Sampling Strategy and Potential Utility of Indels for DNA Barcoding of Closely Related Plant Species: A Case Study in Taxus

Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling Strategy
2.2. DNA Extraction, PCR and Sequencing
2.3. Data Analysis
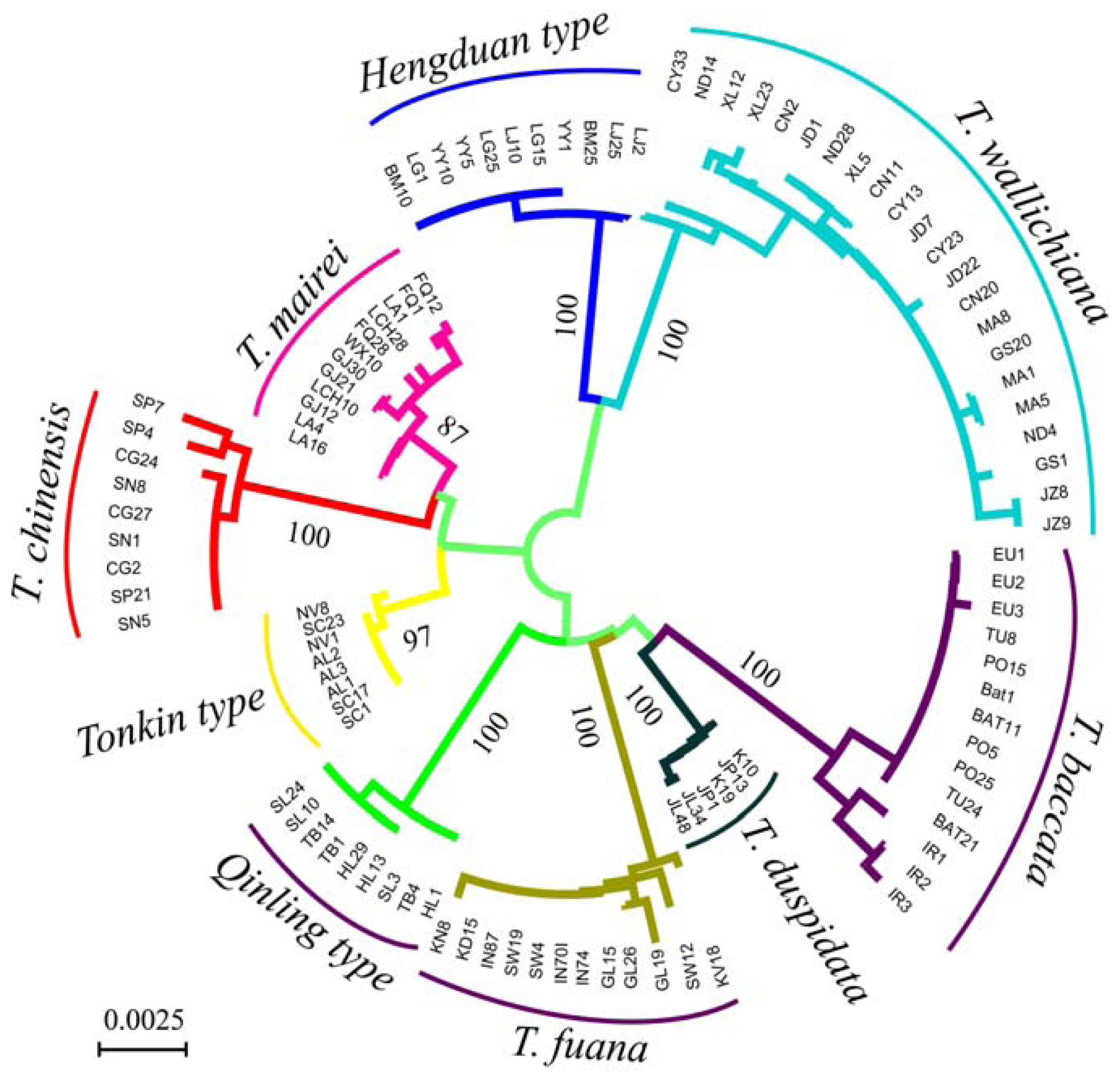
3. Results
3.1. Sequence Characters of the Four Loci
3.2. Genetic Distance and Clustering Relationship
3.3. Indel-Treating Method Comparison
4. Discussion
4.1. Sampling Size of Population/Species for Plant Barcoding
4.2. Utility of Indels for Barcoding and Effect of Different Indel Treatments
Supplementary Information
ijms-13-08740-s001.pdfAcknowledgements
References
- Hebert, P.D.N; Cywinska, A; Ball, S.L; deWaard, J.R. Biological identifications through DNA barcodes. Proc. R. Soc. Biol. Sci. Ser. B 2003, 270, 313–321. [Google Scholar]
- Lahaye, R; van der Bank, M; Bogarin, D; Warner, J; Pupulin, F; Gigot, G; Maurin, O; Duthoit, S; Barraclough, T.G; Savolainen, V. DNA barcoding the floras of biodiversity hotspots. Proc. Natl. Acad. Sci. USA 2008, 105, 2923–2928. [Google Scholar]
- Dalton, D.L; Kotze, A. DNA barcoding as a tool for species identification in three forensic wildlife cases in South Africa. Forensic Sci. Int 2011, 207, e51–e54. [Google Scholar]
- Kress, W.J; Erickson, D.L; Jones, F.A; Swenson, N.G; Perez, R; Sanjur, O; Bermingham, E. Plant DNA barcodes and a community phylogeny of a tropical forest dynamics plot in Panama. Proc. Natl. Acad. Sci. USA 2009, 106, 18621–18626. [Google Scholar]
- Valentini, A; Pompanon, F; Taberlet, P. DNA barcoding for ecologists. Trends Ecol. Evol 2009, 24, 110–117. [Google Scholar]
- Kress, W.J; Wurdack, K.J; Zimmer, E.A; Weigt, L.A; Janzen, D.H. Use of DNA barcodes to identify flowering plants. Proc. Natl. Acad. Sci. USA 2005, 102, 8369–8374. [Google Scholar]
- Fazekas, A.J; Burgess, K.S; Kesanakurti, P.R; Graham, S.W; Newmaster, S.G; Husband, B.C; Percy, D.M; Hajibabaei, M; Barrett, S.C. Multiple multilocus DNA barcodes from the plastid genome discriminate plant species equally well. PLoS One 2008, 3, e2802. [Google Scholar]
- Hollingsworth, P.M; Graham, S.W; Little, D.P. Choosing and using a plant DNA barcode. PLoS One 2011, 6, e19254. [Google Scholar]
- China Plant BOL Group. Comparative analysis of a large dataset indicates that internal transcribed spacer (ITS) should be incorporated into the core barcode for seed plants. Proc. Natl. Acad. Sci. USA 2011, 108, 19641–19646.
- Pennisi, E. Wanted: A barcode for plants. Science 2007, 318, 190–191. [Google Scholar]
- CBOL Plant Working Group. A DNA barcode for land plants. Proc. Natl. Acad. Sci. USA 2009, 106, 12794–12797.
- Kress, W.J; Erickson, D.L. A two-locus global DNA barcode for land plants: The coding rbcL gene complements the non-coding trnH-psbA spacer region. PloS One 2007, 2, e508. [Google Scholar]
- T Taberlet, P; Coissac, E; Pompanon, F; Gielly, L; Miquel, C; Valentini, A; Vermat, T; Corthier, G; Brochmann, C; Willerslev, E. Power and limitations of the chloroplast trnL (UAA) intron for plant DNA barcoding. Nucleic Acids Res 2007, 35, e14. [Google Scholar]
- Hollingsworth, M.L; Clark, A.A; Forrest, L.L; Richardson, J; Pennington, R.T; Long, D.G; Cowan, R; Chase, M.W; Gaudeul, M; Hollingsworth, P.M. Selecting barcoding loci for plants: Evaluation of seven candidate loci with species-level sampling in three divergent groups of land plants. Mol. Ecol. Resour 2009, 9, 439–457. [Google Scholar]
- Gonzalez, M.A; Baraloto, C; Engel, J; Mori, S.A; Pétronelli, P; Riéra, B; Roger, A; Thébaud, C; Chave, J. Identification of Amazonian trees with DNA barcodes. PLoS One 2009, 4, e7483. [Google Scholar]
- Bruni, I; de Mattia, F; Galimberti, A; Galasso, G; Banfi, E; Casiraghi, M; Labra, M. Identification of poisonous plants by DNA barcoding approach. Int. J. Leg. Med 2010, 124, 595–603. [Google Scholar]
- Muellner, A.N; Schaefer, H; Lahaye, R. Evaluation of candidate DNA barcoding loci for economically important timber species of the mahogany family (Meliaceae). Mol. Ecol. Resour 2011, 11, 450–460. [Google Scholar]
- Matz, M.V; Nielsen, R. A likelihood ratio test for species membership based on DNA sequence data. Philos. Trans. R Soc. Lond. B Biol. Sci 2005, 360, 1969–1974. [Google Scholar]
- Meyer, C.P; Paulay, G. DNA barcoding: Error rates based on comprehensive sampling. PLoS Biol 2005, 3, 2229–2238. [Google Scholar]
- Wiemers, M; Fiedler, K. Does the DNA barcoding gap exist?—A case study in blue butterflies (Lepidoptera: Lycaenidae). Front. Zool 2007, 4, 8. [Google Scholar]
- Zhang, A.B; He, L.J; Crozier, R.H; Muster, C; Zhu, C.D. Estimating sample sizes for DNA barcoding. Mol. Phylogenet. Evol 2010, 54, 1035–1039. [Google Scholar]
- Simmons, M.P; Ochoterena, H. Gaps as characters in sequence-based phylogenetic analyses. Syst. Biol 2000, 49, 369–381. [Google Scholar]
- Simmons, M.P; Muller, K; Norton, A.P. The relative performance of indel-coding methods in simulations. Mol. Phylogenet. Evol 2007, 44, 724–740. [Google Scholar]
- Dwivedi, B; Gadagkar, S.R. Phylogenetic inference under varying proportions of indel-induced alignment gaps. BMC Evol. Biol 2009, 9, 211. [Google Scholar]
- Graham, S.W; Reeves, P.A; Burns, A.C.E; Olmstead, R.G. Microstructural changes in noncoding chloroplast DNA: Interpretation, evolution, and utility of indels and inversions in basal angiosperm phylogenetic inference. Int. J. Plant Sci. 2000, 161, S83–S96. [Google Scholar]
- Farrington, L; MacGillivray, P; Faast, R; Austin, A. Investigating DNA barcoding options for the identification of Caladenia (Orchidaceae) species. Aust. J. Bot 2009, 57, 276–286. [Google Scholar]
- Monaghan, M.T; Balke, M; Pons, J; Vogler, A.P. Beyond barcodes: Complex DNA taxonomy of a South Pacific Island radiation. Proc. R. Soc. Biol. Sci. Ser. B 2006, 273, 887–893. [Google Scholar]
- Newmaster, S.G; Fazekas, A.J; Steeves, R.A.D; Janovec, J. Testing candidate plant barcode regions in the Myristicaceae. Mol. Ecol. Resour 2008, 8, 480–490. [Google Scholar]
- Liu, J; Möller, M; Gao, L.M; Zhang, D.Q; Li, D.Z. DNA barcoding for the discrimination of Eurasian yews (Taxus L., Taxaceae) and the discovery of cryptic species. Mol. Ecol. Resour 2011, 11, 89–100. [Google Scholar]
- Möller, M; Gao, L.M; Mill, R.R; Li, D.Z; Hollingsworth, M.L; Gibby, M. Morphometric analysis of the Taxus wallichiana complex (Taxaceae) based on herbarium material. Bot. J. Linn. Soc. 2007, 155, 307–335. [Google Scholar]
- Liu, J; Gao, L.M. Comparative analysis of three different methods of total DNA extraction used in Taxus. Guihaia 2011, 31, 244–249. [Google Scholar]
- Larkin, M.A; Blackshields, G; Brown, N.P; Chenna, R; McGettigan, P.A; McWilliam, H; Valentin, F; Wallace, I.M; Wilm, A; Lopez, R; et al. Clustal W and clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar]
- ES-Computing. EditPlus Text Editor 3.20. Available online: http://www.editplus.com accessed on 12 July 2012.
- Tamura, K; Dudley, J; Nei, M; Kumar, S. MEGA4: Molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol. Biol. Evol 2007, 24, 1596–1599. [Google Scholar]
- Müller, K. Incorporating information from length-mutational events into phylogenetic analysis. Mol. Phylogenet. Evol 2006, 38, 667–676. [Google Scholar]
- Nielsen, R; Matz, M. Statistical approaches for DNA barcoding. Syst. Biol 2006, 55, 162–169. [Google Scholar]
- Egan, A.N; Crandall, K.A. Incorporating gaps as phylogenetic characters across eight DNA regions: Ramifications for North American Psoraleeae (Leguminosae). Mol. Phylogenet. Evol 2008, 46, 532–546. [Google Scholar]
- Provan, J; Powell, W; Hollingsworth, P.M. Chloroplast microsatellites: New tools for studies in plant ecology and evolution. Trends Ecol. Evol 2001, 16, 142–147. [Google Scholar]
- Beatty, G.E; Provan, J. Refugial persistence and postglacial recolonization of North America by the cold-tolerant herbaceous plant. Orthilia secunda. Mol. Ecol 2010, 19, 5009–5021. [Google Scholar]
- Beatty, G.E; Provan, J. Phylogeographic analysis of North American populations of the parasitic herbaceous plant Monotropa hypopitys L. reveals a complex history of range expansion from multiple late glacial refugia. J. Biogeogr 2011, 38, 1585–1599. [Google Scholar]
- Yu, W.B; Huang, P.H; Ree, R.H; Liu, M.L; Li, D.Z; Wang, H. DNA barcoding of Pedicularis L. (Orobanchaceae): Evaluating four universal barcode loci in a large and hemiparasitic genus. J. Syst. Evol 2011, 49, 425–437. [Google Scholar]
- Nicolè, S; Erickson, D.L; Ambrosi, D; Bellucci, E; Lucchin, M; Papa, R; Kress, W.J; Barcaccia, G. Biodiversity studies in Phaseolus species by DNA barcoding. Genome 2011, 54, 529–545. [Google Scholar]
- Wolfe, K.H; Li, W.H; Sharp, P.M. Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc. Natl. Acad. Sci. USA 1987, 84, 9054–9058. [Google Scholar]
- Drouin, G; Daoud, H; Xia, J. Relative rates of synonymous substitutions in the mitochondrial, chloroplast and nuclear genomes of seed plants. Mol. Phylogenet. Evol 2008, 49, 827–831. [Google Scholar]


{kind=link}
| Data set | Indel treating method | No. of indel | Aligned length | No. (%) VC | No. (%) PIC | Mean interspecific distance | Mean intraspecific distance |
|---|---|---|---|---|---|---|---|
| matK | - | 0 | 1533 | 15 (0.98) | 15 (0.98) | 0.0027 (0.00065–0.0046) | 0.00006 (0–0.00024) |
| trnL-trnF | - | 11 | 869 | 25 (2.88) | 22 (2.53) | 0.0063 (0.00083–0.0063) | 0.00034 (0–0.0011) |
| SIC | 880 | 36 (4.09) | 29 (3.30) | - | |||
| MCIC | 874 | 30 (3.43) | 26 (2.98) | - | |||
| trnH-psbA | - | 13 | 1321 | 17 (1.29) | 13 (0.98) | 0.0075 (0–0.013) | 0.00046 (0–0.0014) |
| SIC | 1334 | 30 (2.25) | 22 (1.65) | - | |||
| MCIC | 1330 | 26 (1.96) | 21 (1.57) | - | |||
| ITS | - | 5 | 1143 | 51 (4.46) | 44 (3.85) | 0.010 (0.0046–0.015) | 0.00042 (0–0.00096) |
| matK | trnL-trnF | trnH-psbA | ITS | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Lineage | N | NH | Within population distance | Between population distance | Intraspecific distance | NH | Within population distance | Between population distance | Intraspecific distance | NH | Within population distance | Between population distance | Intraspecific distance | NH | Within population distance | Between population distance | Intraspecific distance |
| Hengduan type | 11 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 2 | 0–0.00088 | 0–0.00088 | 0.00038 |
| Qinling type | 9 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 3 | 0–0.00188 | 0–0.00188 | 0.00094 | 1 | 0 | 0 | 0 |
| Taxus baccata | 14 | 3 | 0 | 0–0.00067 | 0.00024 | 4 | 0–0.00124 | 0–0.00369 | 0.00095 | 2 | 0 | 0–0.0387 | 0.0014 | 2 | 0–0.00351 | 0–0.00357 | 0.00050 |
| T. chinensis | 9 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 4 | 0–0.00527 | 0–0.00527 | 0.0019 |
| T. cuspidata | 6 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 2 | 0 | 0–0.00088 | 0.00047 |
| T. fuana | 12 | 3 | 0–0.00065 | 0–0.00130 | 0.00031 | 2 | 0–0.00249 | 0–0.00249 | 0.00041 | 3 | 0–0.00376 | 0–0.00564 | 0.00094 | 1 | 0 | 0 | 0 |
| T. mairei | 12 | 1 | 0 | 0 | 0 | 3 | 0–0.00125 | 0–0.00125 | 0.00060 | 1 | 0 | 0 | 0 | 6 | 0–0.00264 | 0–0.00264 | 0.00096 |
| T. wallichiana | 22 | 1 | 0 | 0 | 0 | 8 | 0–0.00124 | 0–0.00369 | 0.0011 | 10 | 0–0.00362 | 0–0.00519 | 0.00084 | 3 | 0–0.00176 | 0–0.00176 | 0.00026 |
| Tonkin type | 8 | 1 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| Region | Indel coding schemes | Hengduan type | Qinling type | T. baccata | T. chinensis | T. cuspidata | T. fuana | T. mairei | T. wallichiana | Tonkin type | Resolution (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| trnL-trnF | CD | 77 | n.d. | 97 | 83 | 76 | 76 | n.d. | 87 | 77 | 77.8 (7/9) |
| PWD | 80 | n.d. | 98 | 84 | 90 | 79 | n.d. | 86 | 73 | 77.8 (7/9) | |
| SIC | 79 | 58 | 98 | 94 | 74 | 84 | n.d. | 91 | 81 | 88.9 (8/9) | |
| MCIC | 72 | 77 | 99 | 97 | 80 | 86 | 40 | 91 | 89 | 100 (9/9) | |
| trnH-psbA | CD | n.d. | n.d. | n.d. | n.d. | 62 | n.d. | n.d. | n.d. | n.d. | 11.1 (1/9) |
| PWD | n.d. | n.d. | n.d. | 64 | 95 | n.d. | n.d. | 93 | n.d. | 33.3 (3/9) | |
| SIC | 58 | n.d. | n.d. | 64 | 96 | n.d. | n.d. | 93 | n.d. | 44.4 (4/9) | |
| MCIC | 55 | n.d. | n.d. | 64 | 96 | n.d. | n.d. | 98 | n.d. | 44.4 (4/9) |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liu, J.; Provan, J.; Gao, L.-M.; Li, D.-Z. Sampling Strategy and Potential Utility of Indels for DNA Barcoding of Closely Related Plant Species: A Case Study in Taxus. Int. J. Mol. Sci. 2012, 13, 8740-8751. https://doi.org/10.3390/ijms13078740
Liu J, Provan J, Gao L-M, Li D-Z. Sampling Strategy and Potential Utility of Indels for DNA Barcoding of Closely Related Plant Species: A Case Study in Taxus. International Journal of Molecular Sciences. 2012; 13(7):8740-8751. https://doi.org/10.3390/ijms13078740
Chicago/Turabian StyleLiu, Jie, Jim Provan, Lian-Ming Gao, and De-Zhu Li. 2012. "Sampling Strategy and Potential Utility of Indels for DNA Barcoding of Closely Related Plant Species: A Case Study in Taxus" International Journal of Molecular Sciences 13, no. 7: 8740-8751. https://doi.org/10.3390/ijms13078740