 ± 0.3 (14) | 2.2 ± 0.1 z (8) | 0.37 | ||
| BB3103 (20 μM) | 5.5 ± 0.2 (12) | 2.1 ± 0.3 z (10) | 0.38 | ||
| 3AB (10 mM) | 5.8 ± 0.1 (16) | 1.7 ± 0.1 z (10) | 0.29 | ||
| DEVD (1 μM) | 5.5 ± 0.1 (20) | 2.0 ± 0.2 z (10) | 0.36 | ||
| 500 J UVB-irradiated cells | |||||
| Control | 4.8 ± 0.2 (24) | 4.0 ± 0.1 z (44) | 0.83 | 1.9 ± 0.1 z (32) | 0.40 |
| Bestatin (10 μM) | 4.5 ± 0.6 (8) | 4.5 ± 0.2 (14) | 1.00 | 1.8 ± 0.1 z (10) | 0.40 |
| BB3103 (20 μM) | 4.2 ± 0.2 (10) | 3.9 ± 0.1 (16) | 0.93 | 2.2 ± 0.2 z (12) | 0.52 |
| 3AB (10 mM) | 4.8 ± 0.3 (16) | 4.4 ± 0.1 z (20) | 0.92 | 2.2 ± 0.1 a,z(14) | 0.46 |
| DEVD (1 μM) | 3.2 ± 0.2 a (12) | 3.1 ± 0.1 a (20) | 0.97 | 1.6 ± 0.2 z (10) | 0.50 |
| P9-Derived Peptide Fragment (pmol/mg cell protein/15 min) | |||||
|---|---|---|---|---|---|
| Treatment | Cell Population | P1 | P2 | P3 | P5 |
| 0 μM Bestatin | |||||
| Control | Viable (10) | 139.0 ± 10.9 | 15.1 ± 1.2 | 5.1 ± 0.8 | 7.3 ± 1.2 |
| Apoptotic | |||||
| Necrotic (7) | 125.4 ± 25.3 | 26.3 ± 8.7 | 18.1 ± 3.3 z | 21.2 ± 4.6 z | |
| Bestatin (10 μM) | Viable (3) | 113.8 ± 9.7 a | 29.2 ± 6.1 a | 3.7 ± 1.0 | 8.2 ± 0.7 |
| Apoptotic | |||||
| Necrotic (2) | 111.6 ± 62.6 | 21.7 ± 9.2 | 14.9 ± 8.0 | 14.5 ± 2.5 | |
| BB3103 (20 μM) | Viable (2) | 104.4 ± 10.2 a | 10.2 ± 2.6 | 4.7 ± 1.5 | 10.8 ± 6.5 |
| Apoptotic | |||||
| Necrotic (2) | 82.6 ± 60.7 | 10.6 ± 8.2 a | 12.2 ± 5.0 | 17.6 ± 8.8 | |
| 3AB (1 mM) | Viable (3) | 106.9 ± 23.3 | 12.9 ± 4.9 | 5.8 ± 1.3 | 7.1 ± 0.7 |
| Apoptotic | |||||
| Necrotic (2) | 36.4 ± 24.8 a | 15.9 ± 3.0 | 8.0 ± 3.7 | 21.8 ± 16.4 | |
| DEVD (1 μM) | Viable (4) | 172.4 ± 21.8 a | 14.3 ± 2.4 | 3.9 ± 1.3 | 6.8 ± 2.1 |
| Apoptotic | |||||
| Necrotic (2) | 259.0 ± 76.8 a | 33.3 ± 3.7 a | 15.2 ± 0.7 a | 16.0 ± 12.9 | |
| 10 μM Bestatin | |||||
| Control | Viable (10) | 14.5 ± 2.2 | 38.5 ± 3.3 | 50.8 ± 3.1 | 10.2 ± 1.5 |
| Apoptotic | |||||
| Necrotic (7) | 23.8 ± 3.3 z | 17.9 ± 3.2 z | 47.8 ± 9.1 | 22.1 ± 5.2 z | |
| Bestatin (10 μM) | Viable (3) | 21.5 ± 4.1 a | 39.7 ± 9.1 | 58.2 ± 12.3 | 12.1 ± 1.6 |
| Apoptotic | |||||
| Necrotic (2) | 45.0 ± 26.6 a | 32.6 ± 19.2 | 62.2 ± 34.4 | 23.4 ± 7.5 a | |
| BB3103 (20 μM) | Viable (2) | 33.4 ± 22.1 | 14.1 ± 2.2 a | 69.2 ± 36.6 | 18.1 ± 9.4 |
| Apoptotic | |||||
| Necrotic (2) | 31.1 ± 23.6 | 8.5 ± 6.4 a | 45.9 ± 31.6 | 18.4 ± 11.5 | |
| 3AB (1 mM) | Viable (3) | 13.9 ± 2.5 | 30.2 ± 1.8 a | 51.5 ± 7.5 | 12.6 ± 1.6 |
| Apoptotic | |||||
| Necrotic (2) | 21.3 ± 16.2 | 26.2 ± 18.8 | 40.8 ± 28.3 | 20.2 ± 16.0 | |
| DEVD (1 μM) | Viable (4) | 9.5 ± 2.6 a | 42.3 ± 6.6 | 52.3 ± 6.5 | 6.4 ± 2.9 a |
| Apoptotic | |||||
| Necrotic (2) | 23.1 ± 20.3 | 67.4 ± 1.0 a | 151.0 ± 10.8 a | 24.2 ± 1.1 a | |
| P9-Derived Peptide Fragment (pmol/mg cell protein/15 min) | |||||
|---|---|---|---|---|---|
| Treatment | Cell Population | P1 | P2 | P3 | P5 |
| 0 μM Bestatin | |||||
| Control | Viable (8) | 154.1 ± 14.9 | 21.1 ± 3.5 | 12.9 ± 5.1 | 9.9 ± 1.8 |
| Apoptotic (16) | 275.9 ± 20.6 z | 28.8 ± 2.9 z | 8.1 ± 1.1 | 9.7 ± 1.5 | |
| Necrotic (10) | 6.7 ± 1.3 z | 5.2 ± 0.9 z | 5.1 ± 0.7 z | 7.6 ± 1.6 | |
| Bestatin (10 μM) | Viable (2) | 92.8 ± 40.0 a | 14.3 ± 1.2 a | 10.7 ± 8.8 | 8.7 ± 5.6 |
| Apoptotic (4) | 323.4 ± 105.7 | 59.2 ± 20.3 a | 9.0 ± 3.4 | 8.2 ± 1.2 | |
| Necrotic (3) | 7.2 ± 3.3 | 8.8 ± 3.1 | 9.5 ± 3.8 a | 1.8 ± 0.4 a | |
| BB3103 (20 μM) | Viable (2) | 149.9 ± 5.4 | 5.9 ± 1.6 a | 6.5 ± 2.7 | 12.6 ± 8.3 |
| Apoptotic (4) | 221.5 ± 22.4 a | 7.0 ± 1.0 a | 7.5 ± 3.1 | 15.3 ± 3.9 a | |
| Necrotic (3) | 5.4 ± 1.5 | 4.2 ± 1.6 | 4.0 ± 0.3 | 6.7 ± 1.7 | |
| 3AB (1 mM) | Viable (4) | 82.4 ± 6.3 a | 11.1 ± 0.7 a | 4.0 ± 0.7 a | 8.4 ± 0.9 |
| Apoptotic (4) | 107.3 ± 10.7 a | 13.5 ± 2.5 a | 7.6 ± 0.3 | 11.2 ± 1.5 | |
| Necrotic (3) | 32.5 ± 5.8 | 4.2 ± 1.7 | 7.0 ± 2.9 | 7.6 ± 2.4 | |
| DEVD (1 μM) | Viable (2) | 231.0 ± 41.4 a | 30.8 ± 3.1 | 22.2 ± 3.8 | 21.0 ± 5.5 a |
| Apoptotic (5) | 350.7 ± 17.2 a | 40.0 ± 2.7 a | 10.5 ± 2.8 | 12.9 ± 2.6 | |
| Necrotic (2) | 8.1 ± 5.9 | 6.3 ± 3.5 | 7.2 ± 1.5 | 5.4 ± 1.5 | |
| 10 μM Bestatin | |||||
| Control | Viable (8) | 27.7 ± 7.5 | 29.5 ± 4.2 | 67.7 ± 9.7 | 15.4 ± 2.4 |
| Apoptotic (16) | 33.5 ± 4.6 | 89.1 ± 10.8 z | 138.8 ± 12.5 z | 17.2 ± 1.8 | |
| Necrotic (10) | 3.3 ± 0.6 z | 4.1 ± 0.5 z | 6.8 ± 1.4 z | 4.5 ± 0.8 z | |
| Bestatin (10 μM) | Viable (2) | 36.5 ± 19.3 | 20.4 ± 0.3 a | 50.5 ± 17.1 | 18.2 ± 8.4 |
| Apoptotic (4) | 47.1 ± 11.1 | 131.8 ± 48.7 | 175.3 ± 55.7 | 15.3 ± 2.8 | |
| Necrotic (3) | 3.1 ± 1.5 | 3.9 ± 1.6 | 8.2 ± 3.8 | 4.7 ± 1.8 | |
| BB3103 (20 μM) | Viable (2) | 39.9 ± 13.0 | 5.4 ± 0.9 a | 58.3 ± 10.7 | 24.9 ± 10.3 |
| Apoptotic (4) | 72.5 ± 12.9 a | 9.9 ± 1.2 a | 119.4 ± 9.7 a | 34.1 ± 9.0 a | |
| Necrotic (3) | 3.8 ± 1.5 | 4.4 ± 1.8 | 7.7 ± 0.3 | 8.1 ± 3.0 | |
| 3AB (1 mM) | Viable (4) | 11.0 ± 1.5 a | 17.9 ± 2.0 a | 34.0 ± 3.6 a | 11.6 ± 1.7 |
| Apoptotic (4) | 18.3 ± 0.9 a | 24.2 ± 5.8 a | 56.7 ± 5.7 a | 14.8 ± 2.4 | |
| Necrotic (3) | 8.4 ± 3.6 | 6.5 ± 0.7 | 14.5 ± 5.2 a | 9.6 ± 1.0 a | |
| DEVD (1 μM) | Viable (2) | 58.9 ± 9.1 a | 58.0 ± 0.2 a | 129.4 ± 11.1 a | 27.4 ± 1.0 a |
| Apoptotic (5) | 37.4 ± 12.0 | 150.0 ± 11.6 a | 204.7 ± 26.8 a | 15.7 ± 4.5 | |
| Necrotic (2) | 12.1 ± 6.5 a | 7.5 ± 4.3 | 21.8 ± 13.8 a | 12.9 ± 9.0 | |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Piva, T.J.; Davern, C.M.; Hall, P.M.; Winterford, C.M.; Ellem, K.A.O. Increased Activity of Cell Surface Peptidases in HeLa Cells Undergoing UV-Induced Apoptosis Is Not Mediated by Caspase 3. Int. J. Mol. Sci. 2012, 13, 2650-2675. https://doi.org/10.3390/ijms13032650
Piva TJ, Davern CM, Hall PM, Winterford CM, Ellem KAO. Increased Activity of Cell Surface Peptidases in HeLa Cells Undergoing UV-Induced Apoptosis Is Not Mediated by Caspase 3. International Journal of Molecular Sciences. 2012; 13(3):2650-2675. https://doi.org/10.3390/ijms13032650
Chicago/Turabian StylePiva, Terrence J., Catherine M. Davern, Paula M. Hall, Clay M. Winterford, and Kay A. O. Ellem. 2012. "Increased Activity of Cell Surface Peptidases in HeLa Cells Undergoing UV-Induced Apoptosis Is Not Mediated by Caspase 3" International Journal of Molecular Sciences 13, no. 3: 2650-2675. https://doi.org/10.3390/ijms13032650