Localization of Axonal Motor Molecules Machinery in Neurodegenerative Disorders

{kind=link}
{kind=link}
Abstract
:1. Introduction
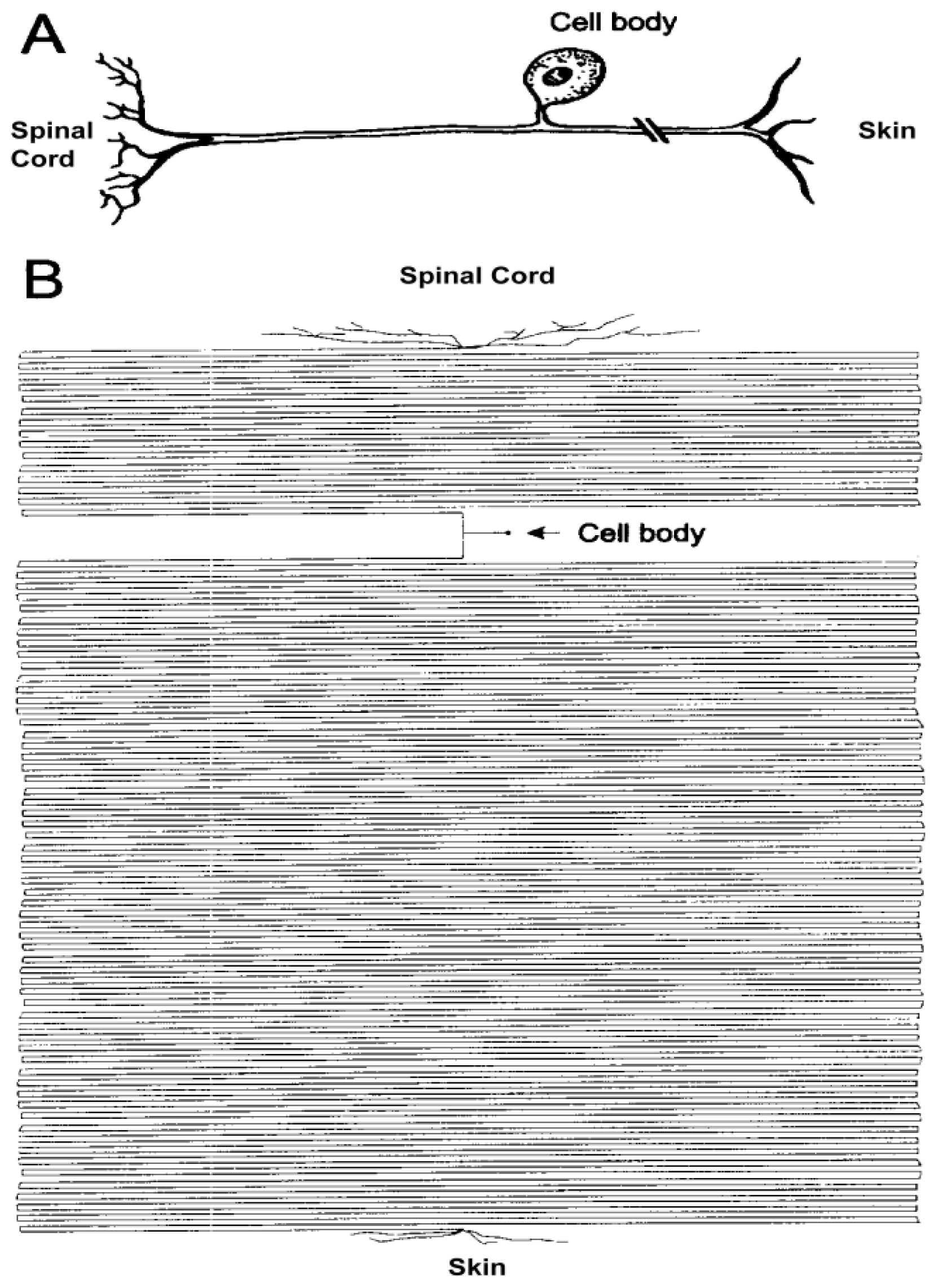
2. Axon: The Most Sensitive Neuronal Domain
3. Transport Machinery: Roads, Vehicles and Traffic Regulation
4. Cytoskeleton Composition of Axons Is Related to Environmental Stimuli
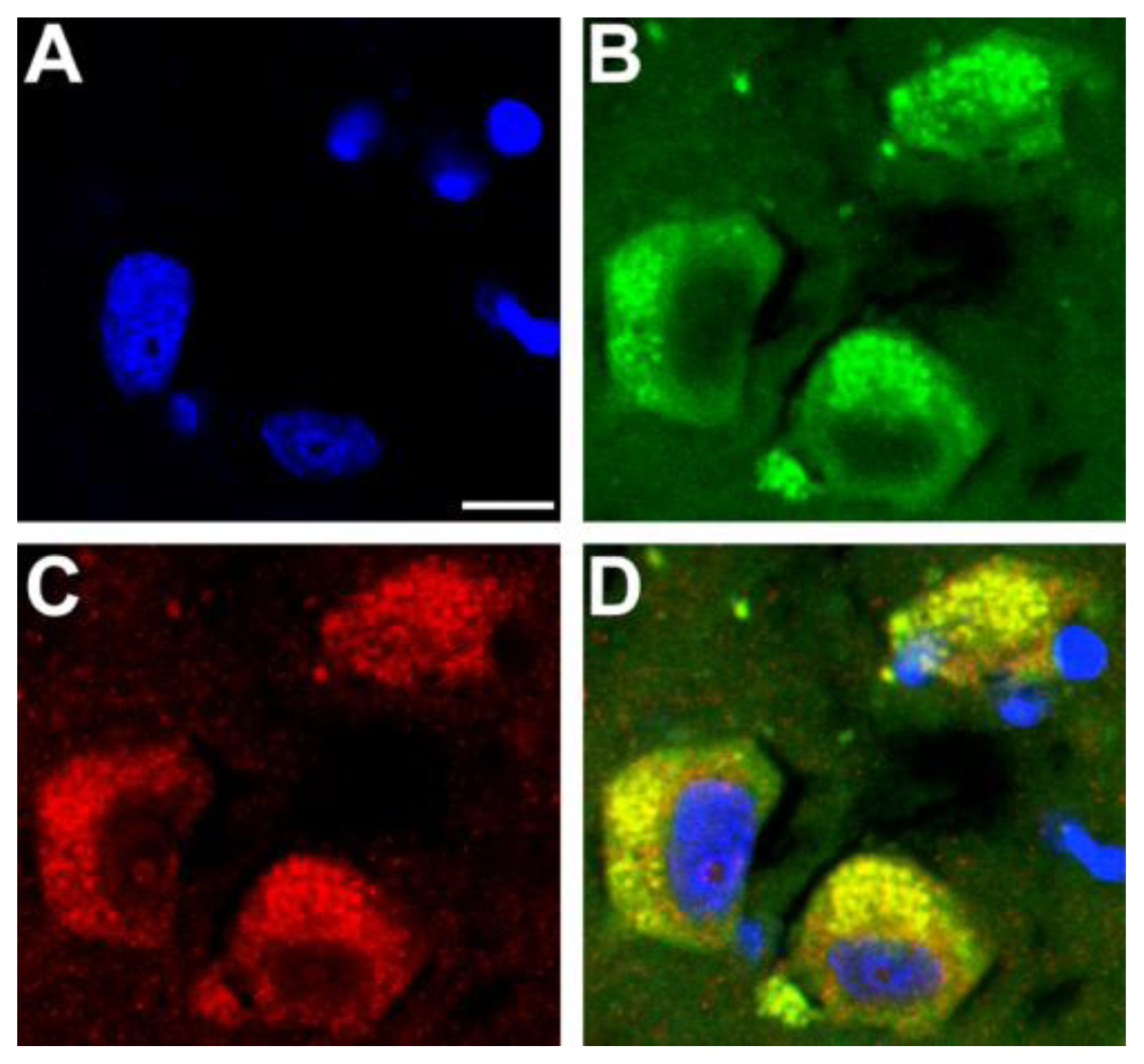
5. Adult Onset Neurodegenerative Diseases Involve Axonopathy and Transport Deficit
6. Conclusions
Acknowledgements
References
- Perlson, E.; Maday, S.; Fu, M.; Moughamian, A.J.; Holzbaur, L.F. Retrograde Axonal Transport: Pathways to Cell Death. Trends Neurosci 2010, 33, 335–344. [Google Scholar]
- Morfini, G.A.; Burns, M.; Binder, L.I.; Kanaan, N.M.; LaPointe, N.; Bosco, D.A.; Brown, R.H., Jr; Brown, H.; Tiwari, A.; Hayward, L.; et al. Axonal transport defects in neurodegenerative diseases. J. Neurosci 2009, 29, 12776–12786. [Google Scholar]
- de Vos, K.J.; Grierson, A.J.; Ackerley, S.; Miller, C.C. Role of axonal transport in neurodegenerative diseases. Annu. Rev. Neurosci 2008, 31, 151–173. [Google Scholar]
- Devor, M. Unexplained peculiarities of the dorsal root ganglion. Pain 1999, 82, S27–S35. [Google Scholar]
- Coleman, M.P.; Freeman, M.-R. Wallerian degeneration, wld(s), and nmnat. Annu. Rev. Neurosci 2010, 33, 245–267. [Google Scholar]
- Fjell, A.M.; Walhovd, K.B. Structural brain changes in aging: courses, causes and cognitive consequences. Rev. Neurosci 2010, 21, 187–221. [Google Scholar]
- Pakkenberg, B.; Gundersen, H.J. Neocortical neuron number in humans: effect of sex and age. J. Comp. Neurol 1997, 384, 312–320. [Google Scholar]
- Duyckaerts, C.; Delatour, B.; Potier, M.C. Classification and basic pathology of Alzheimer disease. Acta Neuropathol 2009, 118, 5–36. [Google Scholar]
- Coleman, M. Axon degeneration mechanisms: commonality amid diversity. Nat. Rev. Neurosci 2005, 6, 889–898. [Google Scholar]
- Waller, A. Experiments on the section of glossopharyngeal and hypoglossal nerves of the frog and observations of the alternatives produced thereby in the structure of their primitive fibres. Phil. Trans. R. Soc. Lond. B 1850, 140, 423–429. [Google Scholar]
- Nave, K.A.; Trapp, B.D. Axon-glial signaling and the glial support of axon function. Annu. Rev. Neurosci 2008, 31, 535–561. [Google Scholar]
- Beirowski, B.; Adalbert, R.; Wagner, D.; Grumme, D.S.; Addicks, K.; Ribchester, R.R.; Coleman, M.P. The progressive nature of Wallerian degeneration in wild-type and slow Wallerian degeneration (WldS) nerves. BMC Neurosci 2005, 6, 6:1–6:27. [Google Scholar]
- Beirowski, B.; Nógrádi, A.; Babetto, E.; Garcia-Alias, G.; Coleman, M.P. Mechanisms of axonal spheroid formation in central nervous system Wallerian degeneration. J. Neuropathol. Exp. Neurol 2010, 69, 455–472. [Google Scholar]
- van den Heuvel, C.; Blumbergs, P.C.; Finnie, J.W.; Manavis, J.; Jones, N.R.; Reilly, P.L.; Pereira, R.A. Upregulation of amyloid precursor protein messenger RNA in response to traumatic brain injury: an ovine head impact model. Exp. Neurol 1999, 159, 441–450. [Google Scholar]
- Medana, I.M.; Esiri, M.M. Axonal damage: a key predictor of outcome in human CNS diseases. Brain 2003, 126, 515–530. [Google Scholar]
- van Den Heuvel, C.; Thornton, E.; Vink, R. Traumatic brain injury and Alzheimer’s disease: a review. Prog. Brain Res 2007, 161, 303–316. [Google Scholar]
- Geddes, J.F.; Whitwell, H.L.; Graham, D.I. Traumatic or diffuse axonal injury Author’s response. Neuropathol. Appl. Neurobiol 2000, 26, 491. [Google Scholar]
- Florenzano, F.; Viscomi, M.T.; Cavaliere, F.; Volonté, C.; Molinari, M. Cerebellar lesion up-regulates P2X1 and P2X2 purinergic receptors in precerebellar nuclei. Neurosci 2002, 115, 425–434. [Google Scholar]
- Florenzano, F.; Viscomi, M.T.; Cavaliere, F.; Volonté, C.; Molinari, M. The Role of Ionotropic Purinergic Receptors (P2X) in Mediating Plasticity Responses in the Central Nervous System. Adv. Exp. Med. Biol 2006, 557, 77–100. [Google Scholar]
- Viscomi, M.T.; Florenzano, F.; Latini, L.; Amantea, D.; Bernardi, G.; Molinari, M. Methylprednisolone treatment delays remote cell death after focal brain lesion. Neuroscience 2008, 154, 1267–1282. [Google Scholar]
- Florenzano, F.; Viscomi, M.T.; Volonté, C.; Molinari, M. Do ATP and NO interact in the CNS. Prog. Neurobiol 2008, 84, 40–56. [Google Scholar]
- Conde, C.; Cáceres, A. Microtubule assembly, organization and dynamics in axons and dendrites. Nat. Rev. Neurosci 2009, 10, 319–332. [Google Scholar]
- Hirokawa, N. Kinesin and dynein superfamily proteins and the mechanism of organelle transport. Science 1998, 279, 519–526. [Google Scholar]
- Janke, C.; Kneussel, M. Tubulin post-translational modifications: encoding functions on the neuronal microtubule cytoskeleton. Trends Neurosci 2010, 33, 362–372. [Google Scholar]
- Hirokawa, N.; Noda, Y. Intracellular transport and kinesin superfamily proteins, KIFs: structure, function, and dynamics. Physiol. Rev 2008, 88, 1089–1118. [Google Scholar]
- Verhey, K.J.; Kaul, N.; Soppina, V. Kinesin assembly and movement in cells. Annu. Rev. Biophys 2011, 40, 267–288. [Google Scholar]
- Vale, R.D. The molecular motor toolbox for intracellular transport. Cell 2003, 112, 467–480. [Google Scholar]
- Hirokawa, N.; Niwa, S.; Tanaka, Y. Molecular motors in neurons: transport mechanisms and roles in brain function, development, and disease. Neuron 2010, 68, 610–638. [Google Scholar]
- de Boer, S.R.; You, Y.; Szodorai, A.; Kaminska, A.; Pigino, G.; Nwabuisi, E.; Wang, B.; Estrada-Hernandez, T.; Kins, S.; Brady, S.T.; et al. Conventional kinesin holoenzymes are composed of heavy and light chain homodimers. Biochemistry 2008, 47, 4535–4543. [Google Scholar]
- Morfini, G.; Szebenyi, G.; Brown, H.; Pant, H.C.; Pigino, G.; DeBoer, S.; Beffert, U.; Brady, S.T. A novel CDK5-dependent pathway for regulating GSK3 activity and kinesin-driven motility in neurons. EMBO J 2004, 23, 2235–2245. [Google Scholar]
- Morfini, G.; Pigino, G.; Beffert, U.; Busciglio, J.; Brady, S.T. Fast axonal transport misregulation and Alzheimer’s disease. Neuromolecular Med 2002, 2, 89–99. [Google Scholar]
- Wagey, R.T.; Krieger, C. Abnormalities of protein kinases in neurodegenerative diseases. Prog. Drug Res 1998, 51, 133–183. [Google Scholar]
- Badiola, N.; Suárez-Calvet, M.; Lleó, A. Tau phosphorylation and aggregation as a therapeutic target in tauopathies. CNS Neurol. Disord. Drug Targets 2010, 9, 727–740. [Google Scholar]
- Hollenbeck, P.J.; Saxton, W.M. The axonal transport of mitochondria. J. Cell. Sci 2005, 118, 5411–5419. [Google Scholar]
- Seo, A.Y.; Joseph, A.M.; Dutta, D.; Hwang, J.C.Y.; Aris, J.P.; Leeuwenburgh, C. New insights into the role of mitochondria in aging: mitochondrial dynamics and more. J. Cell. Sci 2010, 123, 2533–2542. [Google Scholar]
- Rintoul, G.L.; Reynolds, I.J. Mitochondrial trafficking and morphology in neuronal injury. Biochim. Biophys. Acta 2010, 1802, 143–150. [Google Scholar]
- Eckert, A.; Schmitt, K.; Götz, J. Mitochondrial dysfunction—the beginning of the end in Alzheimer’s disease Separate and synergistic modes of tau and amyloid-β toxicity. Alzheimers Res. Ther 2011, 3, 2–15. [Google Scholar]
- Amadoro, G.; Corsetti, V.; Ciotti, M.T.; Florenzano, F.; Capsoni, S.; Amato, G.; Calissano, P. Endogenous Aβ causes cell death via early tau hyperphosphorylation. Neurobiol. Aging 2011, 32, 969–990. [Google Scholar]
- Amadoro, G.; Corsetti, V.; Atlante, A.; Florenzano, F.; Capsoni, S.; Bussani, R.; Mercanti, D.; Calissano, P. Interaction between NH2-tau fragment and Abeta in AD mitochondria contributes to the synaptic deterioration. Neurobiol. Aging 2012, 33, 833.e1–833.e25. [Google Scholar]
- Fadić, R.; Vergara, J.; Alvarez, J. Microtubules and caliber of central and peripheral processes of sensory axons. J. Comp. Neurol 1985, 236, 258–264. [Google Scholar]
- Vergara, J.; Serra, M.; Saitua, F.; Iturriaga, R.; Alvarez, J. Axonal microtubules: comparative anatomy in vertebrates, including man. J. Submicrosc. Cytol. Pathol 1991, 23, 357–363. [Google Scholar]
- López, J.M.; Alvarez, J. The Microtubular Pattern Changes at the Spinal Cord-Root Junction and Reverts at the Root-Peripheral Nerve Junction in Sensory and Motor Fibres of the Rat. Eur. J. Neurosci 1990, 2, 873–878. [Google Scholar]
- Court, F.A.; Alvarez, J. Local regulation of the axonal phenotype, a case of merotrophism. Biol. Res 2005, 38, 365–374. [Google Scholar]
- Alvarez, J.; Ramirez, B.U. Axonal microtubules: their regulation by the electrical activity of the nerve. Neurosci. Lett 1979, 15, 19–22. [Google Scholar]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar]
- Bossy-Wetzel, E.; Schwarzenbacher, R.; Lipton, S.A. Molecular pathways to neurodegeneration. Nat. Med 2004, 10, S2–S9. [Google Scholar]
- Bilsland, L.G.; Sahai, E.; Kelly, G.; Golding, M.; Greensmith, L.; Schiavo, G. Deficits in axonal transport precede ALS symptoms in vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 20523–20528. [Google Scholar]
- Double, K.L.; Reyes, S.; Werry, E.L.; Halliday, G.M. Selective cell death in neurodegeneration: why are some neurons spared in vulnerable regions? Prog. Neurobiol 2010, 92, 316–329. [Google Scholar]
- Ballatore, C.; Lee, V.M.; Trojanowski, J.Q. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat. Rev. Neurosci 2007, 8, 663–672. [Google Scholar]
- Fiala, J.C. Mechanisms of amyloid plaque pathogenesis. Acta Neuropathol 2007, 114, 551–571. [Google Scholar]
- Xiao, S.; McLean, J.; Robertson, J. Neuronal intermediate filaments and ALS: a new look at an old question. Biochim. Biophys. Acta 2006, 1762, 1001–1012. [Google Scholar]
- Hirokawa, N.; Takemura, R. Biochemical and molecular characterization of diseases linked to motor proteins. Trends Biochem. Sci 2003, 28, 558–565. [Google Scholar]
- Kamal, A.; Goldstein, L.S. Connecting vesicle transport to the cytoskeleton. Curr. Opin. Cell Biol 2000, 12, 503–508. [Google Scholar]
- Falzone, T.L.; Gunawardena, S.; McCleary, D.; Reis, G.F.; Goldstein, L.S. Kinesin-1 transport reductions enhance human tau hyperphosphorylation, aggregation and neurodegeneration in animal models of tauopathies. Hum. Mol. Genet 2010, 19, 4399–4408. [Google Scholar]


© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Florenzano, F. Localization of Axonal Motor Molecules Machinery in Neurodegenerative Disorders. Int. J. Mol. Sci. 2012, 13, 5195-5206. https://doi.org/10.3390/ijms13045195
Florenzano F. Localization of Axonal Motor Molecules Machinery in Neurodegenerative Disorders. International Journal of Molecular Sciences. 2012; 13(4):5195-5206. https://doi.org/10.3390/ijms13045195
Chicago/Turabian StyleFlorenzano, Fulvio. 2012. "Localization of Axonal Motor Molecules Machinery in Neurodegenerative Disorders" International Journal of Molecular Sciences 13, no. 4: 5195-5206. https://doi.org/10.3390/ijms13045195