Characterization of Aptamer-Protein Complexes by X-ray Crystallography and Alternative Approaches


 ,
,
Abstract
:1. Introduction
2. Structure Determination
2.1. Nucleic Acid Parameters
2.2. Protein Parameters
2.3. Protein: Aptamer Ratio
2.4. Crystallization Screens
3. Examples from Literature
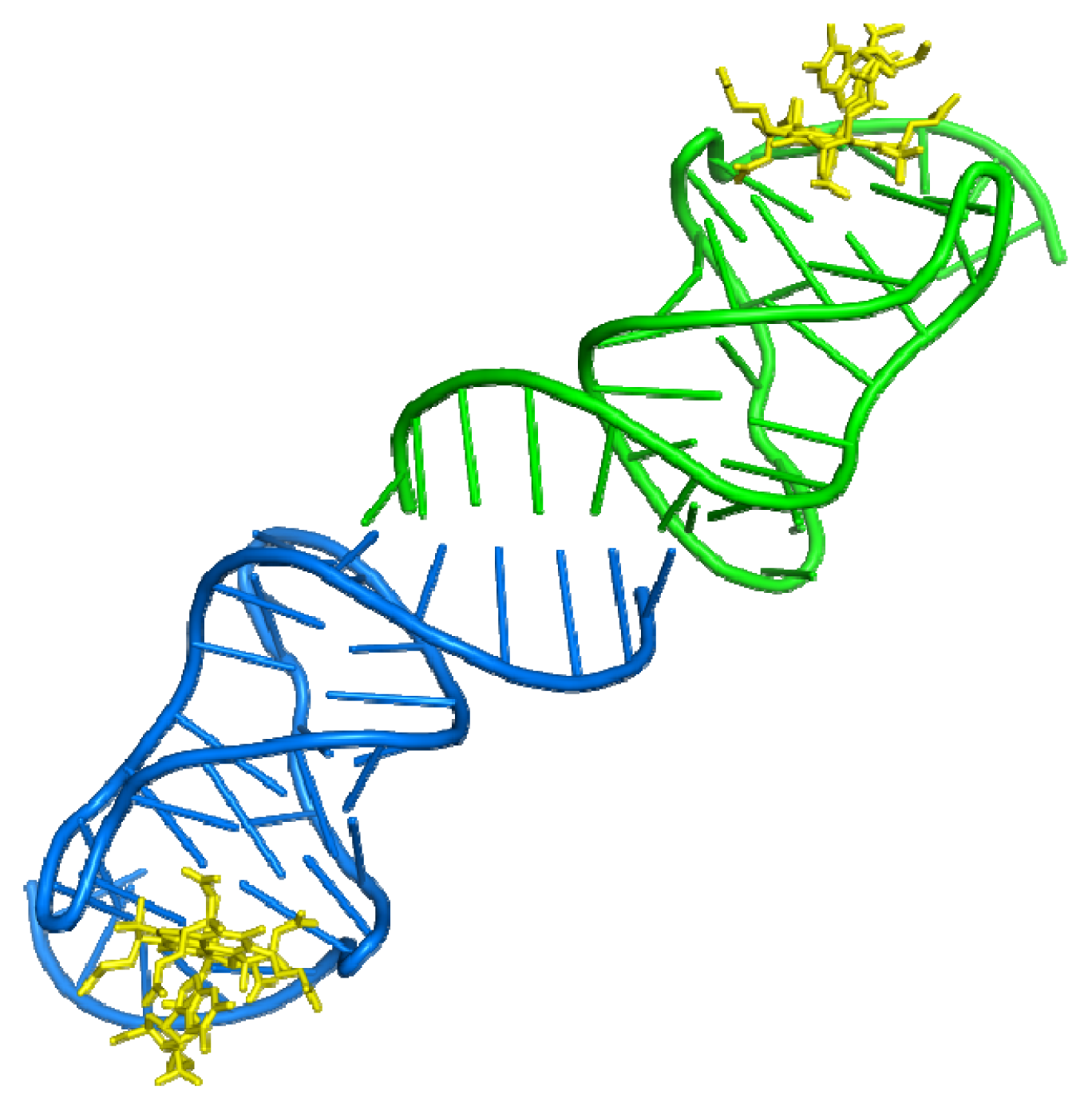
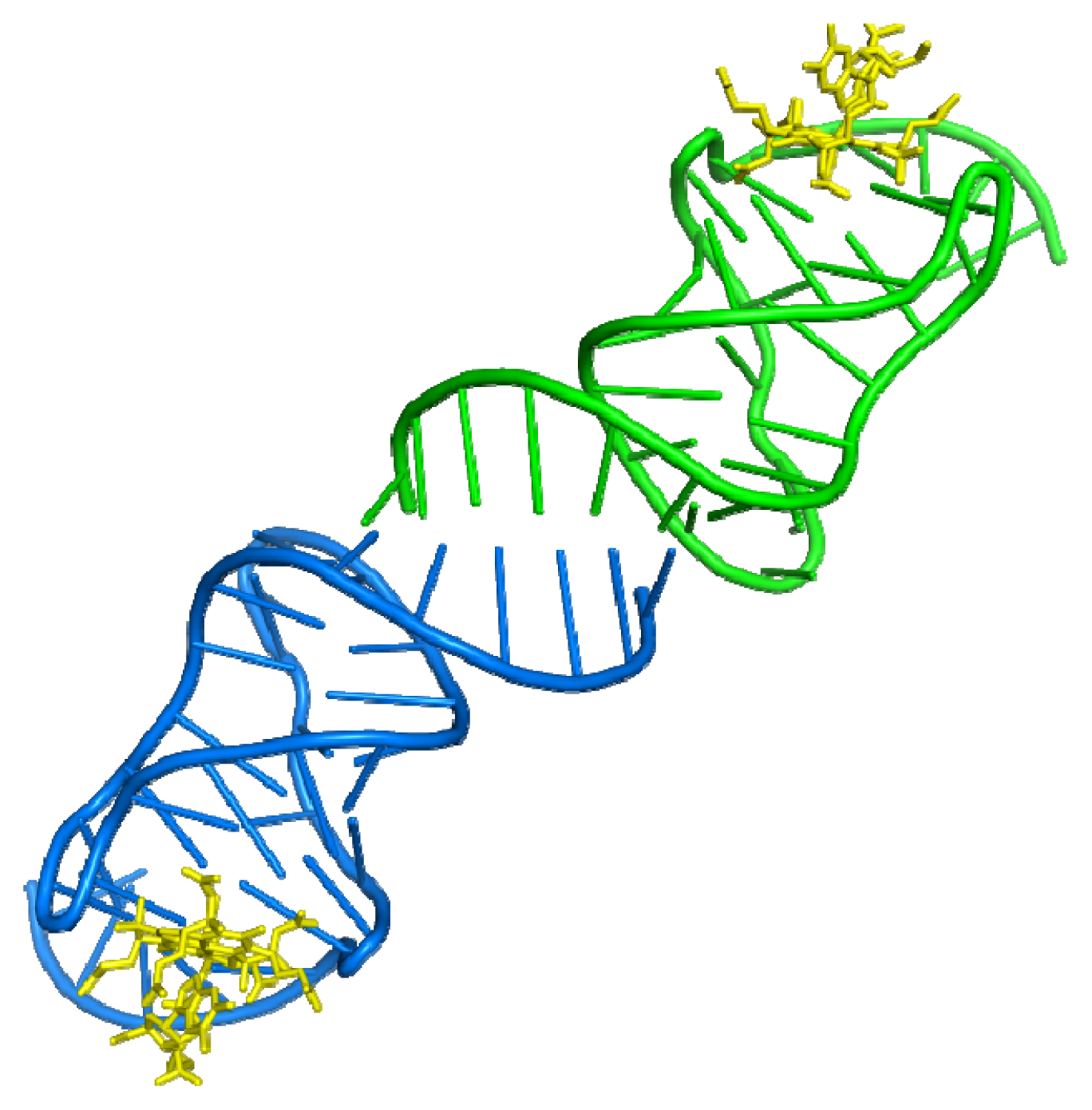
3.1. Aptamer-Thrombin Complex
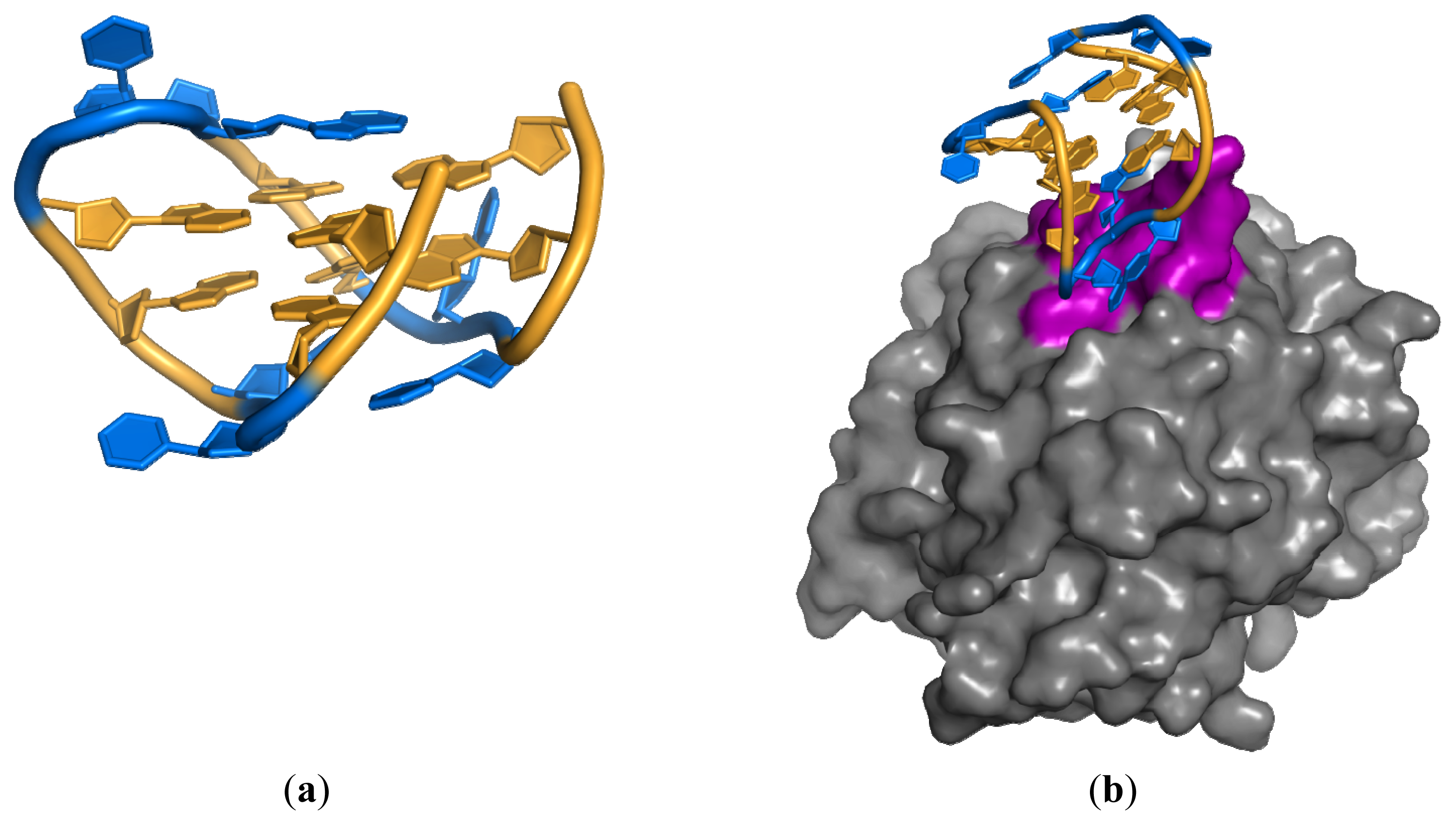
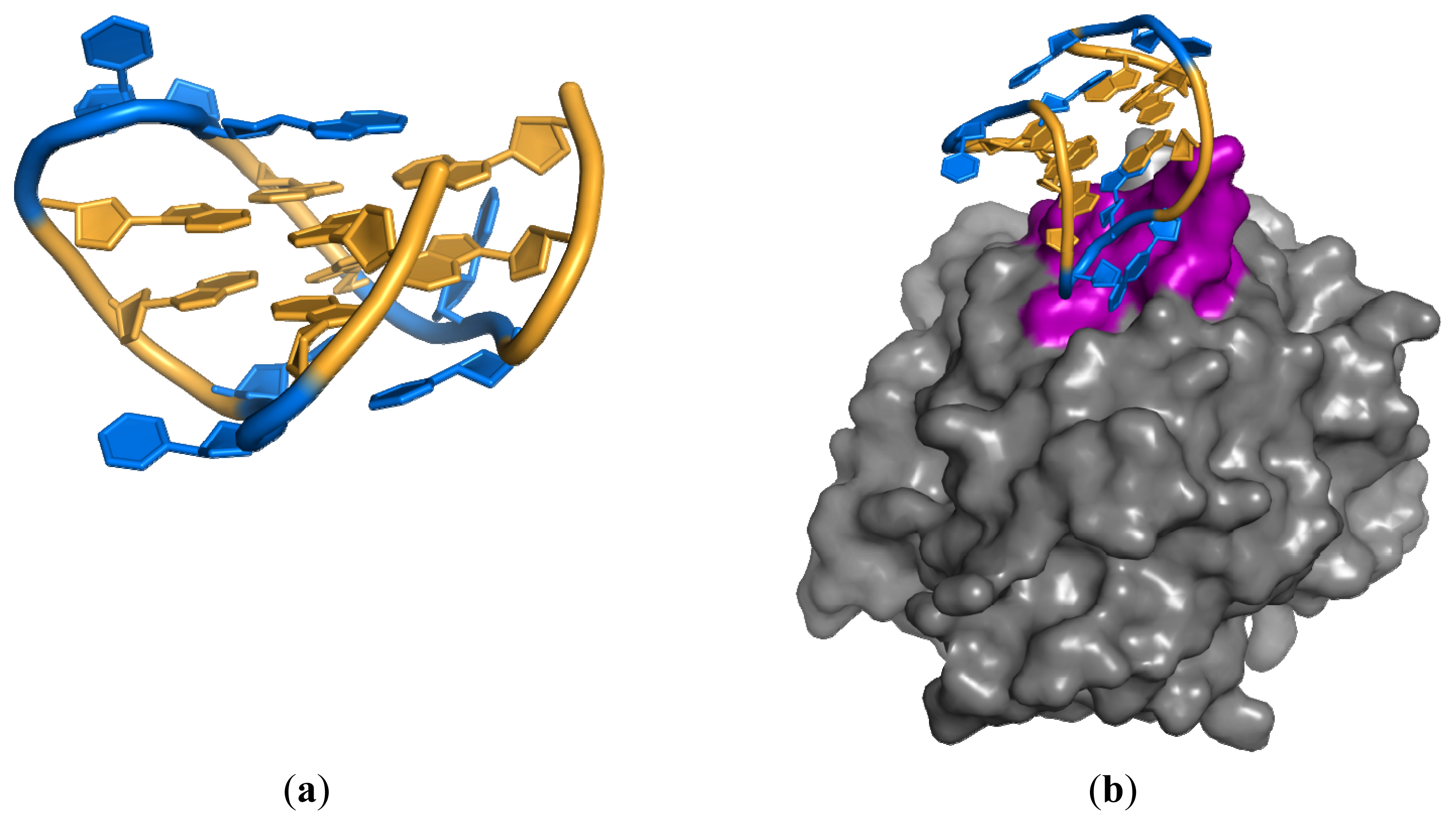
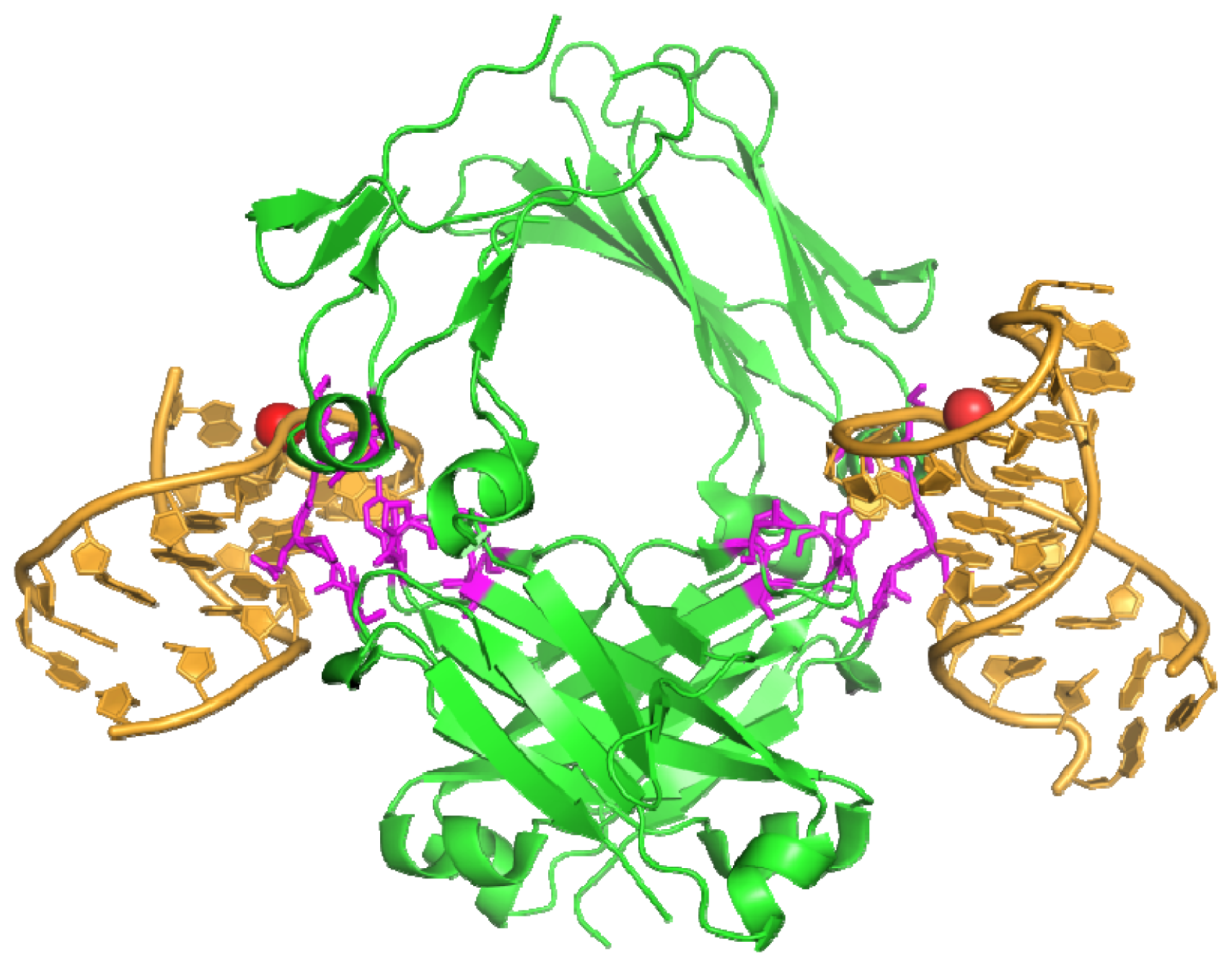
3.2. Anti-IgG Fc RNA Aptamer
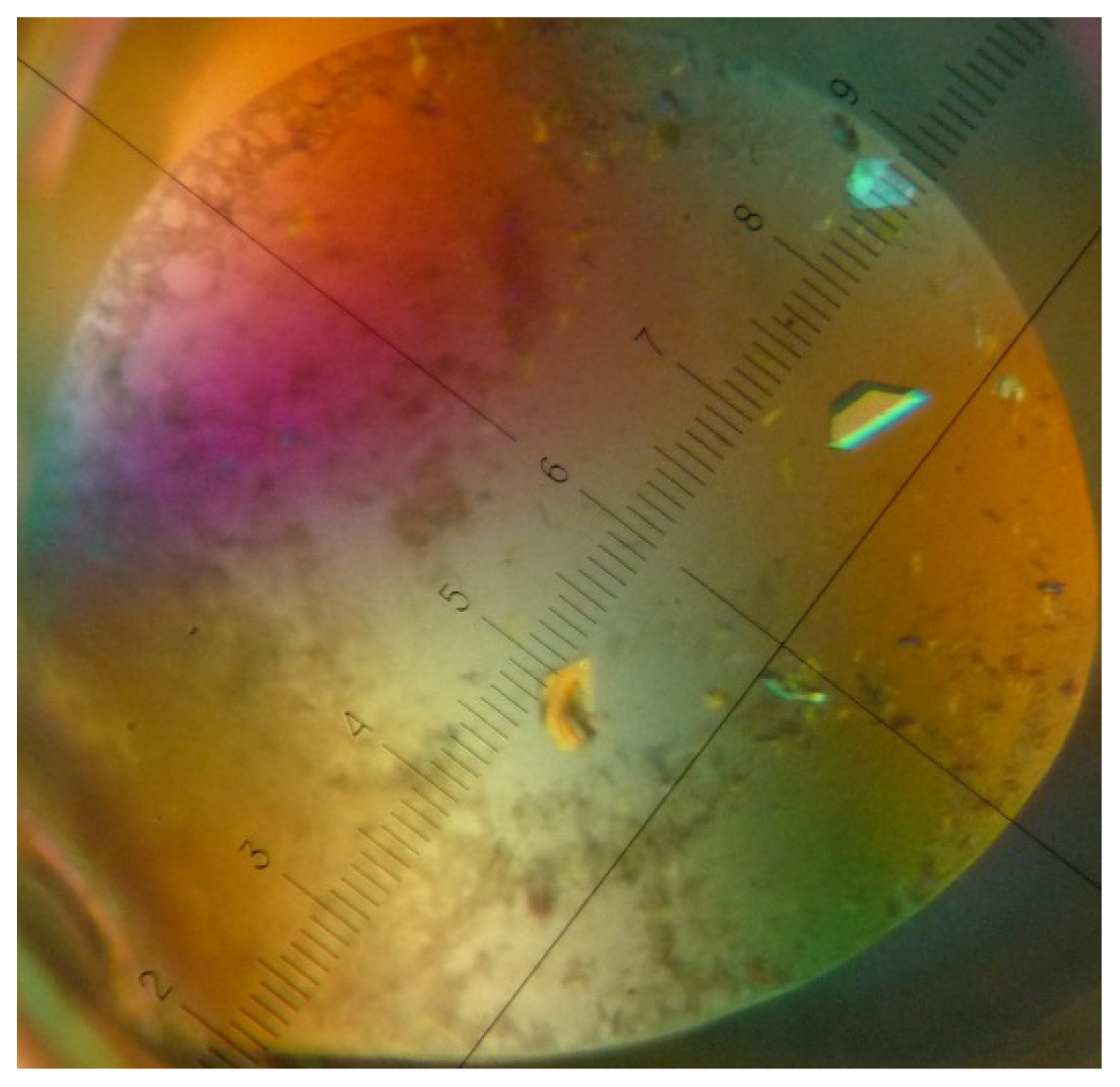
4. Crystallization of a Streptavidin-Aptamer Complex
5. Alternative Methods of Investigating Aptamer Target Interactions
5.1. Computational Approaches
5.2. Experimental Approaches
6. Conclusions and Future Outlook
Supplementary Materials
ijms-13-10537-s001.pdfAcknowledgements
References
- Ellington, A.D.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar]
- Tuerk, C.; Gold, L. Systemic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar]
- Paige, J.S.; Wu, K.Y.; Jaffrey, S.R. RNA mimics of green fluorescent protein. Science 2011, 333, 642–646. [Google Scholar]
- Mehta, J.; van Dorst, B.; Rouah-Martin, E.; Herrebout, W.; Scippo, M.L.; Blust, R.; Robbens, J. In vitro selection and characterization of DNA aptamers recognizing chloramphenicol. J. Biotechnol 2011, 155, 361–369. [Google Scholar]
- Wochner, A.; Menger, M.; Orgel, D.; Cech, B.; Rimmele, M.; Erdmann, V.A.; Glökler, J. A DNA aptamer with high affinity and specificity for therapeutic anthracyclines. Anal. Biochem 2008, 373, 34–42. [Google Scholar]
- Shum, K.T.; Lui, E.L.H.; Wong, S.C.K.; Yeung, P.; Sam, L.; Wang, Y.; Watt, R.M.; Tanner, J.A. Aptamer-mediated inhibition of mycobacterium tuberculosis polyphosphate kinase 2. Biochemistry 2011, 50, 3261–3271. [Google Scholar]
- Stoltenburg, R.; Reinemann, C.; Strehlitz, B. FluMag-SELEX as an advantageous method for DNA aptamer selection. Anal. Bioanal. Chem 2005, 383, 83–91. [Google Scholar]
- Cao, X.X.; Li, S.H.; Chen, L.C.; Ding, H.M.; Xu, H.; Huang, Y.P.; Li, J.; Liu, N.L.; Cao, W.H.; Zhu, Y.J.; et al. Combining use of a panel of ssDNA aptamers in the detection of Staphylococcus aureus. Nucleic Acids Res 2009, 37, 4621–4628. [Google Scholar]
- Lee, Y.J.; Han, S.R.; Maeng, J.S.; Cho, Y.J.; Lee, S.W. In vitro selection of Escherichia coli O157:H7-specific RNA aptamer. Biochem. Biophys. Res. Commun 2012, 417, 414–420. [Google Scholar]
- Mascini, M.; Palchetti, I.; Tombelli, S. Nucleic acid and peptide aptamers: Fundamentals and bioanalytical aspects. Angew. Chem. Int. Ed 2012, 51, 1316–1332. [Google Scholar]
- Bing, T.; Yang, X.; Mei, H.; Cao, Z.; Shangguan, D. Conservative secondary structure motif of streptavidin-binding aptamers generated by different laboratories. Bioorg. Med. Chem 2010, 18, 1798–1805. [Google Scholar]
- Ruigrok, V.J.B.; van Duijn, E.; Barendregt, A.; Dyer, K.; Tainer, J.A.; Stoltenburg, R.; Strehlitz, B.; Levisson, M.; Smidt, H.; van der Oost, J. Kinetic and stoichiometric characterisation of streptavidin-binding aptamers. ChemBioChem 2012, 13, 829–836. [Google Scholar]
- Wilson, C.; Nix, J.; Szostak, J. Functional requirements for specific ligand recognition by a biotin- binding RNA pseudoknot. Biochemistry 1998, 37, 14410–14419. [Google Scholar]
- Kaur, H.; Yung, L.-Y.L. Probing high affinity sequences of DNA aptamer against VEGF 165. PLoS One 2012, 7, e31196. [Google Scholar]
- Lupold, S.E.; Hicke, B.J.; Lin, Y.; Coffey, D.S. Identification and characterization of nuclease-stabilized RNA molecules that bind human prostate cancer cells via the prostate-specific membrane antigen. Cancer Res 2002, 62, 4029–4033. [Google Scholar]
- Hermann, T.; Patel, D.J. Adaptive recognition by nucleic acid aptamers. Science 2000, 287, 820–825. [Google Scholar]
- Hianik, T.; Ostatná, V.; Sonlajtnerova, M.; Grman, I. Influence of ionic strength, pH and aptamer configuration for binding affinity to thrombin. Bioelectrochemistry 2007, 70, 127–133. [Google Scholar]
- Poniková, S.; Tlučková, K.; Antalík, M.; Víglaský, V.; Hianik, T. The circular dichroism and differential scanning calorimetry study of the properties of DNA aptamer dimers. Biophys. Chem 2011, 155, 29–35. [Google Scholar]
- Reinstein, O.; Neves, M.A.D.; Saad, M.; Boodram, S.N.; Lombardo, S.; Beckham, S.A.; Brouwer, J.; Audette, G.F.; Groves, P.; Wilce, M.C.J.; et al. Engineering a structure switching mechanism into a steroid-binding aptamer and hydrodynamic analysis of the ligand binding mechanism. Biochemistry 2011, 50, 9368–9376. [Google Scholar]
- Huang, R.H.; Fremont, D.H.; Diener, J.L.; Schaub, R.G.; Sadler, J.E. A structural explanation for the antithrombotic activity of ARC1172, a DNA aptamer that binds von willebrand factor domain A1. Structure 2009, 17, 1476–1484. [Google Scholar]
- Padmanabhan, K.; Padmanabhan, K.P.; Ferrara, J.D.; Sadler, J.E.; Tulinsky, A. The structure of α-thrombin inhibited by a 15-mer single-stranded DNA aptamer. J. Biol. Chem 1993, 268, 17651–17654. [Google Scholar]
- Padmanabhan, K.; Tulinsky, A. An ambiguous structure of a DNA 15-mer thrombin complex. Acta Crystallogr. Sect. D 1996, 52, 272–282. [Google Scholar]
- Long, S.B.; Long, M.B.; White, R.R.; Sullenger, B.A. Crystal structure of an RNA aptamer bound to thrombin. RNA 2008, 14, 2504–2512. [Google Scholar]
- Krauss, I.R.; Merlino, A.; Giancola, C.; Randazzo, A.; Mazzarella, L.; Sica, F. Thrombin-aptamer recognition: A revealed ambiguity. Nucleic Acids Res 2011, 39, 7858–7867. [Google Scholar]
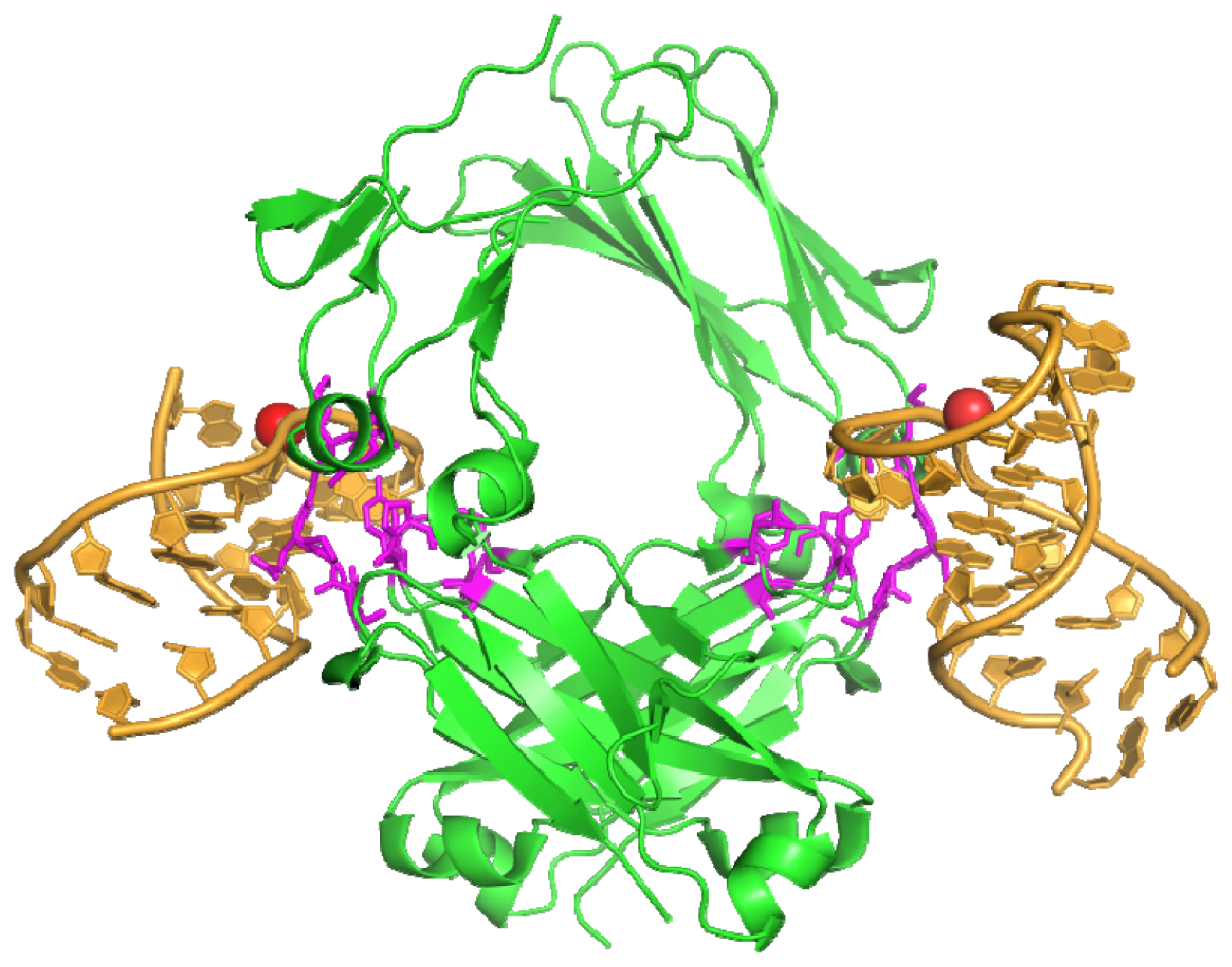
- Huang, D.B.; Vu, D.; Cassiday, L.A.; Zimmerman, J.M.; Maher Iii, L.J.; Ghosh, G. Crystal structure of NF-κb (p50)2 complexed to a high-affinity RNA aptamer. Proc. Natl. Acad. Sci. USA 2003, 100, 9268–9273. [Google Scholar]
- Moorthy, A.K.; Huang, D.B.; Wang, V.Y.F.; Vu, D.; Ghosh, G. X-ray Structure of a NF-κB p50/RelB/DNA complex reveals assembly of multiple dimers on tandem κB sites. J. Mol. Biol 2007, 373, 723–734. [Google Scholar]
- Someya, T.; Baba, S.; Fujimoto, M.; Kawai, G.; Kumasaka, T.; Nakamura, K. Crystal structure of Hfq from Bacillus subtilis in complex with SELEX-derived RNA aptamer: Insight into RNA-binding properties of bacterial Hfq. Nucleic Acids Res 2012, 40, 1856–1867. [Google Scholar]
- Nomura, Y.; Sugiyama, S.; Sakamoto, T.; Miyakawa, S.; Adachi, H.; Takano, K.; Murakami, S.; Inoue, T.; Mori, Y.; Nakamura, Y.; et al. Conformational plasticity of RNA for target recognition as revealed by the 2.15 Å crystal structure of a human IgG-aptamer complex. Nucleic Acids Res 2010, 38, 7822–7829. [Google Scholar]
- Convery, M.A.; Rowsell, S.; Storehouse, N.J.; Ellington, A.D.; Hirao, I.; Murray, J.B.; Peabody, D.S.; Phillips, S.E.V.; Stockley, P.G. Crystal structure of an RNA aptamer-protein complex at 2.8 Å resolution. Nat. Struct. Biol 1998, 5, 133–139. [Google Scholar]
- Rowsell, S.; Stonehouse, N.J.; Convery, M.A.; Adams, C.J.; Ellington, A.D.; Hirao, I.; Peabody, D.S.; Stockley, P.G.; Phillips, S.E.V. Crystal structures of a series of RNA aptamers complexed to the same protein target. Nat. Struct. Biol 1998, 5, 970–975. [Google Scholar]
- Horn, W.T.; Convery, M.A.; Stonehouse, N.J.; Adams, C.J.; Liljas, L.; Phillips, S.E.V.; Stockley, P.G. The crystal structure of a high affinity RNA stem-loop complexed with the bacteriophage MS2 capsid: Further challenges in the modeling of ligand-RNA interactions. RNA 2004, 10, 1776–1782. [Google Scholar]
- Baugh, C.; Grate, D.; Wilson, C. 2.8 Å Crystal structure of the malachite green aptamer. J. Mol. Biol. 2000, 301, 117–128. [Google Scholar]
- Sussman, D.; Nix, J.; Wilson, C. The structural basis for molecular recognition by the vitamin B12 RNA aptamer. Nat. Struct. Biol 2000, 7, 53–57. [Google Scholar]
- Tereshko, V.; Skripkin, E.; Patel, D.J. Encapsulating streptomycin within a small 40-mer RNA. Chem. Biol 2003, 10, 175–187. [Google Scholar]
- Nix, J.; Sussman, D.; Wilson, C. The 1.3 Å crystal structure of a biotin-binding pseudoknot and the basis for RNA molecular recognition. J. Mol. Biol 2000, 296, 1235–1244. [Google Scholar]
- Snyder, D.A.; Chen, Y.; Denissova, N.G.; Acton, T.; Aramini, J.M.; Ciano, M.; Karlin, R.; Liu, J.; Manor, P.; Rajan, P.A.; et al. Comparisons of NMR spectral quality and success in crystallization demonstrate that NMR and X-ray crystallography are complementary methods for small protein structure determination. J. Am. Chem. Soc 2005, 127, 16505–16511. [Google Scholar]
- Yee, A.A.; Savchenko, A.; Ignachenko, A.; Lukin, J.; Xu, X.; Skarina, T.; Evdokimova, E.; Liu, C.S.; Semesi, A.; Guido, V.; et al. NMR and X-ray crystallography, complementary tools in structural proteomics of small proteins. J. Am. Chem. Soc 2005, 127, 16512–16517. [Google Scholar]
- Chayen, N.E.; Saridakis, E. Protein crystallization: From purified protein to diffraction-quality crystal. Nat. Methods 2008, 5, 147–153. [Google Scholar]
- Renault, L.; Chou, H.T.; Chiu, P.L.; Hill, R.M.; Zeng, X.; Gipson, B.; Zhang, Z.Y.; Cheng, A.; Unger, V.; Stahlberg, H. Milestones in electron crystallography. J. Comput. Aided Mol. Des 2006, 20, 519–527. [Google Scholar]
- Hoggan, D.B.; Chao, J.A.; Prasad, G.S.; Stout, C.D.; Williamson, J.R. Combinatorial crystallization of an RNA-protein complex. Acta Crystallogr. Sect. D 2003, 59, 466–473. [Google Scholar]
- Hollis, T. Crystallization of protein-DNA complexes. Methods Mol. Biol 2007, 363, 225–237. [Google Scholar]
- Sugiyama, S.; Nomura, Y.; Sakamoto, T.; Kitatani, T.; Kobayashi, A.; Miyakawa, S.; Takahashi, Y.; Adachi, H.; Takano, K.; Murakami, S.; et al. Crystallization and preliminary X-ray diffraction studies of an RNA aptamer in complex with the human IgG Fc fragment. Acta Crystallogr. Sect. F 2008, 64, 942–944. [Google Scholar]
- Friedmann, D.; Messick, T.; Marmorstein, R. Crystallization of macromolecules. Curr. Protoc. Protein Sci 2011, 66, 17.4.1–17.4.26. [Google Scholar]
- McPherson, A. Introduction to protein crystallization. Methods 2004, 34, 254–265. [Google Scholar]
- Doudna, J.A.; Grosshans, C.; Gooding, A.; Kundrot, C.E. Crystallization of ribozymes and small RNA motifs by a sparse matrix approach. Proc. Natl. Acad. Sci. USA 1993, 90, 7829–7833. [Google Scholar]
- Ke, A.; Doudna, J.A. Crystallization of RNA and RNA-protein complexes. Methods 2004, 34, 408–414. [Google Scholar]
- Garber, M.; Gongadze, G.; Meshcheryakov, V.; Nikonov, O.; Nikulin, A.; Perederina, A.; Piendl, W.; Serganov, A.; Tishchenko, S. Crystallization of RNA/protein complexes. Acta Crystallogr. Sect. D 2002, 58, 1664–1669. [Google Scholar]
- Bock, L.C.; Griffin, L.C.; Latham, J.A.; Vermaas, E.H.; Toole, J.J. Selection of single-stranded DNA molecules that bind and inhibit human thrombin. Nature 1992, 355, 564–566. [Google Scholar]
- Skrzypczak-Jankun, E.; Carperos, V.E.; Ravichandran, K.G.; Tulinsky, A.; Westbrook, M.; Maraganore, J.M. Structure of the hirugen and hirulog 1 complexes of α-thrombin. J. Mol. Biol 1991, 221, 1379–1393. [Google Scholar]
- Macaya, R.F.; Schultze, P.; Smith, F.W.; Roe, J.A.; Feigon, J. Thrombin-binding DNA aptamer forms a unimolecular quadruplex structure in solution. Proc. Natl. Acad. Sci. USA 1993, 90, 3745–3749. [Google Scholar]
- Wang, K.Y.; McCurdy, S.; Shea, R.G.; Swaminathan, S.; Bolton, P.H. A DNA aptamer which binds to and inhibits thrombin exhibits a new structural motif for DNA. Biochemistry 1993, 32, 1899–1904. [Google Scholar]
- Kelly, J.A.; Feigon, J.; Yeates, T.O. Reconciliation of the X-ray and NMR structures of the thrombin-binding aptamer d(GGTTGGTGTGGTTGG). J. Mol. Biol 1996, 256, 417–422. [Google Scholar]
- Miyakawa, S.; Nomura, Y.; Sakamoto, T.; Yamaguchi, Y.; Kato, K.; Yamazaki, S.; Nakamura, Y. Structural and molecular basis for hyperspecificity of RNA aptamer to human immunoglobulin G. RNA 2008, 14, 1154–1163. [Google Scholar]
- Murai, R.; Yoshikawa, H.Y.; Kawahara, H.; Maki, S.; Sugiyama, S.; Kitatani, T.; Adachi, H.; Takano, K.; Matsumura, H.; Murakami, S.; et al. Effect of solution flow produced by rotary shaker on protein crystallization. J. Cryst. Growth 2008, 310, 2168–2172. [Google Scholar]
- Jiang, X.; Egli, M. Use of Chromophoric Ligands to Visually Screen Co-Crystals of Putative Protein-Nucleic Acid Complexes. Curr. Protoc. Nucleic Acid Chem 2011, 7, 7.15.1–7.15.8. [Google Scholar]
- Gold, L.; Janjic, N.; Jarvis, T.; Schneider, D.; Walker, J.J.; Wilcox, S.K.; Zichi, D. Aptamers and the RNA world, past and present. Cold Spring Harb. Perspect. Biol 2012, 4. [Google Scholar] [CrossRef]
- Win, M.N.; Klein, J.S.; Smolke, C.D. Codeine-binding RNA aptamers and rapid determination of their binding constants using a direct coupling surface plasmon resonance assay. Nucleic Acids Res 2006, 34, 5670–5682. [Google Scholar]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 2003, 31, 3406–3415. [Google Scholar]
- Rivas, E.; Eddy, S.R. A dynamic programming algorithm for RNA structure prediction including pseudoknots. J. Mol. Biol 1999, 285, 2053–2068. [Google Scholar]
- Kikin, O.; D’Antonio, L.; Bagga, P.S. QGRS Mapper: A web-based server for predicting G-quadruplexes in nucleotide sequences. Nucleic Acids Res 2006, 34, W676–W682. [Google Scholar]
- Laing, C.; Schlick, T. Computational approaches to RNA structure prediction, analysis, and design. Curr. Opin. Struct. Biol 2011, 21, 306–318. [Google Scholar]
- Parisien, M.; Major, F. The MC-Fold and MC-Sym pipeline infers RNA structure from sequence data. Nature 2008, 452, 51–55. [Google Scholar]
- Sekiya, S.; Noda, K.; Nishikawa, F.; Yokoyama, T.; Kumar, P.K.R.; Nishikawa, S. Characterization and application of a novel RNA aptamer against the mouse prion protein. J. Biochem (Tokyo) 2006, 139, 383–390. [Google Scholar]
- Lebruska, L.L.; Maher Iii, L.J. Selection and characterization of an RNA decoy for transcription factor NF-κB. Biochemistry 1999, 38, 3168–3174. [Google Scholar]
- Hwang, J.; Fauzi, H.; Fukuda, K.; Sekiya, S.; Kakiuchi, N.; Shimotohno, K.; Taira, K.; Kusakabe, I.; Nishikawa, S. The RNA aptamer-binding site of hepatitis C virus NS3 protease. Biochem. Biophys. Res. Commun 2000, 279, 557–562. [Google Scholar]
- Yan, X.; Maier, C.S. Hydrogen/Deuterium Exchange Mass Spectrometry. In Mass Spectrometry of Proteins and Peptides; Lipton, M.S., Paša-Tolic, L., Eds.; Humana Press: New York, NY, USA, 2009; Volume 492, pp. 255–271. [Google Scholar]
- Reinemann, C.; Stoltenburg, R.; Strehlitz, B. Investigations on the specificity of DNA aptamers binding to ethanolamine. Anal. Chem 2009, 81, 3973–3978. [Google Scholar]
- Kwan, A.H.; Mobli, M.; Gooley, P.R.; King, G.F.; MacKay, J.P. Macromolecular NMR spectroscopy for the non-spectroscopist. FEBS J 2011, 278, 687–703. [Google Scholar]
- Orlova, E.V.; Saibil, H.R. Structural analysis of macromolecular assemblies by electron microscopy. Chem. Rev 2011, 111, 7710–7748. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | DNA/RNA | PDB entry code | Ref. |
|---|---|---|---|
| Aptamer-protein complexes | |||
| von Willebrand Factor Domain A1 | DNA | 3HXO 3HXQ | [20] |
| Alpha-thrombin (human) | DNA | 1HUT | [21] |
| Alpha-thrombin (human) | DNA | 1HAO 1HAP | [22] |
| Alpha-thrombin (human) | RNA | 3DD2 | [23] |
| Alpha-thrombin (human) | DNA | 3QLP | [24] |
| NF-κB(p50)2 | RNA | 1OOA | [25] |
| NF-κB P50-RelB | DNA | 2V2T | [26] |
| YmaH (Hfq) | RNA | 3HSB 3AHU | [27] |
| a human IgG | RNA | 3AGV | [28] |
| Enterobacterio phage MS2 coat protein complex | RNA | 6MSF | [29] |
| Enterobacterio phage MS2 | RNA | 5MSF 7MSF | [30] |
| Enterobacterio phage MS2 | RNA | 1U1Y | [31] |
| Aptamer-small molecule complexes | |||
| Malachite green | RNA | 1F1T | [32] |
| Vitamin B12 | RNA | 1ET4 1DDY | [33] |
| Streptomycin | RNA | 1NTB 1NTA | [34] |
| Biotin | RNA | 1F27 | [35] |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ruigrok, V.J.B.; Levisson, M.; Hekelaar, J.; Smidt, H.; Dijkstra, B.W.; Van der Oost, J. Characterization of Aptamer-Protein Complexes by X-ray Crystallography and Alternative Approaches. Int. J. Mol. Sci. 2012, 13, 10537-10552. https://doi.org/10.3390/ijms130810537
Ruigrok VJB, Levisson M, Hekelaar J, Smidt H, Dijkstra BW, Van der Oost J. Characterization of Aptamer-Protein Complexes by X-ray Crystallography and Alternative Approaches. International Journal of Molecular Sciences. 2012; 13(8):10537-10552. https://doi.org/10.3390/ijms130810537
Chicago/Turabian StyleRuigrok, Vincent J. B., Mark Levisson, Johan Hekelaar, Hauke Smidt, Bauke W. Dijkstra, and John Van der Oost. 2012. "Characterization of Aptamer-Protein Complexes by X-ray Crystallography and Alternative Approaches" International Journal of Molecular Sciences 13, no. 8: 10537-10552. https://doi.org/10.3390/ijms130810537