Comparison of Different Ranking Methods in Protein-Ligand Binding Site Prediction

Abstract
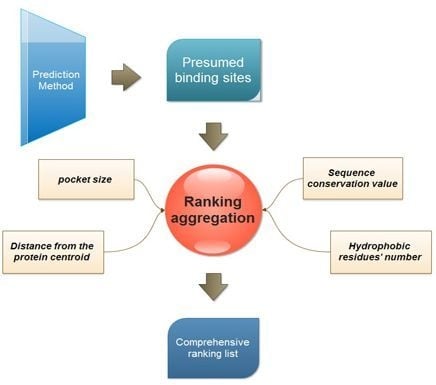
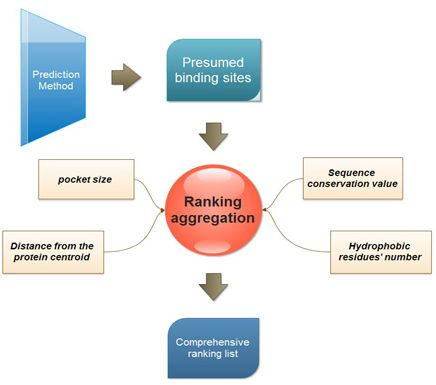
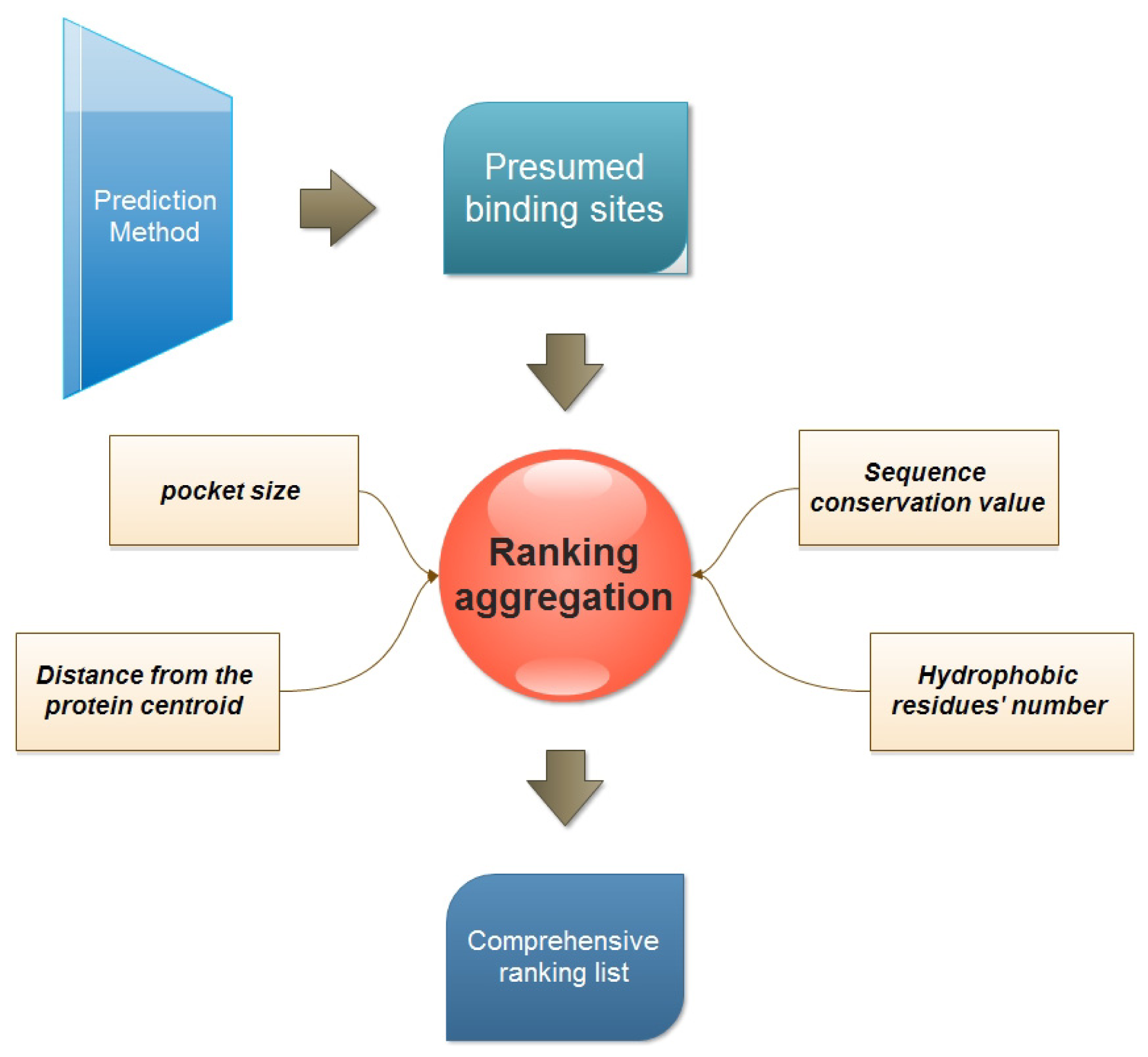
:
1. Introduction
2. Results and Discussion
2.1. Individual Property Comparison
2.2. Ranking Aggregation from a Multi-View Perspective
3. Methods
3.1. Four Properties Used for Ranking
- Pocket size. This is one of the most popular ranking properties. In this study, the volume of every presumed binding site is calculated with the Qhull program [25].
- Distance of binding site from the protein centroid. This property is considered to reflect the depth of a presumed binding site. And the distance is defined as the Euclidian distance between the protein centroid and the geometric center of the presumed binding site.where (xb, yb, zb) is the coordination of the predicted binding site center, and (xp, yp, zp) is the center of the protein.
- Sequence conservation value. The sequence conservation information is achieved by the ConSurf-DB [26], which provides the pre-calculated evolutionary conservation profiles for proteins with known structures in the PDB. In ConSurf-DB, every residue in every corresponding protein is evaluated with a normalized conservation score so that its average over all residues is zero and the standard deviation is one. Low (negative) scores indicate the conserved positions while the high scores indicate the variable ones. In our study, the candidate binding sites are ranked according to the conservation score of all residues in the same binding site.
- The number of hydrophobic residues. Due to the importance of hydrophobicity in protein-ligand binding sites [27,28], the number of hydrophobic residues in each presumed binding site is also calculated. The hydrophobic residues include ALA, VAL, LEU, ILE, PRO, PHE, TRP and MET. The following equation is used to calculate hydrophobic residues:
3.2. Multi-View Ranking Aggregation
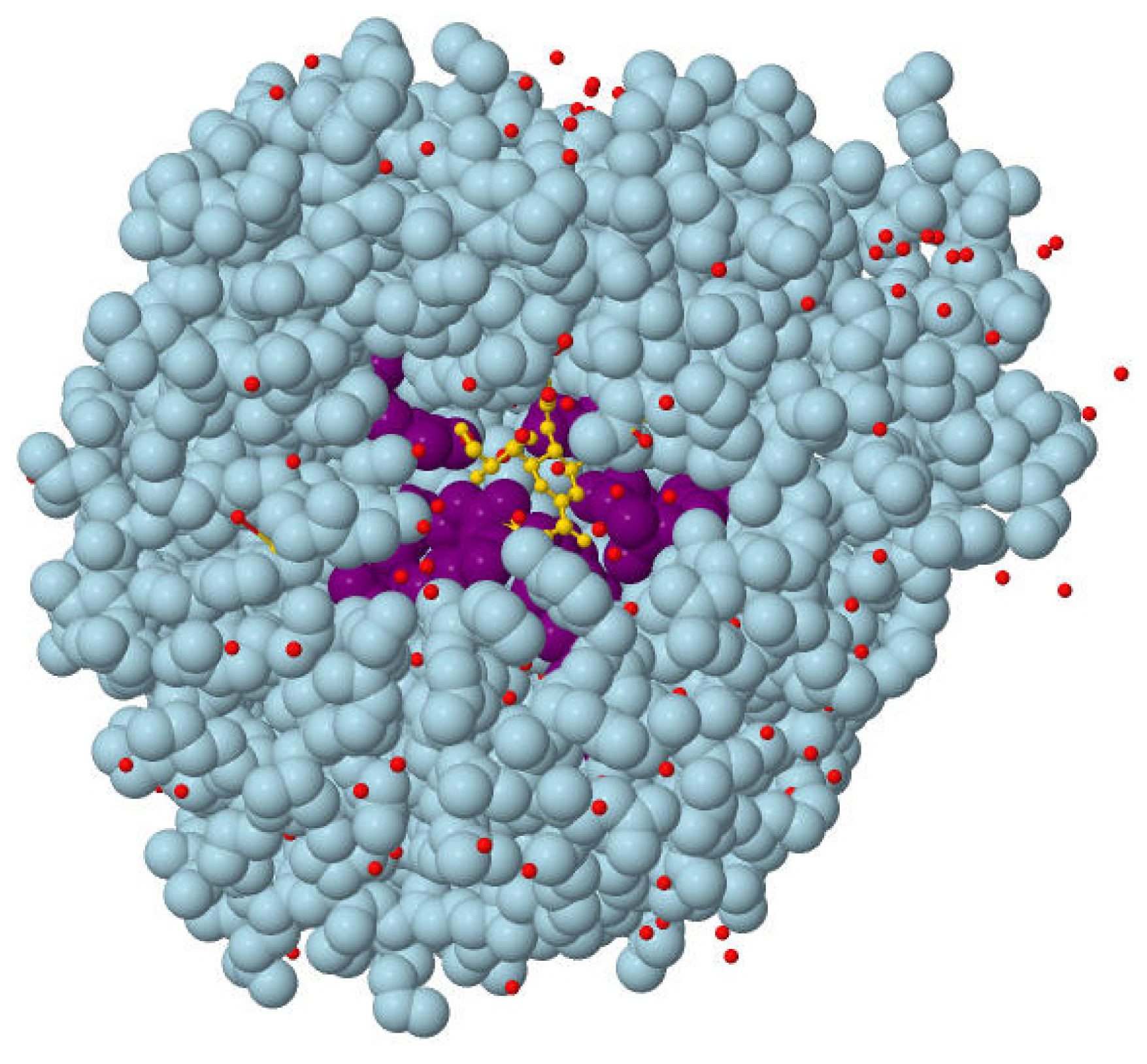
3.3. Test Dataset and Evaluation of the Pocket Prediction
4. Conclusions
Acknowledgments
References
- Zhu, R.; Hu, L.; Li, H.; Su, J.; Cao, Z.; Zhang, W. Novel natural inhibitors of CYP1A2 identified by in silico and in vitro screening. Int. J. Mol. Sci 2011, 12, 3250–3262. [Google Scholar]
- Zhu, R.; Liu, Q.; Tang, J.; Li, H.; Cao, Z. Investigations on inhibitors of hedgehog signal pathway: A quantitative structure-activity relationship study. Int. J. Mol. Sci 2011, 12, 3018–3033. [Google Scholar]
- Henrich, S.; Salo-Ahen, O.M.; Huang, B.; Rippmann, F.F.; Cruciani, G.; Wade, R.C. Computational approaches to identifying and characterizing protein binding sites for ligand design. J. Mol. Recognit 2010, 23, 209–219. [Google Scholar]
- Dai, T.; Liu, Q.; Gao, J.; Cao, Z.; Zhu, R. A new protein-ligand binding sites prediction method based on the integration of protein sequence conservation information. BMC Bioinforma 2011, 12(Suppl 14), S9. [Google Scholar]
- Levitt, D.G.; Banaszak, L.J. POCKET: A computer graphics method for identifying and displaying protein cavities and their surrounding amino acids. J. Mol. Graph 1992, 10, 229–234. [Google Scholar]
- Hendlich, M.; Rippmann, F.; Barnickel, G. LIGSITE: Automatic and efficient detection of potential small molecule-binding sites in proteins. J. Mol. Graph. Model 1997, 15. [Google Scholar]
- Brady, G.P., Jr; Stouten, P.F. Fast prediction and visualization of protein binding pockets with PASS. J. Comput. Aided Mol. Des 2000, 14, 383–401. [Google Scholar]
- Laskowski, R.A. SURFNET: A program for visualizing molecular surfaces, cavities, and intermolecular interactions. J. Mol. Graph 1995, 13, 323–330. [Google Scholar]
- Weisel, M.; Proschak, E.; Schneider, G. PocketPicker: Analysis of ligand binding-sites with shape descriptors. Chem. Cent. J 2007, 1, 7. [Google Scholar]
- Laurie, A.T.; Jackson, R.M. Q-SiteFinder: An energy-based method for the prediction of protein-ligand binding sites. Bioinformatics 2005, 21, 1908–1916. [Google Scholar]
- Zhou, Y.Q.; Liang, S.D.; Zhang, C.; Liu, S. Protein binding site prediction using an empirical scoring function. Nucleic Acids Res 2006, 34, 3698–3707. [Google Scholar]
- Sonavane, S.; Chakrabarti, P. Prediction of active site cleft using support vector machines. J. Chem. Inf. Model 2010, 50, 2266–2273. [Google Scholar]
- Manning, J.R.; Jefferson, E.R.; Barton, G.J. The contrasting properties of conservation and correlated phylogeny in protein functional residue prediction. BMC Bioinforma 2008, 9, 51. [Google Scholar]
- Caffrey, D.R.; Somaroo, S.; Hughes, J.D.; Mintseris, J.; Huang, E.S. Are protein-protein interfaces more conserved in sequence than the rest of the protein surface? Protein Sci 2004, 13, 190–202. [Google Scholar]
- Prymula, K.; Jadczyk, T.; Roterman, I. Catalytic residues in hydrolases: Analysis of methods designed for ligand-binding site prediction. J. Comput. Aided Mol. Des 2011, 25, 117–133. [Google Scholar]
- Huang, B.; Schroeder, M. LIGSITEcsc: Predicting ligand binding sites using the Connolly surface and degree of conservation. BMC Struct. Biol 2006, 6, 19. [Google Scholar]
- Huang, B. MetaPocket: A meta approach to improve protein ligand binding site prediction. OMICS 2009, 13, 325–330. [Google Scholar]
- Le Guilloux, V.; Schmidtke, P.; Tuffery, P. Fpocket: An open source platform for ligand pocket detection. BMC Bioinforma 2009, 10, 168. [Google Scholar]
- Savir, Y.; Tlusty, T. Conformational proofreading: The impact of conformational changes on the specificity of molecular recognition. PLoS One 2007, 2, e468. [Google Scholar]
- Nayal, M.; Honig, B. On the nature of cavities on protein surfaces: Application to the identification of drug-binding sites. Proteins 2006, 63, 892–906. [Google Scholar]
- An, J.; Totrov, M.; Abagyan, R. Comprehensive identification of “druggable” protein ligand binding sites. Genome Inform 2004, 15, 31–41. [Google Scholar]
- Zhong, S.; MacKerell, A.D., Jr. Binding response: A descriptor for selecting ligand binding site on protein surfaces. J. Chem. Inf. Model 2007, 47, 2303–2315. [Google Scholar]
- Crennell, S.J.; Garman, E.F.; Philippon, C.; Vasella, A.; Laver, W.G.; Vimr, E.R.; Taylor, G.L. The structures of Salmonella typhimurium LT2 neuraminidase and its complexes with three inhibitors at high resolution. J. Mol. Biol 1996, 259, 264–280. [Google Scholar]
- Jmol: An open-source Java viewer for chemical structures in 3D. Available online: http://www.jmol.org/ accessed on 29 March 2012.
- Barber, C.B.; Dobkin, D.P.; Huhdanpaa, H. The Quickhull algorithm for convex hulls. ACM Trans. Math. Softw 1996, 22, 469–483. [Google Scholar]
- Goldenberg, O.; Erez, E.; Nimrod, G.; Ben-Tal, N. The ConSurf-DB: Pre-calculated evolutionary conservation profiles of protein structures. Nucleic Acids Res 2009, 37, D323–D327. [Google Scholar]
- Wang, L.; Berne, B.J.; Friesner, R.A. Ligand binding to protein-binding pockets with wet and dry regions. Proc. Natl. Acad. Sci. USA 2011, 108, 1326–1330. [Google Scholar]
- Guharoy, M.; Chakrabarti, P. Conserved residue clusters at protein-protein interfaces and their use in binding site identification. BMC Bioinforma 2010, 11, 286. [Google Scholar]
- Kang, H.; Sheng, Z.; Zhu, R.; Huang, Q.; Liu, Q.; Cao, Z. Virtual drug screen schema based on multiview similarity integration and ranking aggregation. J. Chem. Inf. Model 2012, 52, 834–843. [Google Scholar]
- Fagin, R.; Kumar, R.; Sivakumar, D. Comparing top k lists. SIAM J. Discret. Math 2003, 17, 134–160. [Google Scholar]
- Pihur, V.; Datta, S. Weighted rank aggregation of cluster validation measures: A Monte Carlo cross-entropy approach. Bioinformatics 2007, 23, 1607–1615. [Google Scholar]
- Matthews, B.W. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim. Biophys. Acta 1975, 405, 442–451. [Google Scholar]
- Holland, R.C.; Down, T.A.; Pocock, M.; Prlic, A.; Huen, D.; James, K.; Foisy, S.; Drager, A.; Yates, A.; Heuer, M.; et al. BioJava: An open-source framework for bioinformatics. Bioinformatics 2008, 24, 2096–2097. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
| Bound | Unbound/bound | |||||
|---|---|---|---|---|---|---|
| Methods | TOP1 | MCC for TOP1 | TOP3 | TOP1 | MCC for TOP1 | TOP3 |
| Conservation score | 59% | 0.53 | 73% | 57 | 0.53 | 72 |
| Distance | 48% | 0.53 | 66% | 56 | 0.53 | 70 |
| Volume | 47% | 0.50 | 69% | 44 | 0.53 | 59 |
| Hydrophobic | 39% | 0.51 | 62% | 30 | 0.51 | 48 |
| SURFNET (Control) | 42% | ~ | 57% | ~ | ~ | ~ |
| Bound | Unbound/bound | |||||
|---|---|---|---|---|---|---|
| Methods | TOP1 | MCC * for TOP1 | TOP3 | TOP1 | MCC for TOP1 | TOP3 |
| CON + DIS | 57% | 0.52 | 74% | 61 | 0.53 | 74 |
| VOL + DIS | 52% | 0.51 | 73% | 54 | 0.53 | 74 |
| CON + VOL | 52% | 0.52 | 72% | 48 | 0.54 | 65 |
| VOL + HYDRO | 46% | 0.50 | 67% | 39 | 0.53 | 61 |
| DIS + HYDRO | 47% | 0.51 | 68% | 44 | 0.49 | 63 |
| CON + HYDRO | 53% | 0.51 | 70% | 39 | 0.53 | 61 |
| DIS + CON + HYDRO | 53% | 0.50 | 72% | 48 | 0.51 | 67 |
| VOL + CON + HYDRO | 51% | 0.52 | 71% | 41 | 0.55 | 63 |
| VOL + DIS + HYDRO | 50% | 0.52 | 71% | 46 | 0.50 | 67 |
| VOL + DIS + CON | 54% | 0.51 | 73% | 52 | 0.53 | 74 |
| VOL + DIS + CON + HYDRO | 53% | 0.52 | 72% | 48 | 0.53 | 67 |
| Rank | VOL | DIS | REG | CONS |
|---|---|---|---|---|
| 1 | Pocket 0 | Pocket 12 | *Pocket 9 | *Pocket 9 |
| 2 | *Pocket 9 | *Pocket 9 | Pocket 0 | Pocket 5 |
| 3 | Pocket 5 | Pocket 0 | Pocket 10 | Pocket 0 |
| 4 | Pocket 10 | Pocket 7 | Pocket 12 | Pocket 2 |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gao, J.; Liu, Q.; Kang, H.; Cao, Z.; Zhu, R. Comparison of Different Ranking Methods in Protein-Ligand Binding Site Prediction. Int. J. Mol. Sci. 2012, 13, 8752-8761. https://doi.org/10.3390/ijms13078752
Gao J, Liu Q, Kang H, Cao Z, Zhu R. Comparison of Different Ranking Methods in Protein-Ligand Binding Site Prediction. International Journal of Molecular Sciences. 2012; 13(7):8752-8761. https://doi.org/10.3390/ijms13078752
Chicago/Turabian StyleGao, Jun, Qi Liu, Hong Kang, Zhiwei Cao, and Ruixin Zhu. 2012. "Comparison of Different Ranking Methods in Protein-Ligand Binding Site Prediction" International Journal of Molecular Sciences 13, no. 7: 8752-8761. https://doi.org/10.3390/ijms13078752