The above three product channels were divided into five sections: (i) C-H bond activation: dehydrogenation, (ii) C-C bond activation: deethanization, (iii) C-C bond activation: demethanation, (iv) comparison of C-H with C-C bond activation, and (v) activation strain analysis of the direct C-H and C-C bond cleavage.
3.1. C-H Bond Activation: Dehydrogenation
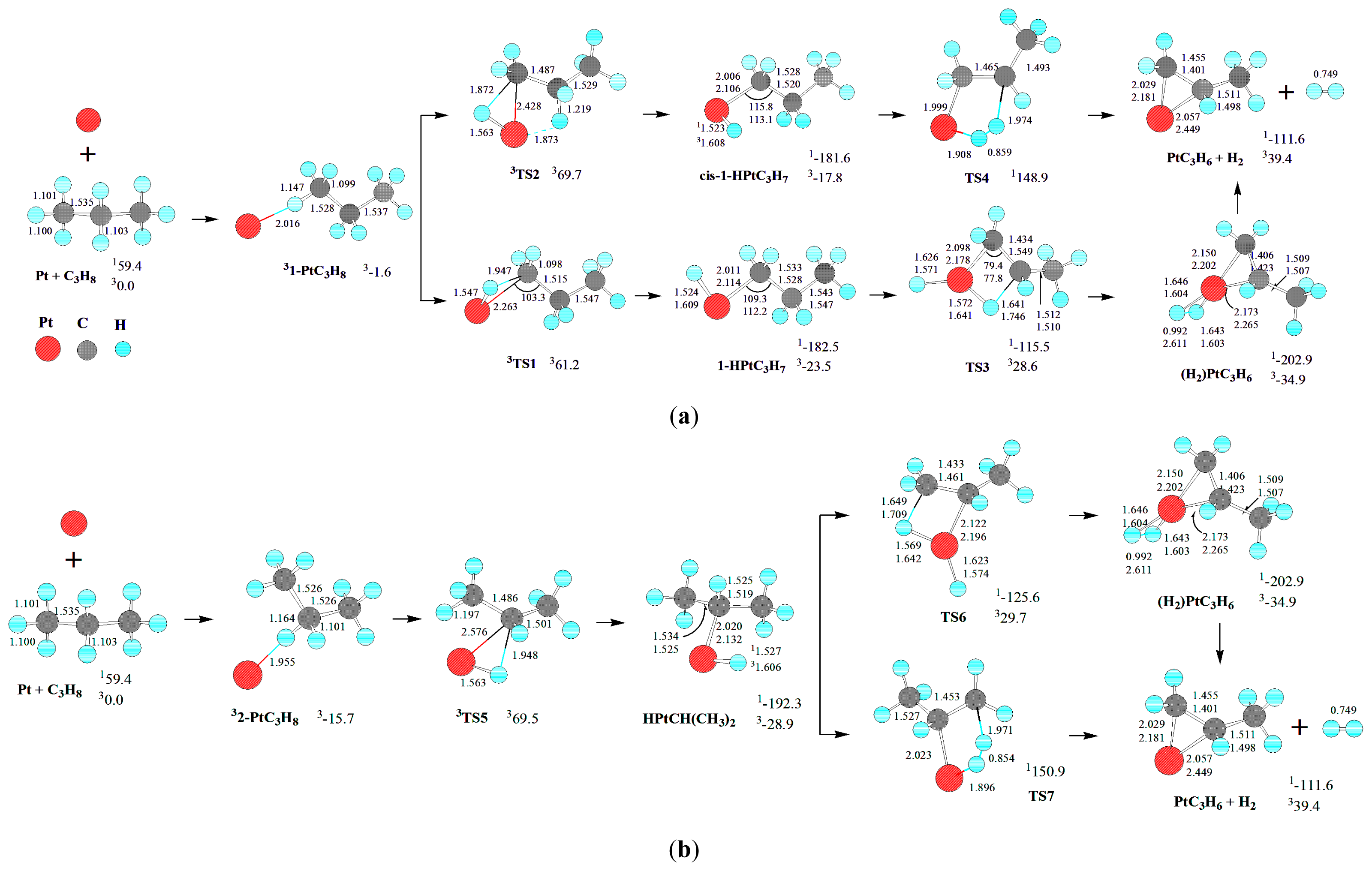
For the dehydrogenation of C
3H
8 by Pt atom, the reaction pathway and the optimized geometric structures of various species are depicted in
Scheme 1.
The triplet state Pt atom,
3D(d
9s
1), is the ground state. The singlet state Pt atom,
1S(d
10), lies 59.4 kJ·mol
−1 above the ground triplet state
3D(d
9s
1), in good agreement with the estimated value of 56.6 kJ·mol
−1 [
26]. The superscript prefixes “
1” and “
3” will be used to indicate the singlet and triplet states, respectively. As depicted in
Scheme 1, there are two primary (
tans-C-H
(1) and
cis-C-H
(1) in the
trans and
cis position with respect to the CH
3 group, respectively) and one secondary (C-H
(2)) C-H bonds in propane. Then, with regard to the initial interaction between Pt atom and C
3H
8, three molecular complexes are considered: (i) Pt atom attacking the H-end of primary
tans-C-H
(1) (1-PtC
3H
8), (ii) Pt atom attacking the H-end of secondary C-H
(2) approaching to Pt atom (2-PtC
3H
8), (iii) Pt atom attacking the H-end of primary
cis-C-H
(1) (3-PtC
3H
8).
As discussed earlier, Pt atom has a triplet ground state (
3D) with excitation energy of 59.4 kJ·mol
−1 to the lowest singlet state (
1S). Considering the initial interaction of Pt atom with C
3H
8, only the triplet ground state
31-PtC
3H
8,
32-PtC
3H
8, and
33-PtC
3H
8 molecular complexes are obtained, whereas we failed to locate the corresponding ones on the singlet PES despite extensive attempts. For
31-PtC
3H
8,
32-PtC
3H
8, and
33-PtC
3H
8, the BSSEs [
46] by BPW91 are 10.1, 10.5, and 10.3 kJ·mol
−1, and the complexation energies corrected by BSSEs are calculated to be 1.6, 15.7, and 3.8 kJ·mol
−1 relative to the reactants Pt(
3D) + C
3H
8, respectively. It is shown that the complex stability increases along
31-PtC
3H
8 <
33-PtC
3H
8 <
32-PtC
3H
8. For
31-PtC
3H
8,
32-PtC
3H
8, and
33-PtC
3H
8, the C-H bond close to Pt atom is elongated to 1.147, 1.164 and 1.147 Å from the 1.100, 1.103, and 1.101 Å of free C
3H
8, while there is a short Pt-H distance of 2.016, 1.955, 2.023 Å, respectively, indicating some molecular interaction between Pt atom and C
3H
8. The minimal energy reaction pathway (MERP) may start at the triplet molecular complexes (
31-PtC
3H
8,
32-PtC
3H
8, and
33-PtC
3H
8) from the corresponding ground triplet reactants.
As shown in
Scheme 1, from these molecular complexes (1-PtC
3H
8, and 2-PtC
3H
8, 3-PtC
3H
8), the C-H bond cleavage may lead to the dehydrogenation product PtC
3H
6 (Pt-propene) + H
2 and Pt(CH
2)
3 (Pt-cyclopropane) + H
2. The change of Gibbs free energies (Δ
G298) for the reactions of Pt(
3D) + C
3H
8→
1PtC
3H
6 (Pt-propene) + H
2 and
1Pt(CH
2)
3 (Pt-cyclopropane) + H
2 are calculated to be −111.6 and −104.5 kJ·mol
−1, respectively. Thereby, the dehydrogenation of C
3H
8 is thermodynamically favorable.
For the formation of PtC
3H
6 (Pt-propene) + H
2, there are five reaction pathways beginning at the three kinds of molecular complexes (two from 1-PtC
3H
8, two from 2-PtC
3H
8, and one from 3-PtC
3H
8), respectively, as shown in
Scheme 1. Alternatively, for the formation of Pt(CH
2)
3 (Pt-cyclopropane) + H
2, there is an unique reaction pathway starting from 3-PtC
3H
8.
First, from 1-PtC3H8, the initial primary C-H bond oxidative insertion via TS1 or TS2 leads to the σ-complex intermediate 1-HPtC3H7 or cis-1-HPtC3H7, respectively. From 1-HPtC3H7, a 1,2-dehydrogenation process takes place via four-center transition state TS3, resulting in a dihydrogen propene complex (H2)PtC3H6. Finally, the molecular complex (H2)PtC3H6 reductively eliminates H2, leaving PtC3H6 behind. Alternatively, from cis-1-HPtC3H7, a 1,2-dehydrogenation process occurs via five-center transition state TS4 directly, leading to the dissociation products PtC3H6 + H2.
Second, from 2-PtC3H8, the initial secondary C-H bond oxidative insertion via TS5 yields the σ-complex intermediate HPtCH(CH3)2. From HPtCH(CH3)2, there are two reaction pathways for the formation of PtC3H6 + H2. On the one hand, a 1,2-dehydrogenation process takes place via four-center transition state TS6, producing the dihydrogen propene complex (H2)PtC3H6. On the other hand, from HPtCH(CH3)2, a 1,2-dehydrogenation process occurs via five-center transition state TS7, directly resulting in the dissociation products PtC3H6 + H2.
Third, from 3-PtC
3H
8, the initial primary C-H bond oxidative insertion via TS8 or TS9 generates a σ-complex intermediate 3-HPtC
3H
7 or
cis-3-HPtC
3H
7, respectively. From 3-HPtC
3H
7, a 1,3-dehydrogenation process takes place via five-member transition state TS10, generating the dihydrogen metallacycle molecular complex H
2Pt(CH
2)
3. Last, the molecular complex dissociates into Pt(CH
2)
3 + H
2. The structure of metallacycle Pt(CH
2)
3 is similar to that of Sc(CH
2)
3+ [
6], TiC
3H
6+ [
7], and NiC
4H
8+ [
47]. Alternatively, from
cis-3-HPtC
3H
7, a 1,2-dehydrogenation process occurs via five-center transition state TS11, producing the dihydrogen propene complex (H
2)PtC
3H
6. Finally, (H
2)PtC
3H
6 reductively eliminates H
2, leaving PtC
3H
6 behind.
For the formation of PtC
3H
6 + H
2, from 1-PtC
3H
8, the MERP should proceed via the minimal energy crossing point (MECP) between
11-HPtC
3H
7 and
31-HPtC
3H
7, with the energy height of the highest point (EHHP) of 61.2 kJ·mol
−1 at
3TS1. From 2-PtC
3H
8, the MERP should proceed via MECP between
1HPtCH(CH
3)
2 and
3HPtCH(CH
3)
2, with the EHHP of 69.5 kJ·mol
−1 at
3TS5. From 3-PtC
3H
8, the MERP should progress via MECP between
1cis-3-HPtC
3H
7 and
3cis-3-HPtC
3H
7, with the EHHP of 162.1 kJ·mol
−1 at TS11. Since the EHHP at
3TS1 from 1-PtC
3H
8 is the lowest among the three reaction channels, this reaction channel for the formation of PtC
3H
6 + H
2 is the most feasible kinetically. Furthermore, these results reveal a high preference of Pt atom for the attack of primary C-H bonds in propane, which is analogous to that of MgO
+ cation for the attack of alkanes [
21]. This feature represents a notable distinction of the transition-metal atom from various transition-metal oxide cations, which show a clear preference for the attack of secondary C-H bonds [
21].
For the formation of Pt(CH2)3 + H2, from 3-PtC3H8, the MERP should advance via MECP between 13-HPtC3H7 and 33-HPtC3H7, with the EHHP of 65.5 kJ·mol−1 at 3TS8.
Moreover, the 11-HPtC3H7, 1cis-1-HPtC3H7, 1HPtCH(CH3)2, 13-HPtC3H7, 1cis-3-HPtC3H7, and 1(H2)PtC3H6 intermediates lie −182.5, −181.6, −192.3, −180.4, −183.3, and −202.9 kJ·mol−1 in a deep energetic well on each MERP, respectively. Then, these intermediates are thermodynamically favored in the dehydrogenation of C3H8. For the intermediates containing –PtH and –Pt-alkyl moieties (11-HPtC3H7, 1cis-1-HPtC3H7, 1HPtCH(CH3)2, 13-HPtC3H7, and 1cis-3-HPtC3H7), the NBO results show that a complete σ-bond has been formed both in Pt-H and in Pt-C.
From Pt + C
3H
8 to the C-H insertion intermediates (
11-HPtC
3H
7,
1cis-1-HPtC
3H
7,
1HPtCH(CH
3)
2,
13-HPtC
3H
7, and
1cis-3-HPtC
3H
7), only the triplet molecular complexes and the triplet TSs are obtained, while we failed to gain the corresponding singlet ones, despite extensive attempts. Furthermore, for Pt atom (10 valence electrons), Pt(
1S) singlet state, has an empty orbital and five doubly occupied nonbonding orbitals, whereas Pt(
3D) triplet state has all of its s and d valence orbitals occupied, with four doubly occupied nonbonding orbitals, and two singly occupied nonbonding orbitals. That is to say, the bonding capacity of Pt(
1S) singlet state to C
3H
8 is stronger than that of Pt(
3D) triplet state. Then, the binding of Pt to C
3H
8 in the C-H insertion intermediates (1-HPtC
3H
7,
cis-1-HPtC
3H
7, HPtCH(CH
3)
2, 3-HPtC
3H
7, and
cis-3-HPtC
3H
7) inverts the energies of the singlet and triplet states from the Pt atom. Therefore, the ground state of the C-H insertion intermediates is the singlet state, as depicted in
Scheme 1. The reaction goes forward from the excited state reactants Pt(
1S) + C
3H
8 to the C-H inserted intermediates, without energy barrier. This can be ascribed to the fact that the singlet state Pt(
1S) has an empty orbital, which should greatly facilitate the interaction with propane and its bond activation, leading smoothly to the formation of two covalent bonds to H and C
3H
7. This phenomenon has also appeared in the analogous Pt + CH
4 system [
26].
3.2. C-C Bond Activation: Deethanization
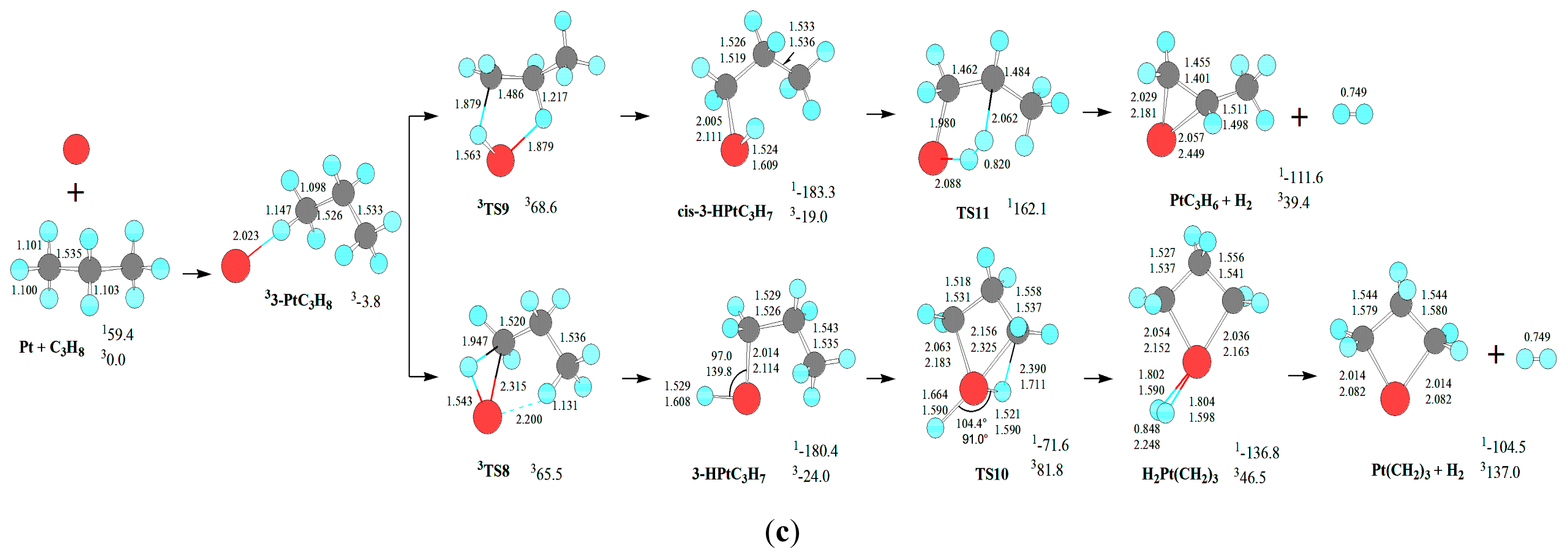
For the deethanization of C
3H
8 by Pt atom, the reaction pathway and the optimized geometric structures of various species are depicted in
Scheme 2. The change of Gibbs free energies (Δ
G298) for the reaction of Pt(
3D) + C
3H
8→
1PtCH
2 + C
2H
6 is calculated to be −45.9 kJ·mol
−1. Thereupon, the deethanization of C
3H
8 is thermodynamically favorable. Then, it is necessary to discuss kinetically the above reaction infra.
As shown in
Scheme 2, for the deethanization of C
3H
8 by Pt atom, there are seven reaction pathways, three from 1-PtC
3H
8, one from 2-PtC
3H
8, and three from 3-PtC
3H
8. These seven reaction pathways are separated into two kinds of reaction pathways, one through the initial C-C bond direct cleavage, and another through the σ-complex assisted C-C σ-bond metathesis. For simplicity, we will primarily discuss the three reaction pathways from 1-PtC
3H
8 infra, which are analogous to those from 2-PtC
3H
8 and 3-PtC
3H
8. The nuances in energy mainly stem from their configuration differences among the three kinds of reaction channels.
As mentioned earlier, from 1-PtC3H8, there are three reaction pathways for the PtCH2 + C2H6 formation. For the first reaction pathway, Pt atom firstly inserts the C-H bond via five-member TS2, resulting in the intermediate cis-1-HPtC3H7. Then, from cis-1-HPtC3H7, a σ-complex assisted C-C σ-bond metathesis takes place via a four-member TS12 with both 1,3-H migration and C-C cleavage, yielding the molecular complex, C2H6PtCH2. Finally, C2H6PtCH2 releases C2H6 molecule, leaving PtCH2 behind. The MERP should proceed via the MECP between 1cis-1-HPtC3H7 and 3cis-1-HPtC3H7, with the highest energy requirement (HER) of 273.2 kJ·mol−1 at the 1cis-1-HPtC3H7→1TS12 reaction step and the EHHP of 91.6 kJ·mol−1 at 1TS12.
For the second and third reaction pathways from 1-PtC3H8, there are two reaction pathways to produce cis-CH3PtC2H5. That is, Pt atom directly inserts the C-C bond via three-member TS13, leading to the intermediate cis-CH3PtC2H5. Alternatively, Pt atom firstly inserts the C-H bond via the three-member TS1, resulting in the intermediate 1-HPtC3H7. Next, from 1-HPtC3H7, a σ-complex assisted C-C σ-bond metathesis occurs via a four-member TS14 with both 1,2-H shift and C-C bond cleavage, also yielding cis-CH3PtC2H5. Then, from CH3PtC2H5, the σ-complex assisted C-H σ-bond metathesis takes place via a four-member TS15 with 1,3-H shift, yielding the molecular complex C2H6PtCH2. As mentioned earlier, C2H6PtCH2 releases C2H6 molecule, staying PtCH2 behind. For the two reaction pathways, each MERP should advance via the MECP between 1cis-CH3PtC2H5 and 3cis-CH3PtC2H5, with the HER of 299.0 kJ·mol−1 at the 1cis-CH3PtC2H5→1TS15 reaction step. The two MERPs involve the EHHP of 156.1 and 120.3 kJ·mol−1 at 3TS13 and 1TS15, respectively.
Comparing these three reaction pathways, one can conclude that the reaction pathway via TS2 involving the first C-H cleavage and via TS12 involving C-C cleavage with synchronous 1,3-H migration is the gross MERP for the 1-PtC3H8→PtCH2 + C2H6 reaction, owing to its comparatively low HER (273.2 vs. 299.0 kJ·mol−1) and low EHHP (91.6 vs. 120.3 and 156.1 kJ·mol−1), with the rate-determining step of 1cis-1-HPtC3H7→1TS12→1C2H6PtCH2.
From 2-PtC3H8 for the formation of PtCH2 + C2H6, only one reaction pathway is obtained, which includes the initial C-C bond cleavage and σ-complex assisted C-H σ-bond metathesis. This reaction pathway is homologous to that via the initial C-C cleavage from 1-PtC3H8 with the HER of 299.0 kJ·mol−1 and the EHHP of 156.1 kJ·mol−1 at 3TS13.
From 3-PtC3H8 for the formation of PtCH2 + C2H6, there are also three reaction pathways. These three reaction pathways are similar to those from 1-PtC3H8. The reaction pathway of σ-complex assisted C-C σ-bond metathesis via 3TS9 and 1TS19 is kinetically most preferable in the three reaction pathways, because of its lowest HER (274.9 vs. 298.7 kJ·mol−1) and lowest EHHP (91.6 vs. 120.3 and 156.1 kJ·mol−1).
In summary, for the formation of the C-C cleavage products PtCH
2 + C
2H
6, the optimal pathway proceeds through the σ-complex
cis-1-HPtC
3H
7 or
cis-3-HPtC
3H
7 from initial C-H bond cleavage, which assists the C-C σ-bond metathesis. This reactivity mode is also complementary for the classical reactivity picture through the direct C-C cleavage intermediate (M = Fe
+ [
48] and Ta
+ [
49]).
3.3. C-C Bond Activation: Demethanation
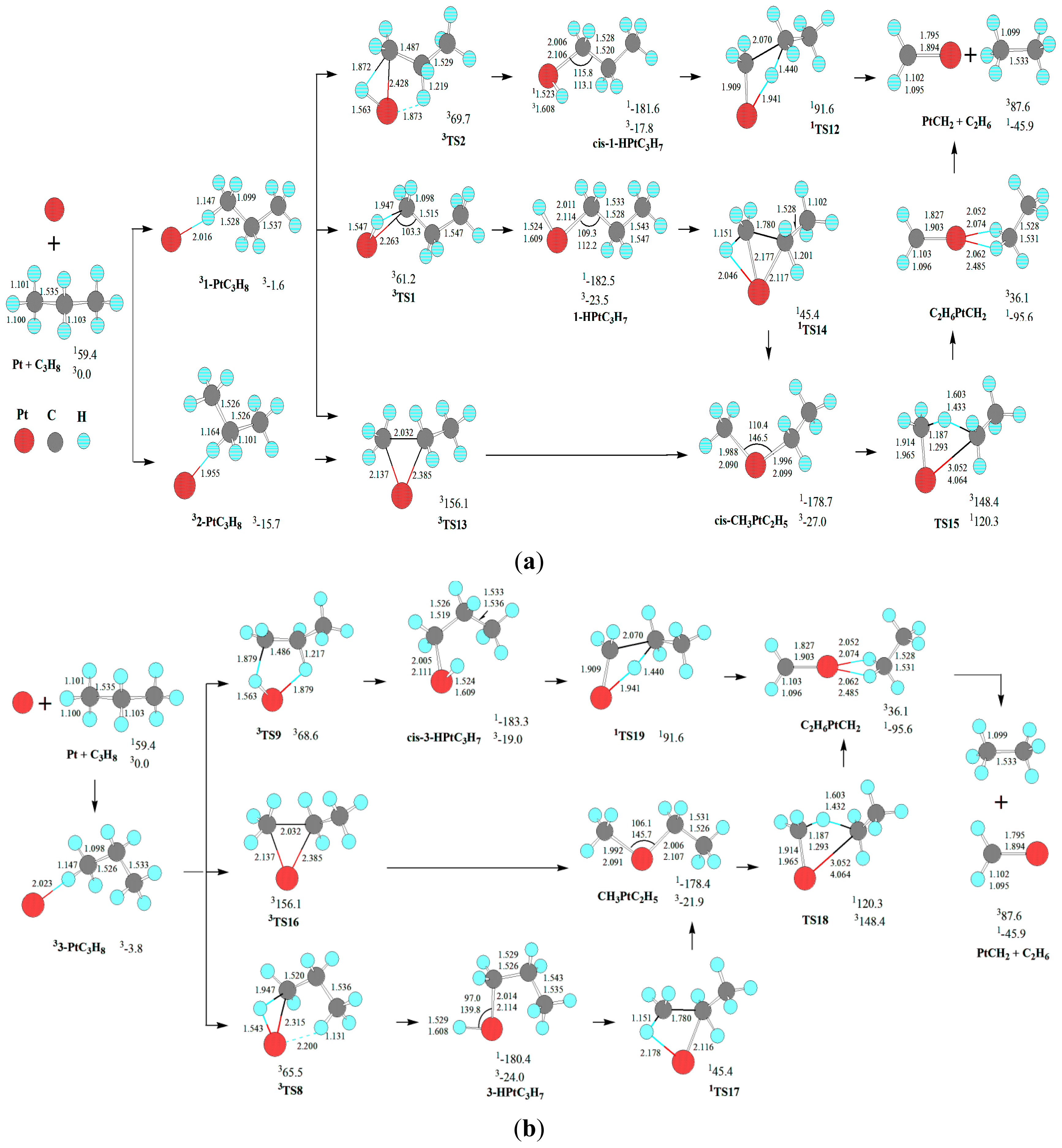
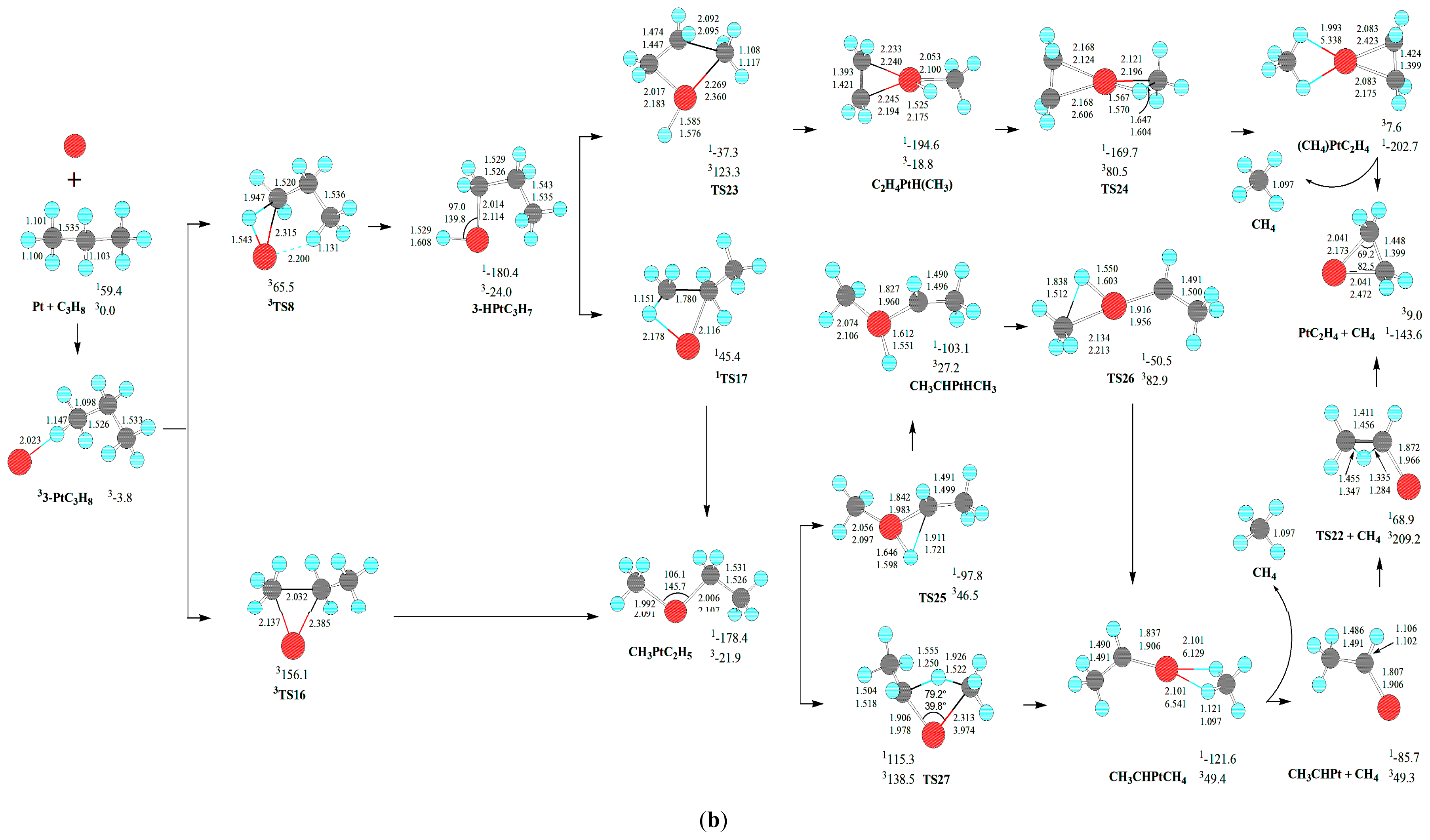
For the demethanation of C
3H
8 by Pt atom, the reaction pathway and the optimized geometric structures of various species are depicted in
Scheme 3. The change of Gibbs free energies (Δ
G298) for the reactions of Pt(3D) + C
3H
8→1PtC
2H
4 + CH
4 are calculated to be −143.6 kJ·mol
−1, which is thermodynamically favorable. Afterwards, we will discuss the kinetics of the above reaction infra. As shown in
Scheme 3, there are four kinds of reaction pathways, which are through
cis-CH
3PtC
2H
5, CH
3PtC
2H
5, HPtCH(CH
3)
2, and C
2H
4PtH(CH
3) intermediates, respectively.
First, from cis-CH3PtC2H5, a σ-complex assisted C-H σ-bond metathesis takes place via a four-member TS20 with 1,4-H shift, yielding the molecular complex CH4PtC2H4. The molecular complex CH4PtC2H4 releases CH4 molecule, leaving PtC2H4 behind. Through cis-CH3PtC2H5, the MERP should go forward via the MECP between 11-HPtC3H7 and 31-HPtC3H7, with the HER of 319.0 kJ·mol−1 at the 1cis-CH3PtC2H5→1TS20 reaction step and the EHHP of 140.3 kJ·mol−1 at 1TS20.
Second, from CH3PtC2H5, a σ-complex assisted C-H σ-bond metathesis occurs via a four-member TS27 with 1,3-H shift, yielding a molecular complex CHCH3PtCH4. Then, the molecular complex CH4PtCHCH3 sets a CH4 molecule free, leaving PtCHCH3 behind. Next, from PtCHCH3, 1,2 H shift occurs via a four-member TS22, staying PtC2H4 behind. Through CH3PtC2H5, the MERP should go forward via the MECP between 13-HPtC3H7 and 33-HPtC3H7, with the HER of 225.8 kJ·mol−1 at the 13-HPtC3H7→1TS17 reaction step and the EHHP of 68.9 kJ·mol−1 at 1TS22.
Third, from HPtCH(CH3)2, a σ-complex assisted C-C σ-bond metathesis occurs via a four-member TS21 with 1,3-H shift, also leading to the molecular complex CHCH3PtCH4. The MERP should go forward via the MECP between 12-HPtC3H7 and 32-HPtC3H7, with the HER of 277.4 kJ·mol−1 at the 1HPtCH(CH3)2→1TS21 reaction step and the EHHP of 85.1 kJ·mol−1 at 1TS21.
Fourth, from 3-HPtC3H7, an oxidative insertion of C-C bond to the platinum center takes place via a four-member TS23, producing a methyl hydrid complex C2H4PtH(CH3). Then, from C2H4PtH(CH3), 1,2-H shift occurs, yielding the molecular complex CH4PtC2H4. Last, the molecular complex CH4PtC2H4 sets a CH4 molecule free, leaving PtC2H4 behind. The MERP should go forward via the MECP between 13-HPtC3H7 and 33-HPtC3H7, with the HER of 143.1 kJ·mol−1 at the 13-HPtC3H7→1TS23 reaction step and the EHHP of 65.5 kJ·mol−1 at 3TS8.
Comparing these four kinds of reaction pathways, one can see that the reaction pathway starting at the 3-PtC
3H
8 involving the crucial intermediate C
2H
4PtH(CH
3) is the most optimal MERP for the Pt + C
3H
8→PtC
2H
4 + CH
4 reaction, thanks to the lowest HER (143.1
vs. 319.0, 225.8, and 277.4 kJ·mol
−1) and lowest EHHP (65.5
vs. 140.3, 85.1, and 68.9 kJ·mol
−1), with the rate-determining step of
13-HPtC
3H
7→
1TS23→C
2H
4PtH(CH
3). Thereby, the optimal pathway proceeds through the σ-complex 3-HPtC
3H
7 from initial C-H bond cleavage, which assists the C-C σ-bond metathesis. That is to say, the optimal C-C bond cleavages are assigned to C-H bond activation as the first step, followed by cleavage of a C-C bond. This reactivity mode is complementary for the classical reactivity picture through the direct C-C cleavage intermediate (M = Fe
+ [
48], and Ta
+ [
49]).
3.4. Comparison of C-H with C-C Bond Activation
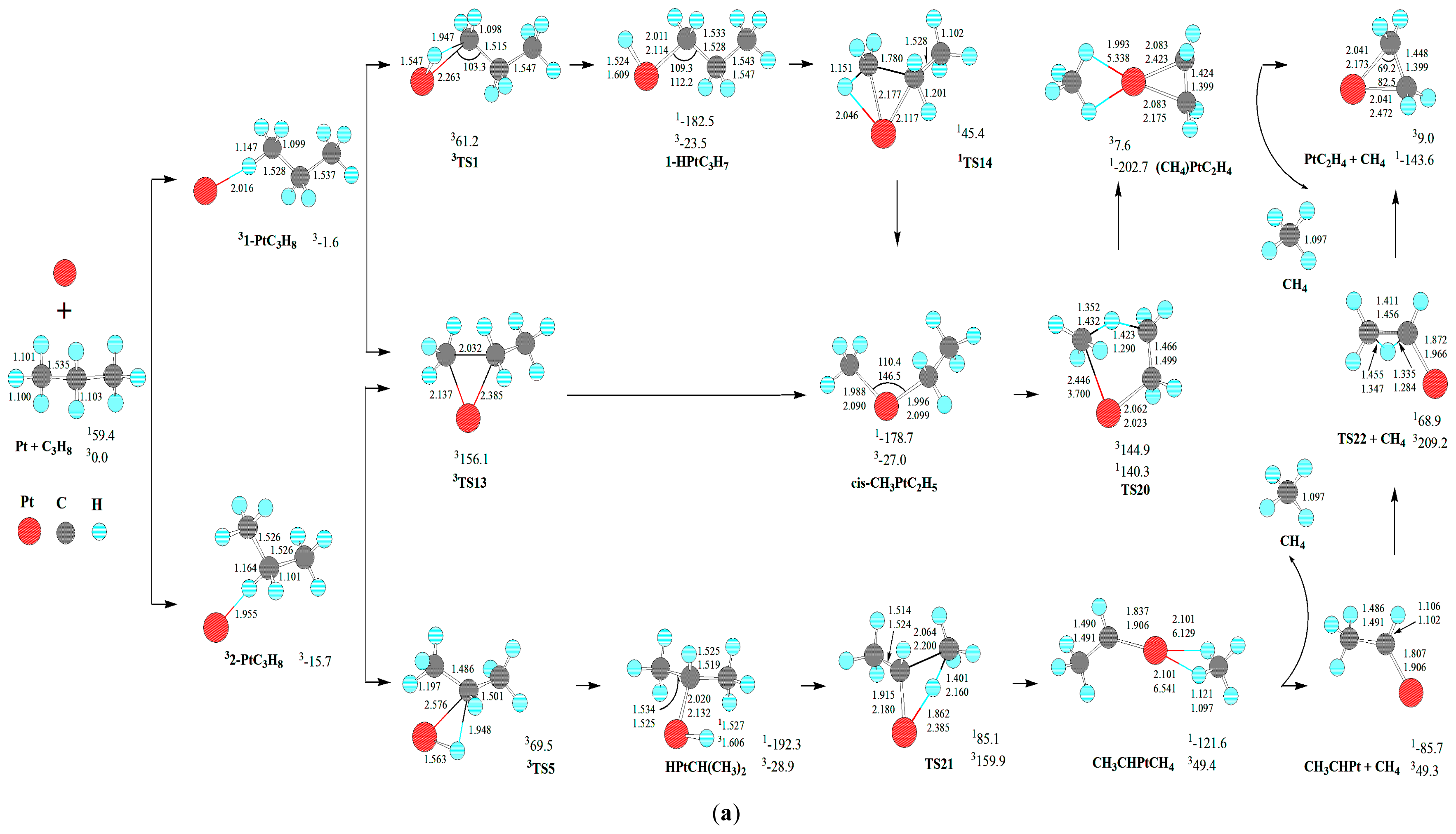
As shown in
Schemes 1–
3, the C-H insertion intermediates (
11-HPtC
3H
7,
1cis-1-HPtC
3H
7,
1HPtCH(CH
3)
2,
13-HPtC
3H
7,
1cis-3-HPtC
3H
7) and the C-C insertion intermediates (
1CH
3PtC
2H
5,
1cis-CH
3PtC
2H
5,
1C
2H
4PtH(CH
3), and
1CH
4PtC
2H
4) deposit in a deep well, respectively. It is indicated that these intermediates are thermodynamically preferred. For the formation of the C-H and the C-C insertion intermediates, the corresponding MERP should involve the HER of about 60~70 and 140~230 kJ·mol
−1, respectively. Thereby, the C-H insertion intermediates are kinetically favored, while the C-C insertion intermediates are kinetically hindered by energy barriers. These results are in qualitative agreement with the experimental results, in which the C-H insertion product is experimentally observed and the C-C insertion product is not formed in observable quantity in Pt + C
2H
6 system [
27].
For the formation of C-C bond cleavage intermediates 1CH3PtC2H5 and 1cis-CH3PtC2H5, one can see that the reaction pathways of the direct C-C activation via 3TS16 and 3TS13 are inferior to those of the σ-complex assisted C-C σ-bond metathesis via 1TS17 and 1TS14 from 3-HPtC3H7 and 1-HPtC3H7, respectively, because of their higher EHHP (156.1 vs. 65.5 and 61.2 kJ·mol−1). This is reminiscent of the important role of σ-complex assistance for the C-C σ-bond metathesis. In other words, the direct C-C bond activation is associated with a sizable barrier, which would prohibit this channel.
A glance to the reaction pathways shown in
Schemes 1–
3 reveals that two kinds of σ-complexes (1-HPtC
3H
7, HPtCH(CH
3)
2, and 3-HPtC
3H
7) and (
cis-1-HPtC
3H
7 and
cis-3-HPtC
3H
7) from initial C-H bond cleavage are crucial for the selective formation of the final C-H and C-C cleavage products.
First, from the identical intermediate 1-HPtC3H7, the reaction step of 11-HPtC3H7→1TS3→1(H2)PtC3H6 is competitive with that of 11-HPtC3H7→1TS14→1cis-CH3PtC2H5. Because 1TS3 lies 160.9 kJ·mol−1 below 1TS14, (H2)PtC3H6 is selectively preferred, whereas cis-CH3PtC2H5 is selectively hampered. In other words, from 1-HPtC3H7, the dehydrogenation process dominates.
Second, from the identical intermediate cis-1-HPtC3H7, the reaction step of 1cis-1-HPtC3H7→1TS12→1C2H6PtCH2 is competitive with that of 1cis-1-HPtC3H7→1TS4→PtC3H6 + H2. Since 1TS12 locates 57.3 kJ·mol−1 below 1TS4, C2H6PtCH2 is selectively favored. That is to say, from cis-1-HPtC3H7, the deethanization process predominates.
Third, from the identical intermediate HPtCH(CH3)2, these reaction steps of HPtCH(CH3)2→1TS6→(H2)PtC3H6, HPtCH(CH3)2→1TS7→PtC3H6 + H2, and HPtCH(CH3)2→1TS21→CH3CHPtCH4 are competitive. As 1TS6 lies 276.5 and 210.7 kJ·mol−1 below 1TS7 and 1TS21, respectively, (H2)PtC3H6 is selectively preferred. Then, from HPtCH(CH3)2, the dehydrogenation process dominates.
Fourth, from the identical intermediate 3-HPtC3H7, these reaction steps of 3-HPtC3H7→1TS10→(H2)Pt(CH2)3, 3-HPtC3H7→1TS17→CH3PtC2H5, and 3-HPtC3H7→1TS23→C2H4PtH(CH3) are competitive. Because 1TS10 locates 117.0 and 34.3 kJ·mol−1 below 1TS17 and 1TS23, respectively, (H2)Pt(CH2)3 is selectively favored, whereas CH3PtC2H5 and C2H4PtH(CH3) are selectively hindered. Thereby, from 3-HPtC3H7, the dehydrogenation process dominates.
Last, from the identical intermediate cis-3-HPtC3H7, these reaction steps of cis-3-HPtC3H7→1TS11 →PtC3H6 + H2 and cis-3-HPtC3H7→1TS19→1C2H6PtCH2 are competitive. Because 1TS19 lies 70.5 kJ·mol−1 below 1TS11, 1C2H6PtCH2 is selectively preferred. Therefore, from cis-3-HPtC3H7, the deethanization process predominates.
In summary, once the σ-complex [1-HPtC3H7, or HPtCH(CH3)2, or 3-HPtC3H7] is formed, the major reaction channel results in the dehydrogenations products. Alternatively, as far as the σ-complex [cis-1-HPtC3H7 or cis-3-HPtC3H7] is concerned, the major reaction channel leads to the deethanization products. Besides, the demethanation process is kinetically ruled out.
To estimate quantitatively the reactivity and selectivity for the two kinds of products [PtC
3H
6 + H
2 and PtCH
2 + C
2H
6], the rate constants have been evaluated according to conventional transition state theory (TST) [
50], including tunneling correction based on Winger’s formulation [
51]. The formation of rate constant
k(T) including tunneling correction coefficient
κ(T) in transition state theory is given by
The rate constant k′(T) is simply given by
where kB is the Bolzmann constant, h is the Planck constant, T is thermodynamic temperature, c0 is standard concentration, and ΔG≠ is Gibbs free energy. The tunneling correction coefficient κ(T) is written in the form of
where kB is the Bolzmann constant, h is the Planck constant, T is thermodynamic temperature, and v≠ is the imaginary frequency of the unbound normal mode at the saddle point. The branching ratio (αi) of product i is calculated by
where ki is the rate constant of product i.
From the identical reactants Pt + C3H8, the formation of PtC3H6 + H2 and PtCH2 + C2H6 are competitive, while their selectivity-controlling steps are Pt(3D) + C3H8→3TS1→31-HPtC3H7 and Pt(3D) + C3H8 → 3TS9→3cis-3-HPtC3H7 on their MERPs, respectively. Thereby, the rate constants were taken into account, where Pt(3D) + C3H8 were taken as reactants, while 3TS1 and 3TS9 served as TSs, respectively. The rate constants for the formation of 31-HPtC3H7 (k1) and 3cis-3-HPtC3H7 (k2) calculated over 300–1100 K temperature range can be fitted by the following expressions (in dm3·mol−1·s−1):
The branching ratios for the formation of PtC3H6 + H2 and PtCH2 + C2H6 are calculated to be 97.7~83.6% and 2.3~16.4%, respectively, over 300–1100 K temperature range. In other words, the dehydrogenation channel is predominant, and the deethanization channel is minor, while the demethanation channel is ruled out.
3.5. Activation Strain Analysis of the direct C-H and C-C Bond Cleavage
To gain insight into how the Pt atom affects the activation barriers of the initial C-H and C-C bond cleavage,
i.e., insight into how this effect depends on the nature of concomitant geometrical deformation and electronic structures of Pt and C
3H
8, the trends in reactivity and competition among the initial C-H and C-C bond mechanisms are analyzed using the activation strain model of chemical reactivity [
30,
31]. In this model, activation energies Δ
E≠ of the TS are divided into the activation strain Δ
E≠strain and the stabilizing TS interaction Δ
E≠int: Δ
E≠ = Δ
E≠strain + Δ
E≠int. The activation strain Δ
E≠strain is the strain energy associated with deforming the reactants from their equilibrium geometry to the geometry they adopt in the TS. The TS interaction Δ
E≠int is the actual interaction energy between the deformed reactants in the TS [
30,
31].
The results of the activation strain analysis are listed in
Table 1. The activation energy Δ
E≠ increases from the initial C–H cleavage TSs (
3TS1,
3TS2,
3TS5,
3TS9, and
3TS8) of 60~70 kJ·mol
−1 to the initial C–C cleavage TSs (
3TS13 and
3TS16) of ~160 kJ·mol
−1. The activation strain Δ
E≠strain decreases from the intial C–H cleavage TSs (
3TS1,
3TS2,
3TS5,
3TS9, and
3TS8) of 290~300 kJ·mol
−1 to the initial C–C cleavage TSs (
3TS13 and
3TS16) of ~200 kJ·mol
−1. The activation strain appears to be related to the bond strength of the activated bond and with the percentage-wise extent of bond stretching in the TS. Typical strengths and lengths of the C-H and C-C bonds are the following: 414 (C-H) and 347 kJ·mol
−1 (C-C), and ~1.1 (C-H) and ~1.5 Å (C-C) [
52]. Moreover, we recall that the percentage-wise extent of bond stretching in the TS for oxidative insertion is 70~80% (C-H) and ~30% (C-C). Hence, both the bond strength and the percentage-wise bond elongation in the TS decreases from C-H to C-C. This correlates nearly with the activation strain Δ
E≠strain, which decreases in the same order. It is indicated that the activation strain prefers the C-C oxidative insertion over the C-H oxidative insertion. That is to say, the activation strain Δ
E≠strain makes in the reverse order as the activation energy Δ
E≠, for the C–H and C–C oxidative insertion. Alternatively, the strength of the TS interaction increases from ~−50 kJ·mol
−1 (C-C) to −220~−240 kJ·mol
−1 (C-H). From C-C to C-H bond activation, the strengthening of the TS interaction varies among 170–190 kJ·mol
−1, whereas the activation strain changes only by ~90 kJ·mol
−1. Thereby, the lowering activation energy from C-C to C-H bond activation stems mainly from the strengthening of the TS interaction.
Thus, the stabilizing interaction ΔE≠int prefers C-H oxidative insertion, whereas the activation strain ΔE≠strain favors C-C oxidative insertion. From C-C to C-H oxidative insertion, the lowering of activation barrier is mainly caused by the TS interaction ΔE≠int becoming more stabilizing.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}