The Role of PPARα in Metformin-Induced Attenuation of Mitochondrial Dysfunction in Acute Cardiac Ischemia/Reperfusion in Rats


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
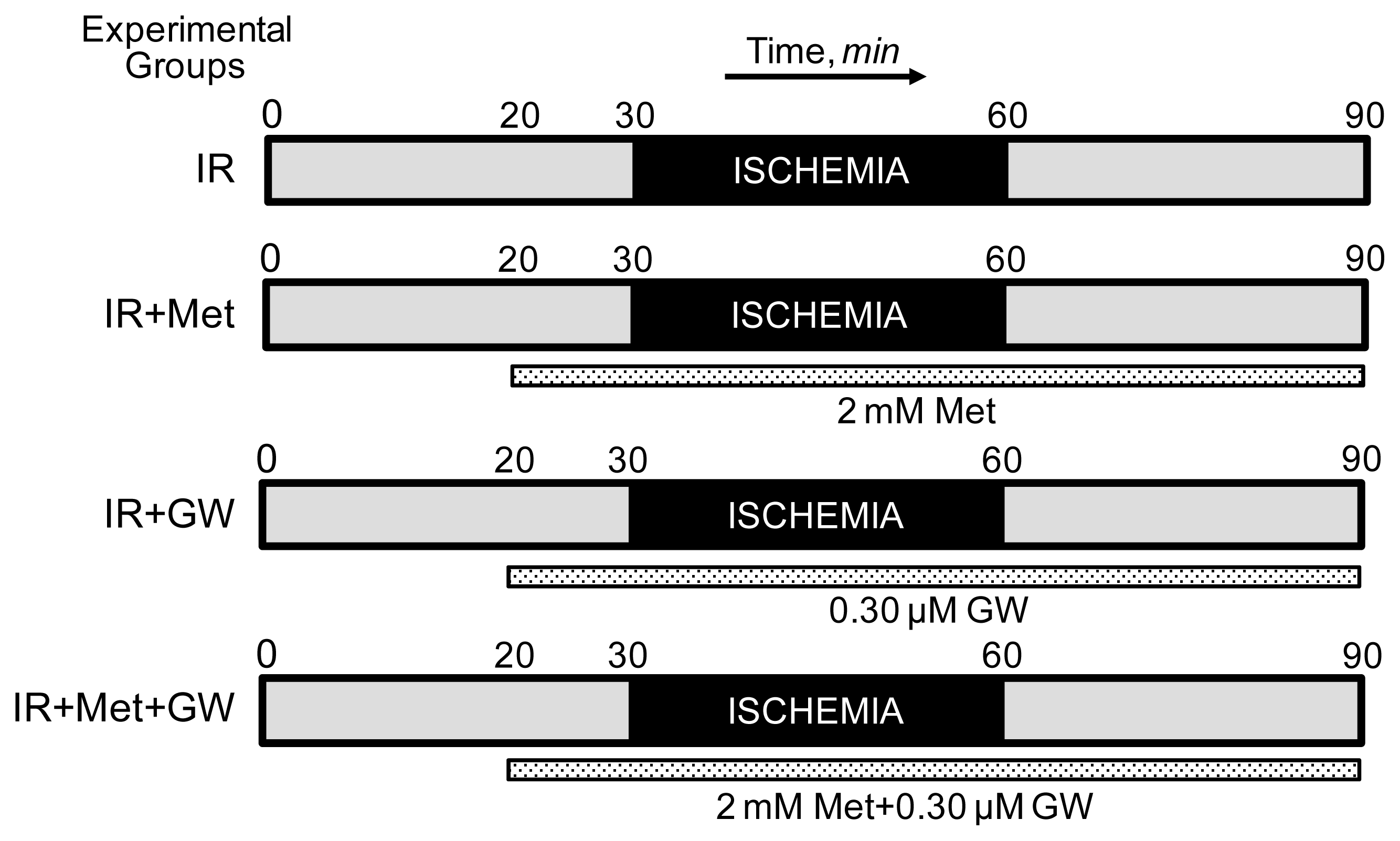
2.1. Langendorff Heart Perfusion and Animal Groups
2.2. Isolation of Mitochondria
2.3. Measurement of the Respiration Rates in Isolated Cardiac Mitochondria
2.4. Measurement of PTP Opening in Isolated Mitochondria
2.5. SDS-PAGE and Western Blotting
2.6. Co-Immunoprecipitation
2.7. Statistical Analysis
3. Results and Discussion
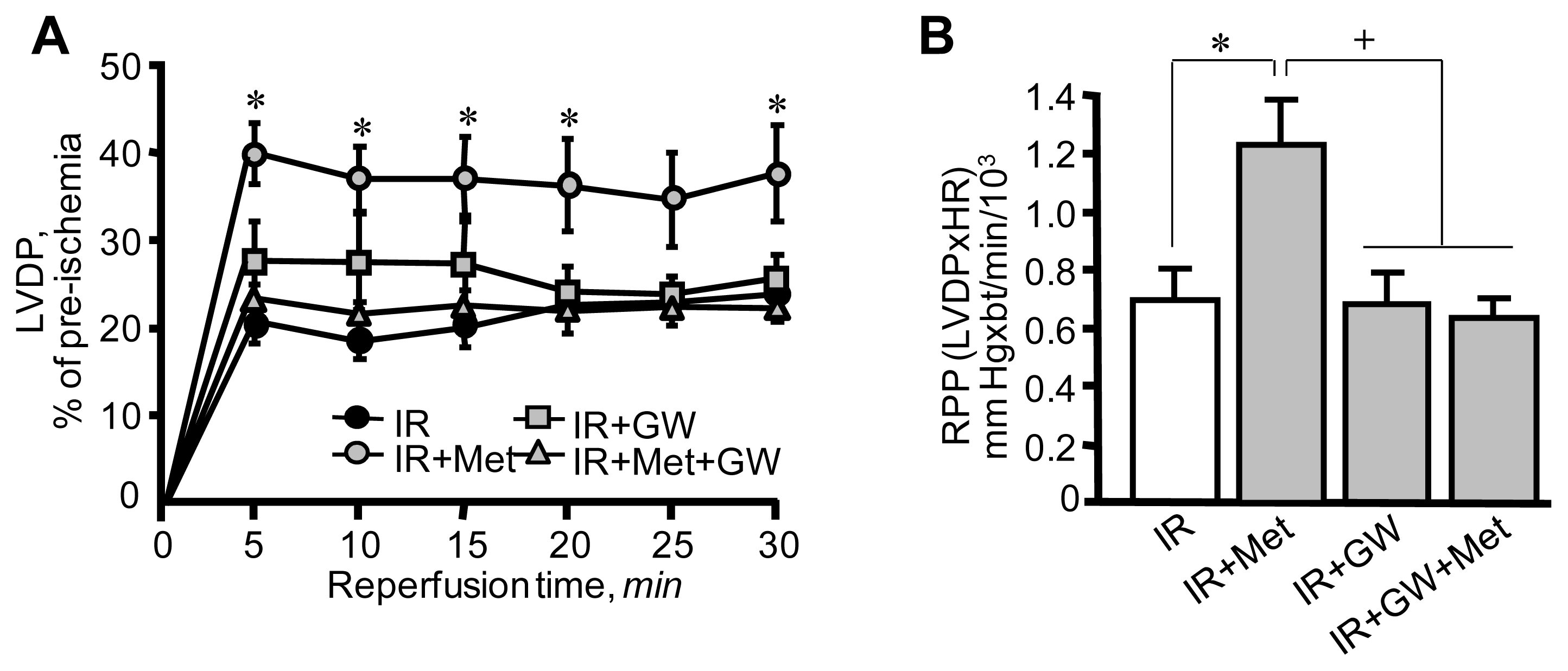
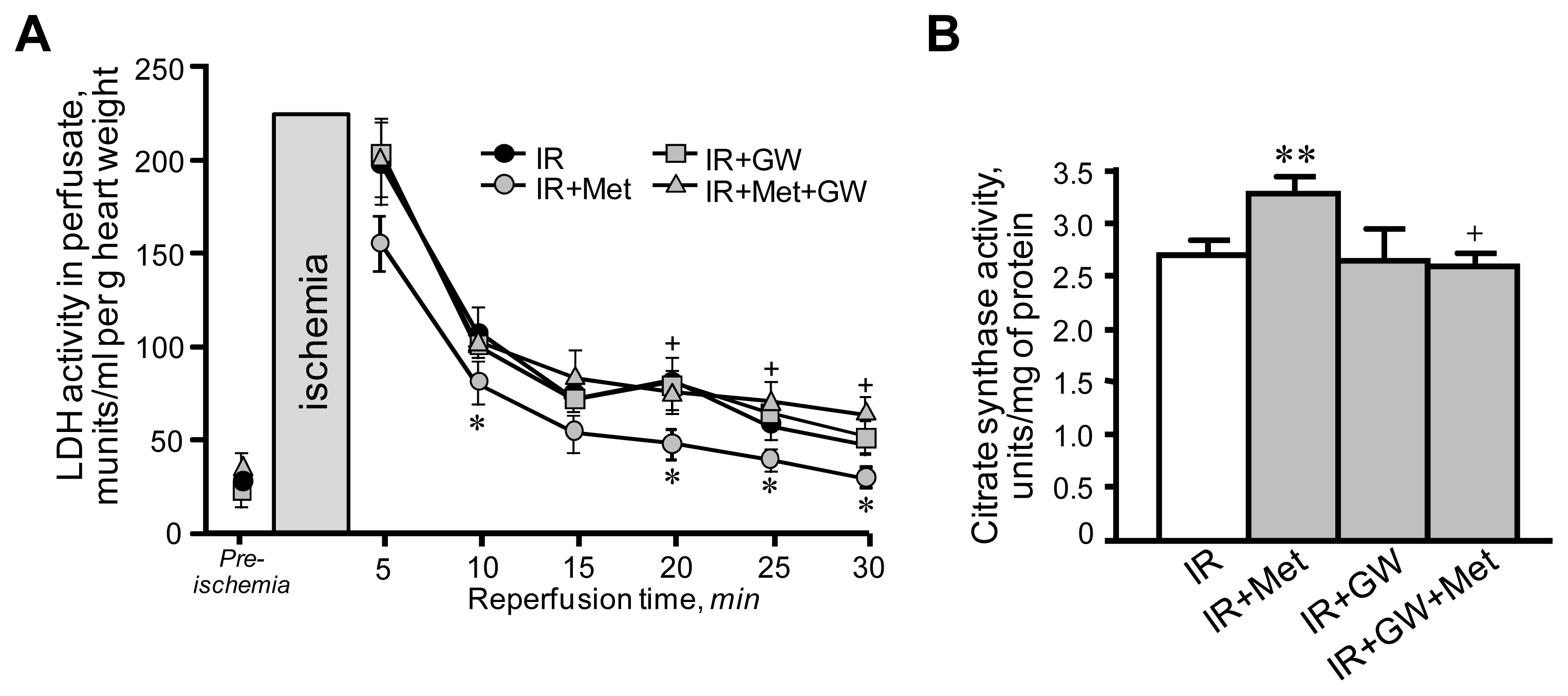
3.1. Metformin-Induced Improvement of Post-Ischemic Cardiac Function Is Attenuated by GW6471
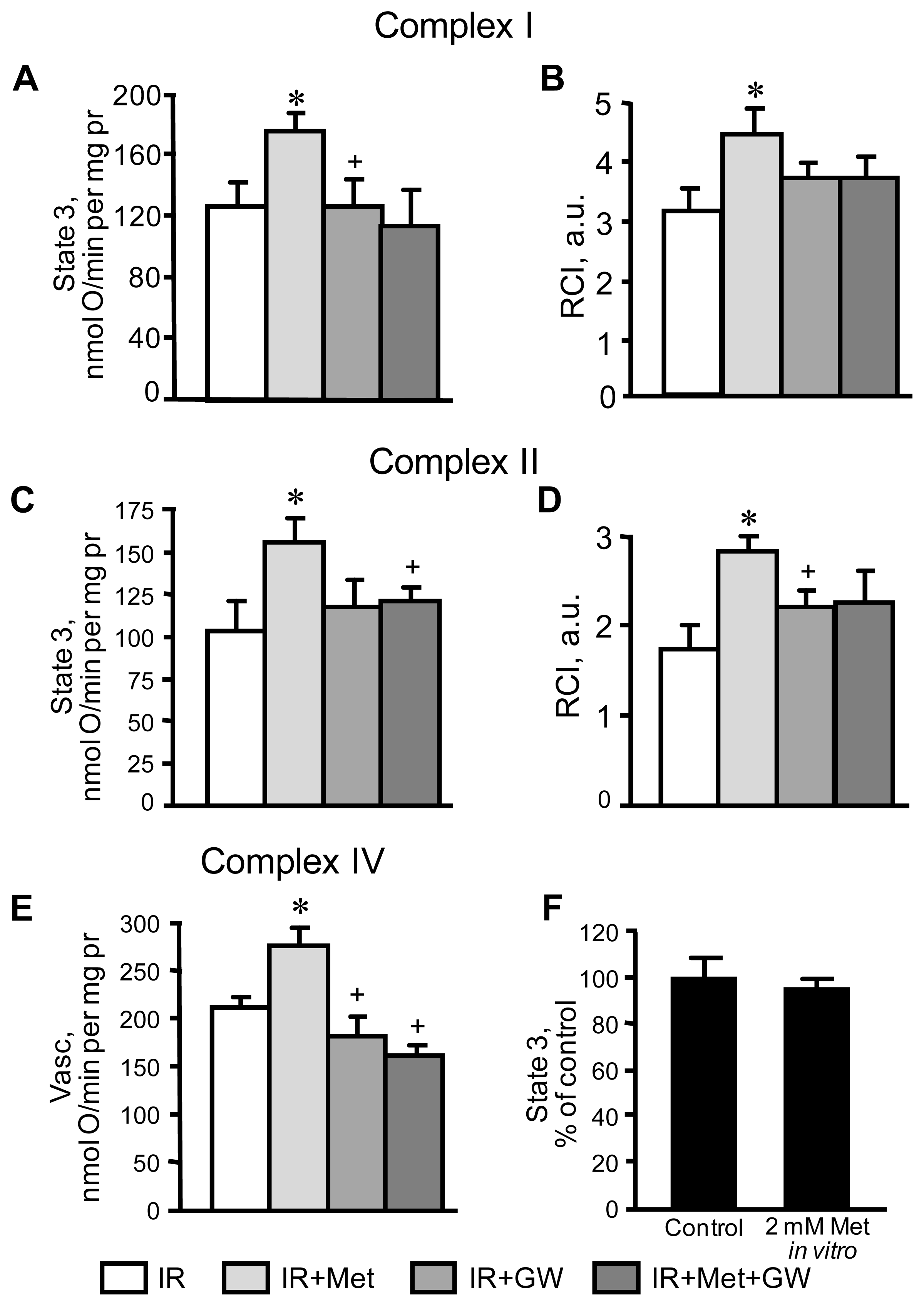
3.2. Beneficial Effects of Metformin on Mitochondria Are Prevented by Inhibition of PPARα
3.3. Attenuation of PTP Opening Induced by Metformin Is Abrogated in the Presence of GW6471
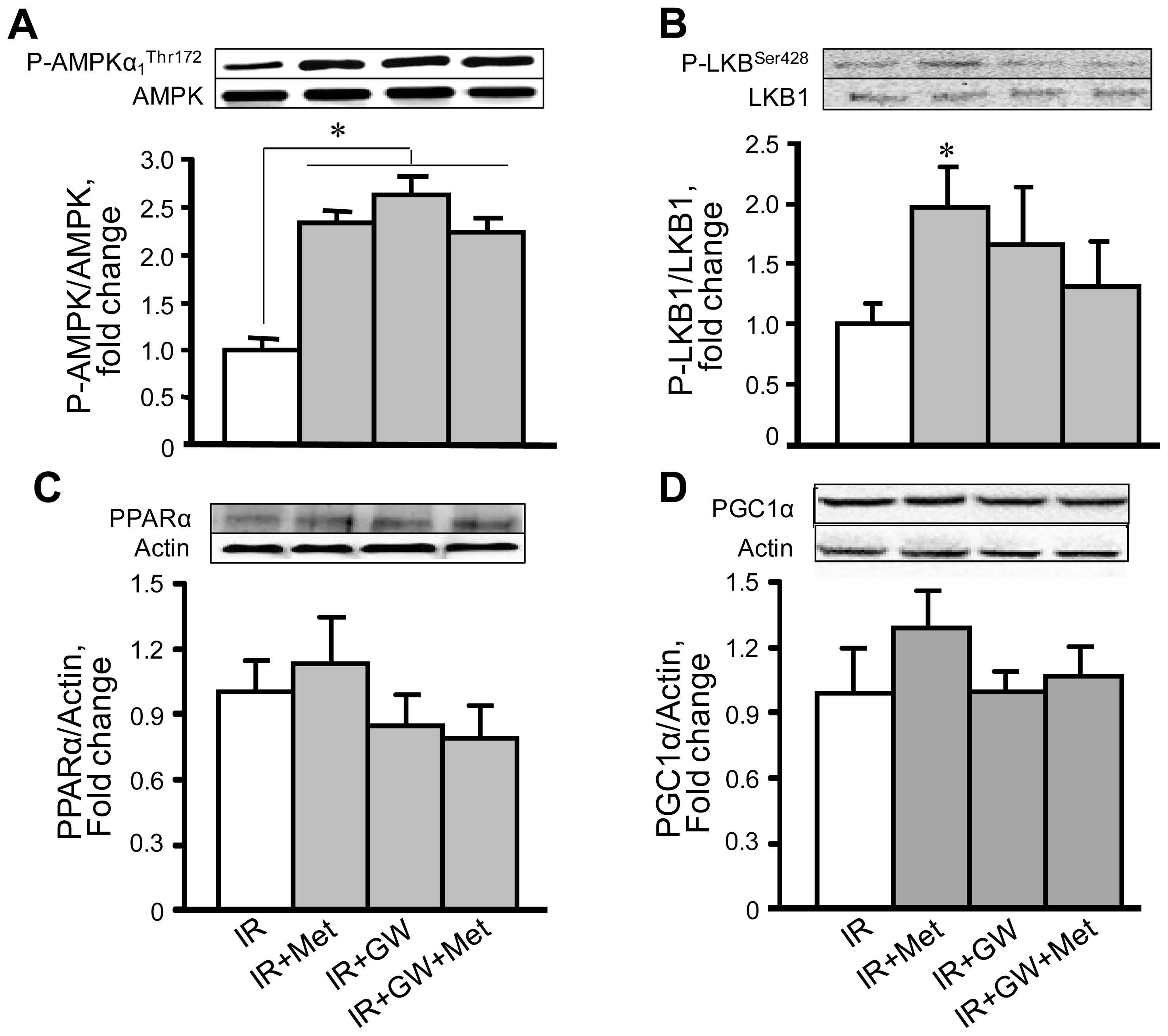
3.4. Metformin Enhances AMPK Phosphorylation with No Changes in PPARα Expression
4. Conclusions
Acknowledgments
- DisclosuresNone declared.
References
- Sanada, S.; Komuro, I.; Kitakaze, M. Pathophysiology of myocardial reperfusion injury: Preconditioning, postconditioning, and translational aspects of protective measures. Am. J. Physiol. Heart Circ. Physiol 2011, 301, H1723–H1741. [Google Scholar]
- Eltzschig, H.K.; Eckle, T. Ischemia and reperfusion—From mechanism to translation. Nat. Med 2011, 17, 1391–1401. [Google Scholar]
- Murphy, E.; Steenbergen, C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol. Rev 2008, 88, 581–609. [Google Scholar]
- Camara, A.K.; Lesnefsky, E.J.; Stowe, D.F. Potential therapeutic benefits of strategies directed to mitochondria. Antioxid. Redox Signal 2010, 13, 279–347. [Google Scholar]
- Heidrich, F.; Schotola, H.; Popov, A.F.; Sohns, C.; Schuenemann, J.; Friedrich, M.; Coskun, K.O.; von Lewinski, D.; Hinz, J.; Bauer, M.; et al. AMPK—Activated protein kinase and its role in energy metabolism of the heart. Curr. Cardiol. Rev 2010, 6, 337–342. [Google Scholar]
- Arad, M.; Seidman, C.E.; Seidman, J.G. AMP-activated protein kinase in the heart: Role during health and disease. Circ. Res 2007, 100, 474–488. [Google Scholar]
- Dyck, J.R.; Lopaschuk, G.D. AMPK alterations in cardiac physiology and pathology: Enemy or ally? J. Physiol 2006, 574, 95–112. [Google Scholar]
- Baron, S.J.; Li, J.; Russell, R.R., 3rd; Neumann, D.; Miller, E.J.; Tuerk, R.; Wallimann, T.; Hurley, R.L.; Witters, L.A.; Young, L.H. Dual mechanisms regulating AMPK kinase action in the ischemic heart. Circ. Res 2005, 96, 337–345. [Google Scholar]
- Bhamra, G.S.; Hausenloy, D.J.; Davidson, S.M.; Carr, R.D.; Paiva, M.; Wynne, A.M.; Mocanu, M.M.; Yellon, D.M. Metformin protects the ischemic heart by the Akt-mediated inhibition of mitochondrial permeability transition pore opening. Basic. Res. Cardiol 2008, 103, 274–284. [Google Scholar]
- Gundewar, S.; Calvert, J.W.; Jha, S.; Toedt-Pingel, I.; Ji, S.Y.; Nunez, D.; Ramachandran, A.; Anaya-Cisneros, M.; Tian, R.; Lefer, D.J. Activation of AMP-activated protein kinase by metformin improves left ventricular function and survival in heart failure. Circ. Res 2009, 104, 403–411. [Google Scholar]
- Calvert, J.W.; Gundewar, S.; Jha, S.; Greer, J.J.; Bestermann, W.H.; Tian, R.; Lefer, D.J. Acute metformin therapy confers cardioprotection against myocardial infarction via AMPK-eNOS-mediated signaling. Diabetes 2008, 57, 696–705. [Google Scholar]
- Ingwall, J.S. Transgenesis and cardiac energetics: New insights into cardiac metabolism. J. Mol. Cell. Cardiol 2004, 37, 613–623. [Google Scholar]
- Kelly, D.P.; Scarpulla, R.C. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev 2004, 18, 357–368. [Google Scholar]
- Bulhak, A.A.; Jung, C.; Ostenson, C.G.; Lundberg, J.O.; Sjoquist, P.O.; Pernow, J. PPAR-alpha activation protects the type 2 diabetic myocardium against ischemia-reperfusion injury: Involvement of the PI3-Kinase/Akt and NO pathway. Am. J. Physiol. Heart Circ. Physiol 2009, 296, H719–H727. [Google Scholar]
- Yue, T.L.; Bao, W.; Jucker, B.M.; Gu, J.L.; Romanic, A.M.; Brown, P.J.; Cui, J.; Thudium, D.T.; Boyce, R.; Burns-Kurtis, C.L.; et al. Activation of peroxisome proliferator-activated receptor-alpha protects the heart from ischemia/reperfusion injury. Circulation 2003, 108, 2393–2399. [Google Scholar]
- Chau, M.D.; Gao, J.; Yang, Q.; Wu, Z.; Gromada, J. Fibroblast growth factor 21 regulates energy metabolism by activating the AMPK-SIRT1-PGC-1alpha pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 12553–12558. [Google Scholar]
- Jager, S.; Handschin, C.; St-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar]
- Meng, R.; Pei, Z.; Zhang, A.; Zhou, Y.; Cai, X.; Chen, B.; Liu, G.; Mai, W.; Wei, J.; Dong, Y. AMPK activation enhances PPARalpha activity to inhibit cardiac hypertrophy via ERK1/2 MAPK signaling pathway. Arch. Biochem. Biophys 2011, 511, 1–7. [Google Scholar]
- Fujita, K.; Maeda, N.; Sonoda, M.; Ohashi, K.; Hibuse, T.; Nishizawa, H.; Nishida, M.; Hiuge, A.; Kurata, A.; Kihara, S.; et al. Adiponectin protects against angiotensin II-induced cardiac fibrosis through activation of PPAR-alpha. Arterioscler. Thromb. Vasc. Biol 2008, 28, 863–870. [Google Scholar]
- Javadov, S.; Choi, A.; Rajapurohitam, V.; Zeidan, A.; Basnakian, A.G.; Karmazyn, M. NHE-1 inhibition-induced cardioprotection against ischaemia/reperfusion is associated with attenuation of the mitochondrial permeability transition. Cardiovasc. Res 2008, 77, 416–424. [Google Scholar]
- Saeedi, R.; Parsons, H.L.; Wambolt, R.B.; Paulson, K.; Sharma, V.; Dyck, J.R.; Brownsey, R.W.; Allard, M.F. Metabolic actions of metformin in the heart can occur by AMPK-independent mechanisms. Am. J. Physiol. Heart Circ. Physiol 2008, 294, H2497–H2506. [Google Scholar]
- Srere, P.A. Citrate synthase. Methods Enzymol 1969, 13, 3–11. [Google Scholar]
- Javadov, S.; Rajapurohitam, V.; Kilic, A.; Zeidan, A.; Choi, A.; Karmazyn, M. Anti-hypertrophic effect of NHE-1 inhibition involves GSK-3beta-dependent attenuation of mitochondrial dysfunction. J. Mol. Cell. Cardiol 2009, 46, 998–1007. [Google Scholar]
- Guigas, B.; Detaille, D.; Chauvin, C.; Batandier, C.; de Oliveira, F.; Fontaine, E.; Leverve, X. Metformin inhibits mitochondrial permeability transition and cell death: A pharmacological in vitro study. Biochem. J 2004, 382, 877–884. [Google Scholar]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J 2000, 348 Pt 3, 607–614. [Google Scholar]
- Kirpichnikov, D.; McFarlane, S.I.; Sowers, J.R. Metformin: An update. Ann. Intern. Med 2002, 137, 25–33. [Google Scholar]
- Paiva, M.A.; Goncalves, L.M.; Providencia, L.A.; Davidson, S.M.; Yellon, D.M.; Mocanu, M.M. Transitory activation of AMPK at reperfusion protects the ischaemic-reperfused rat myocardium against infarction. Cardiovasc. Drugs Ther 2010, 24, 25–32. [Google Scholar]
- Sasaki, H.; Asanuma, H.; Fujita, M.; Takahama, H.; Wakeno, M.; Ito, S.; Ogai, A.; Asakura, M.; Kim, J.; Minamino, T.; et al. Metformin prevents progression of heart failure in dogs: Role of AMP-activated protein kinase. Circulation 2009, 119, 2568–2577. [Google Scholar]
- Yin, M.; van der Horst, I.C.; van Melle, J.P.; Qian, C.; van Gilst, W.H.; Sillje, H.H.; de Boer, R.A. Metformin improves cardiac function in a nondiabetic rat model of post-MI heart failure. Am. J. Physiol. Heart Circ. Physiol 2011, 301, H459–H468. [Google Scholar]
- Zong, H.; Ren, J.M.; Young, L.H.; Pypaert, M.; Mu, J.; Birnbaum, M.J.; Shulman, G.I. AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc. Natl. Acad. Sci. USA 2002, 99, 15983–15987. [Google Scholar]
- Huss, J.M.; Kelly, D.P. Nuclear receptor signaling and cardiac energetics. Circ. Res 2004, 95, 568–578. [Google Scholar]
- Reznick, R.M.; Shulman, G.I. The role of AMP-activated protein kinase in mitochondrial biogenesis. J. Physiol 2006, 574, 33–39. [Google Scholar]
- Zhang, L.; He, H.; Balschi, J.A. Metformin and phenformin activate AMP-activated protein kinase in the heart by increasing cytosolic AMP concentration. Am. J. Physiol. Heart Circ. Physiol 2007, 293, H457–H466. [Google Scholar]
- Bulhak, A.A.; Sjoquist, P.O.; Xu, C.B.; Edvinsson, L.; Pernow, J. Protection against myocardial ischaemia/reperfusion injury by PPAR-alpha activation is related to production of nitric oxide and endothelin-1. Basic Res. Cardiol 2006, 101, 244–252. [Google Scholar]
- Xu, Z.; Ji, X.; Boysen, P.G. Exogenous nitric oxide generates ROS and induces cardioprotection: Involvement of PKG, mitochondrial KATP channels, and ERK. Am. J. Physiol. Heart Circ. Physiol 2004, 286, H1433–H1440. [Google Scholar]
- Dezfulian, C.; Raat, N.; Shiva, S.; Gladwin, M.T. Role of the anion nitrite in ischemia-reperfusion cytoprotection and therapeutics. Cardiovasc. Res 2007, 75, 327–338. [Google Scholar]
- Nadtochiy, S.M.; Burwell, L.S.; Brookes, P.S. Cardioprotection and mitochondrial S-nitrosation: Effects of S-nitroso-2-mercaptopropionyl glycine (SNO-MPG) in cardiac ischemia-reperfusion injury. J. Mol. Cell. Cardiol 2007, 42, 812–825. [Google Scholar]
- Foretz, M.; Hébrard, S.; Leclerc, J.; Zarrinpashneh, E.; Soty, M.; Mithieux, G.; Sakamoto, K.; Andreelli, F.; Viollet, B. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J. Clin. Invest 2010, 120, 2355–2369. [Google Scholar] [Green Version]
- Kalender, A.; Selvaraj, A.; Kim, S.Y.; Gulati, P.; Brûlé, S.; Viollet, B.; Kemp, B.E.; Bardeesy, N.; Dennis, P.; Schlager, J.J.; et al. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab 2010, 11, 390–401. [Google Scholar]






© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Barreto-Torres, G.; Parodi-Rullán, R.; Javadov, S. The Role of PPARα in Metformin-Induced Attenuation of Mitochondrial Dysfunction in Acute Cardiac Ischemia/Reperfusion in Rats. Int. J. Mol. Sci. 2012, 13, 7694-7709. https://doi.org/10.3390/ijms13067694
Barreto-Torres G, Parodi-Rullán R, Javadov S. The Role of PPARα in Metformin-Induced Attenuation of Mitochondrial Dysfunction in Acute Cardiac Ischemia/Reperfusion in Rats. International Journal of Molecular Sciences. 2012; 13(6):7694-7709. https://doi.org/10.3390/ijms13067694
Chicago/Turabian StyleBarreto-Torres, Giselle, Rebecca Parodi-Rullán, and Sabzali Javadov. 2012. "The Role of PPARα in Metformin-Induced Attenuation of Mitochondrial Dysfunction in Acute Cardiac Ischemia/Reperfusion in Rats" International Journal of Molecular Sciences 13, no. 6: 7694-7709. https://doi.org/10.3390/ijms13067694