Damaged DNA Binding Protein 2 in Reactive Oxygen Species (ROS) Regulation and Premature Senescence

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Regulation of DDB2
3. Role of DDB2 in NER
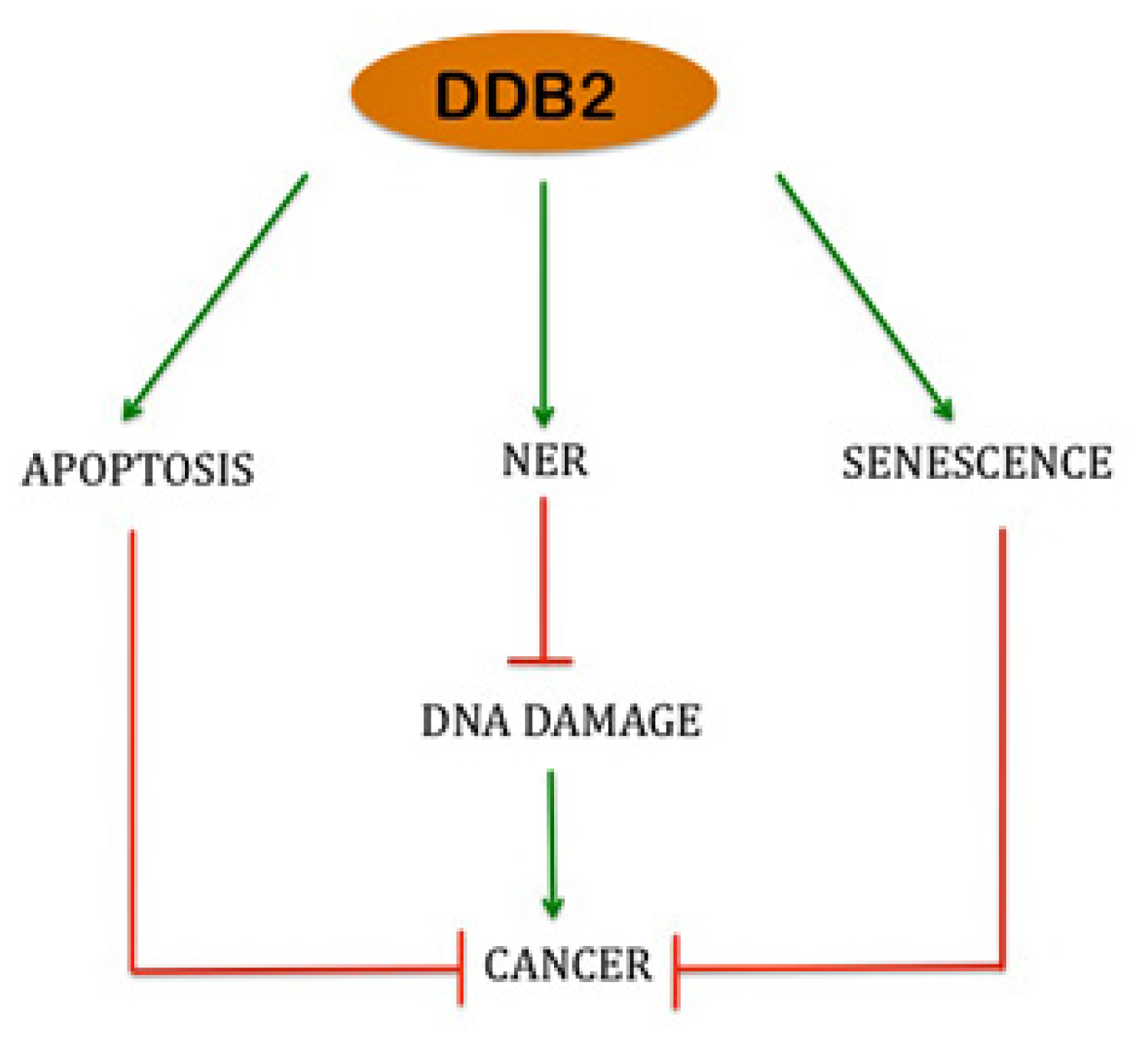
4. Role of DDB2 in Apoptosis
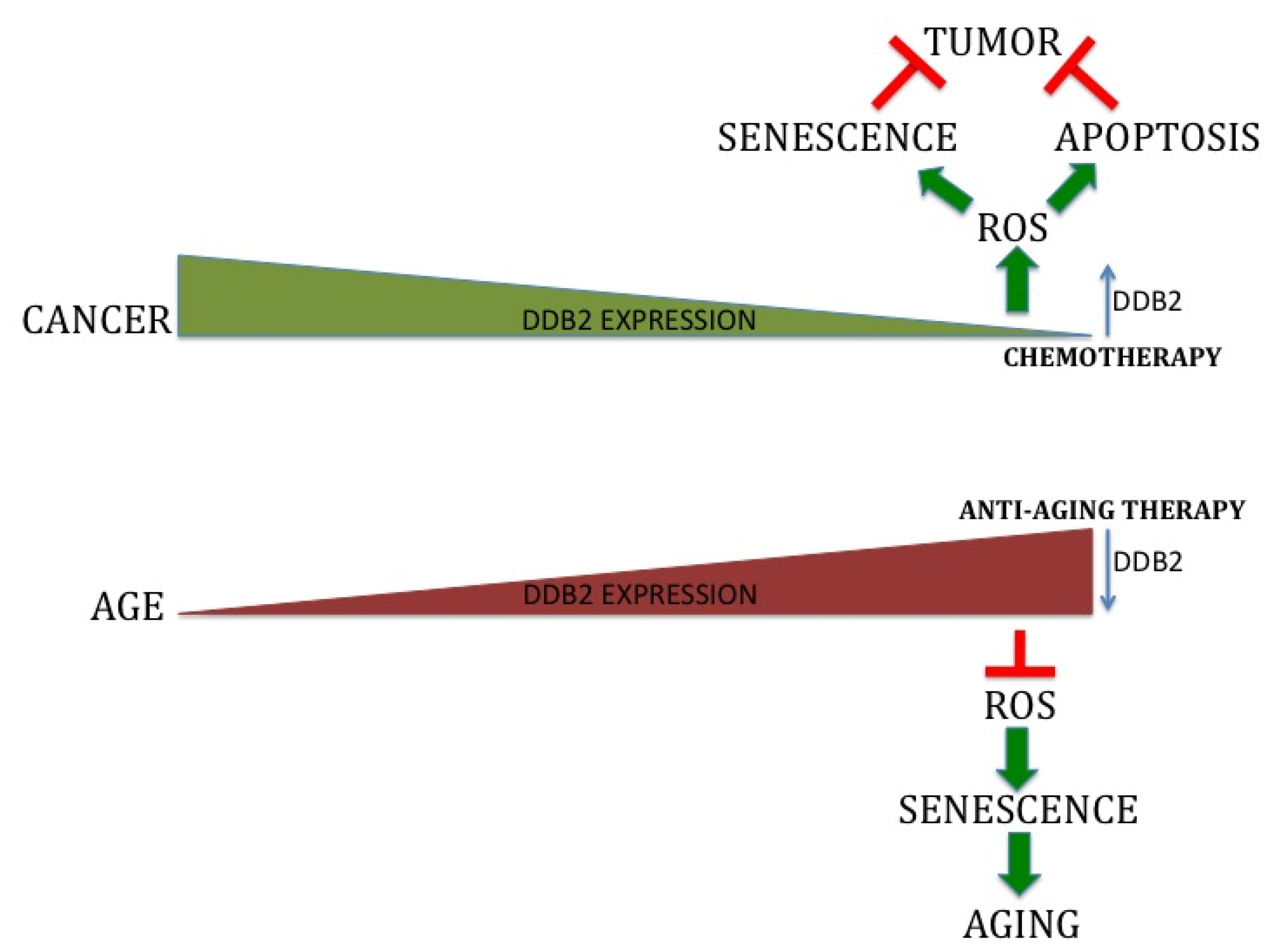
5. Role of DDB2 in Premature Senescence and ROS Regulation
6. Physiological Significance of DDB2 Mediated ROS Regulation
7. Concluding Remarks
Acknowledgement
References
- Tang, J.; Chu, G. Xeroderma pigmentosum complementation group E and UV-damaged DNA-binding protein. DNA Repair 2002, 1, 601–616. [Google Scholar]
- Sancar, A.; Lindsey-Boltz, L.A.; Unsal-Kacmaz, K.; Linn, S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem 2004, 73, 39–85. [Google Scholar]
- Cleaver, J.E. Cancer in xeroderma pigmentosum and related disorders of DNA repair. Nat. Rev. Cancer 2005, 5, 564–573. [Google Scholar]
- Friedberg, E.C. How nucleotide excision repair protects against cancer. Nat. Rev. Cancer 2001, 1, 22–33. [Google Scholar]
- Masutani, C.; Kusumoto, R.; Yamada, A.; Dohmae, N.; Yokoi, M.; Yuasa, M.; Araki, M.; Iwai, S.; Takio, K.; Hanaoka, F. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase eta. Nature 1999, 399, 700–704. [Google Scholar]
- Tang, J.Y.; Hwang, B.J.; Ford, J.M.; Hanawalt, P.C.; Chu, G. Xeroderma pigmentosum p48 gene enhances global genomic repair and suppresses UV-induced mutagenesis. Mol. Cell 2000, 5, 737–744. [Google Scholar]
- Hwang, B.J.; Toering, S.; Francke, U.; Chu, G. p48 Activates a UV-damaged-DNA binding factor and is defective in xeroderma pigmentosum group E cells that lack binding activity. Mol. Cell Biol 1998, 18, 4391–4399. [Google Scholar]
- Keeney, S.; Eker, A.P.; Brody, T.; Vermeulen, W.; Bootsma, D.; Hoeijmakers, J.H.; Linn, S. Correction of the DNA repair defect in xeroderma pigmentosum group E by injection of a DNA damage-binding protein. Proc. Natl. Acad. Sci. USA 1994, 91, 4053–4056. [Google Scholar]
- Itoh, T.; Cado, D.; Kamide, R.; Linn, S. DDB2 gene disruption leads to skin tumors and resistance to apoptosis after exposure to ultraviolet light but not a chemical carcinogen. Proc. Natl. Acad. Sci. USA 2004, 101, 2052–2057. [Google Scholar]
- Yoon, T.; Chakrabortty, A.; Franks, R.; Valli, T.; Kiyokawa, H.; Raychaudhuri, P. Tumor-prone phenotype of the DDB2-deficient mice. Oncogene 2005, 24, 469–478. [Google Scholar]
- Alekseev, S.; Kool, H.; Rebel, H.; Fousteri, M.; Moser, J.; Backendorf, C.; de Gruijl, F.R.; Vrieling, H.; Mullenders, L.H. Enhanced DDB2 expression protects mice from carcinogenic effects of chronic UV-B irradiation. Cancer Res 2005, 65, 10298–12306. [Google Scholar]
- Itoh, T.; Iwashita, S.; Cohen, M.B.; Meyerholz, D.K.; Linn, S. Ddb2 is a haploinsufficient tumor suppressor and controls spontaneous germ cell apoptosis. Hum. Mol. Genet 2007, 16, 1578–1586. [Google Scholar]
- Lee, J.; Zhou, P. DCAFs, the missing link of the CUL4-DDB1 ubiquitin ligase. Mol. Cell 2007, 26, 775–780. [Google Scholar]
- Shiyanov, P.; Nag, A.; Raychaudhuri, P. Cullin 4A associates with the UV-damaged DNA-binding protein DDB. J. Biol. Chem 1999, 274, 35309–35312. [Google Scholar]
- Nag, A.; Bondar, T.; Shiv, S.; Raychaudhuri, P. The xeroderma pigmentosum group E gene product DDB2 is a specific target of cullin 4A in mammalian cells. Mol. Cell Biol 2001, 21, 6738–6747. [Google Scholar]
- Stoyanova, T.; Roy, N.; Kopanja, D.; Bagchi, S.; Raychaudhuri, P. DDB2 decides cell fate following DNA damage. Proc. Natl. Acad. Sci. USA 2009, 106, 10690–10695. [Google Scholar]
- Sugasawa, K.; Okuda, Y.; Saijo, M.; Nishi, R.; Matsuda, N.; Chu, G.; Mori, T.; Iwai, S.; Tanaka, K.; Hanaoka, F. UV-induced ubiquitylation of XPC protein mediated by UV-DDB-ubiquitin ligase complex. Cell 2005, 121, 387–400. [Google Scholar]
- Hwang, B.J.; Ford, J.M.; Hanawalt, P.C.; Chu, G. Expression of the p48 xeroderma pigmentosum gene is p53-dependent and is involved in global genomic repair. Proc. Natl. Acad. Sci. USA 1999, 96, 424–428. [Google Scholar]
- Tan, T.; Chu, G. p53 Binds and activates the xeroderma pigmentosum DDB2 gene in humans but not mice. Mol. Cell Biol 2002, 22, 3247–3254. [Google Scholar]
- Itoh, T.; O’Shea, C.; Linn, S. Impaired regulation of tumor suppressor p53 caused by mutations in the xeroderma pigmentosum DDB2 gene: Mutual regulatory interactions between p48(DDB2) and p53. Mol. Cell Biol 2003, 23, 7540–7553. [Google Scholar]
- Zhao, Q.; Barakat, B.M.; Qin, S.; Ray, A.; El-Mahdy, M.A.; Wani, G.; Arafa, el-S.A.; Mir, S.N.; Wang, Q.E.; Wani, A.A. The p38 mitogen-activated protein kinase augments nucleotide excision repair by mediating DDB2 degradation and chromatin relaxation. J. Biol. Chem. 2008, 283, 32553–32561. [Google Scholar]
- Cong, F.; Tang, J.; Hwang, B.J.; Vuong, B.Q.; Chu, G.; Goff, S.P. Interaction between UV-damaged DNA binding activity proteins and the c-Abl tyrosine kinase. J. Biol. Chem 2002, 277, 34870–34878. [Google Scholar]
- Nichols, A.F.; Itoh, T.; Zolezzi, F.; Hutsell, S.; Linn, S. Basal transcriptional regulation of human damage-specific DNA-binding protein genes DDB1 and DDB2 by Sp1, E2F, N-myc and NF1 elements. Nucleic Acids Res 2003, 31, 562–569. [Google Scholar]
- Groisman, R.; Polanowska, J.; Kuraoka, I.; Sawada, J.; Saijo, M.; Drapkin, R.; Kisselev, A.F.; Tanaka, K.; Nakatani, Y. The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage. Cell 2003, 113, 357–367. [Google Scholar]
- El-Mahdy, M.A.; Zhu, Q.; Wang, Q.E.; Wani, G.; Praetorius-Ibba, M.; Wani, A.A. Cullin 4A-mediated proteolysis of DDB2 protein at DNA damage sites regulates in vivo lesion recognition by XPC. J. Biol. Chem 2006, 281, 13404–13411. [Google Scholar]
- Hanawalt, P.C.; Donahue, B.A.; Sweder, K.S. Repair and transcription: Collision or collusion? Curr. Biol 1994, 4, 518–521. [Google Scholar]
- Mellon, I.; Spivak, G.; Hanawalt, P.C. Selective removal of transcription-blocking DNA damage from the transcribed strand of the mammalian DHFR gene. Cell 1987, 51, 241–249. [Google Scholar]
- De Laat, W.L.; Jaspers, N.G.; Hoeijmakers, J.H. Molecular mechanism of nucleotide excision repair. Genes Dev 1999, 13, 768–785. [Google Scholar]
- Reardon, J.T.; Sancar, A. Recognition and repair of the cyclobutane thymine dimer, a major cause of skin cancers, by the human excision nuclease. Genes Dev 2003, 17, 2539–2551. [Google Scholar]
- Wakasugi, M.; Kawashima, A.; Morioka, H.; Linn, S.; Sancar, A.; Mori, T.; Nikaido, O.; Matsunaga, T. DDB accumulates at DNA damage sites immediately after UV irradiation and directly stimulates nucleotide excision repair. J. Biol. Chem 2002, 277, 1637–1640. [Google Scholar]
- Nag, A.; Datta, A.; Yoo, K.; Bhattacharyya, D.; Chakrabortty, A.; Wang, X.; Slagle, B.L.; Costa, R.H.; Raychaudhuri, P. DDB2 induces nuclear accumulation of the hepatitis B virus X protein independently of binding to DDB1. J. Virol 2001, 75, 10383–10392. [Google Scholar]
- Shiyanov, P.; Hayes, S.A.; Donepudi, M.; Nichols, A.F.; Linn, S.; Slagle, B.L.; Raychaudhuri, P. The naturally occurring mutants of DDB are impaired in stimulating nuclear import of the p125 subunit and E2F1-activated transcription. Mol. Cell Biol 1999, 19, 4935–4943. [Google Scholar]
- Liu, W.; Nichols, A.F.; Graham, J.A.; Dualan, R.; Abbas, A.; Linn, S. Nuclear transport of human DDB protein induced by ultraviolet light. J. Biol. Chem 2000, 275, 21429–21434. [Google Scholar]
- Kapetanaki, M.G.; Guerrero-Santoro, J.; Bisi, D.C.; Hsieh, C.L.; Rapic-Otrin, V.; Levine, A.S. The DDB1-CUL4ADDB2 ubiquitin ligase is deficient in xeroderma pigmentosum group E and targets histone H2A at UV-damaged DNA sites. Proc. Natl. Acad. Sci. USA 2006, 103, 2588–2593. [Google Scholar]
- Wang, H.; Zhai, L.; Xu, J.; Joo, H.Y.; Jackson, S.; Erdjument-Bromage, H.; Tempst, P.; Xiong, Y.; Zhang, Y. Histone H3 and H4 ubiquitylation by the CUL4-DDB-ROC1 ubiquitin ligase facilitates cellular response to DNA damage. Mol. Cell 2006, 22, 383–394. [Google Scholar]
- Datta, A.; Bagchi, S.; Nag, A.; Shiyanov, P.; Adami, G.R.; Yoon, T.; Raychaudhuri, P. The p48 subunit of the damaged-DNA binding protein DDB associates with the CBP/p300 family of histone acetyltransferase. Mutat. Res 2001, 486, 89–97. [Google Scholar]
- Martinez, E.; Palhan, V.B.; Tjernberg, A.; Lymar, E.S.; Gamper, A.M.; Kundu, T.K.; Chait, B.T.; Roeder, R.G. Human STAGA complex is a chromatin-acetylating transcription coactivator that interacts with pre-mRNA splicing and DNA damage-binding factors in vivo. Mol. Cell Biol 2001, 21, 6782–6795. [Google Scholar]
- Luijsterburg, M.S.; Lindh, M.; Acs, K.; Vrouwe, M.G.; Pines, A.; van Attikum, H.; Mullenders, L.H.; Dantuma, N.P. DDB2 promotes chromatin decondensation at UV-induced DNA damage. J. Cell Biol 2012, 197, 267–281. [Google Scholar]
- Abraham, R.T. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev 2001, 15, 2177–2196. [Google Scholar]
- Zhou, B.B.; Elledge, S.J. The DNA damage response: Putting checkpoints in perspective. Nature 2000, 408, 433–439. [Google Scholar]
- Chao, C.; Hergenhahn, M.; Kaeser, M.D.; Wu, Z.; Saito, S.; Iggo, R.; Hollstein, M.; Appella, E.; Xu, Y. Cell type- and promoter-specific roles of Ser18 phosphorylation in regulating p53 responses. J. Biol. Chem 2003, 278, 41028–41033. [Google Scholar]
- Stoyanova, T.; Yoon, T.; Kopanja, D.; Mokyr, M.B.; Raychaudhuri, P. The xeroderma pigmentosum group E gene product DDB2 activates nucleotide excision repair by regulating the level of p21Waf1/Cip1. Mol. Cell Biol 2008, 28, 177–187. [Google Scholar]
- El-Deiry, W.S.; Tokino, T.; Velculescu, V.E.; Levy, D.B.; Parsons, R.; Trent, J.M.; Lin, D.; Mercer, W.E.; Kinzler, K.W.; Vogelstein, B. WAF1, a potential mediator of p53 tumor suppression. Cell 1993, 75, 817–825. [Google Scholar]
- Roy, N.; Stoyanova, T.; Dominguez-Brauer, C.; Park, H.J.; Bagchi, S.; Raychaudhuri, P. DDB2, an essential mediator of premature senescence. Mol. Cell Biol 2010, 30, 2681–2692. [Google Scholar]
- Stoyanova, T.; Roy, N.; Kopanja, D.; Raychaudhuri, P.; Bagchi, S. DDB2 (damaged-DNA binding protein 2) in nucleotide excision repair and DNA damage response. Cell Cycle 2009, 8, 4067–4071. [Google Scholar]
- Gartel, A.L.; Tyner, A.L. The role of the cyclin-dependent kinase inhibitor p21 in apoptosis. Mol. Cancer Ther 2002, 1, 639–649. [Google Scholar]
- Barakat, B.M.; Wang, Q.E.; Han, C.; Milum, K.; Yin, D.T.; Zhao, Q.; Wani, G.; Arafa, el-S.A.; El-Mahdy, M.A.; Wani, A.A. Overexpression of DDB2 enhances the sensitivity of human ovarian cancer cells to cisplatin by augmenting cellular apoptosis. Int. J. Cancer 2010, 127, 977–988. [Google Scholar]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol 2007, 8, 729–740. [Google Scholar]
- Parrinello, S.; Samper, E.; Krtolica, A.; Goldstein, J.; Melov, S.; Campisi, J. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat. Cell Biol 2003, 5, 741–747. [Google Scholar]
- Benhar, M.; Engelberg, D.; Levitzki, A. ROS, stress-activated kinases and stress signaling in cancer. EMBO Rep 2002, 3, 420–425. [Google Scholar]
- Stoyanova, T.; Roy, N.; Bhattacharjee, S.; Kopanja, D.; Valli, T.; Bagchi, S.; Raychaudhuri, P. p21 cooperates with DDB2 protein in suppression of ultraviolet ray-induced skin malignancies. J. Biol. Chem 2012, 287, 3019–3028. [Google Scholar]
- Droge, W. Free radicals in the physiological control of cell function. Physiol. Rev 2002, 82, 47–95. [Google Scholar]
- Li, Y.; Huang, T.T.; Carlson, E.J.; Melov, S.; Ursell, P.C.; Olson, J.L.; Noble, L.J.; Yoshimura, M.P.; Berger, C.; Chan, P.H.; et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat. Genet 1995, 11, 376–381. [Google Scholar]
- Lebovitz, R.M.; Zhang, H.; Vogel, H.; Cartwright, J., Jr; Dionne, L.; Lu, N.; Huang, S.; Matzuk, M.M. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc. Natl. Acad. Sci. USA 1996, 93, 9782–9787. [Google Scholar]
- Ho, Y.S.; Xiong, Y.; Ma, W.; Spector, A.; Ho, D.S. Mice lacking catalase develop normally but show differential sensitivity to oxidant tissue injury. J. Biol. Chem 2004, 279, 32804–32812. [Google Scholar]
- Hayes, S.; Shiyanov, P.; Chen, X.; Raychaudhuri, P. DDB, a putative DNA repair protein, can function as a transcriptional partner of E2F1. Mol. Cell Biol 1998, 18, 240–249. [Google Scholar]
- Kotake, Y.; Zeng, Y.; Xiong, Y. DDB1-CUL4 and MLL1 mediate oncogene-induced p16INK4a activation. Cancer Res 2009, 69, 1809–1814. [Google Scholar]
- Jia, S.; Kobayashi, R.; Grewal, S.I. Ubiquitin ligase component Cul4 associates with Clr4 histone methyltransferase to assemble heterochromatin. Nat. Cell Biol 2005, 7, 1007–1013. [Google Scholar]
- Minig, V.; Kattan, Z.; van Beeumen, J.; Brunner, E.; Becuwe, P. Identification of DDB2 protein as a transcriptional regulator of constitutive SOD2 gene expression in human breast cancer cells. J. Biol. Chem 2009, 284, 14165–14176. [Google Scholar]
- Higa, L.A.; Wu, M.; Ye, T.; Kobayashi, R.; Sun, H.; Zhang, H. CUL4-DDB1 ubiquitin ligase interacts with multiple WD40-repeat proteins and regulates histone methylation. Nat. Cell Biol 2006, 8, 1277–1283. [Google Scholar]
- Barsotti, A.M.; Prives, C. Pro-proliferative FoxM1 is a target of p53-mediated repression. Oncogene 2009, 28, 4295–4305. [Google Scholar]
- Krizhanovsky, V.; Yon, M.; Dickins, R.A.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of activated stellate cells limits liver fibrosis. Cell 2008, 134, 657–667. [Google Scholar]
- Nakamura, S.; Kawai, K.; Takeshita, Y.; Honda, M.; Takamura, T.; Kaneko, S.; Matoba, R.; Matsubara, K. Identification of blood biomarkers of aging by transcript profiling of whole blood. Biochem. Biophys. Res. Commun 2012, 418, 313–318. [Google Scholar]
- Durik, M.; Kavousi, M.; van der Pluijm, I.; Isaacs, A.; Cheng, C.; Verdonk, K.; Loot, A.E.; Oeseburg, H.; Musterd-Bhaggoe, U.; Leijten, F.; et al. Nucleotide excision DNA repair is associated with age-related vascular dysfunction. Circulation 2012, 126, 468–478. [Google Scholar]
- Melquist, S.; Craig, D.W.; Huentelman, M.J.; Crook, R.; Pearson, J.V.; Baker, M.; Zismann, V.L.; Gass, J.; Adamson, J.; Szelinger, S.; et al. Identification of a novel risk locus for progressive supranuclear palsy by a pooled genomewide scan of 500,288 single-nucleotide polymorphisms. Am. J. Hum. Genet 2007, 80, 769–778. [Google Scholar]
- Rodier, F.; Campisi, J. Four faces of cellular senescence. J. Cell Biol 2011, 192, 547–556. [Google Scholar]
- Bagchi, S.; Raychaudhuri, P. Damaged-DNA binding protein-2 drives apoptosis following DNA damage. Cell Div 2010, 5. [Google Scholar] [CrossRef]
- Herman, J.G.; Merlo, A.; Mao, L.; Lapidus, R.G.; Issa, J.P.; Davidson, N.E.; Sidransky, D.; Baylin, S.B. Inactivation of the CDKN2/p16/MTS1 gene is frequently associated with aberrant DNA methylation in all common human cancers. Cancer Res 1995, 55, 4525–4530. [Google Scholar]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov 2009, 8, 579–591. [Google Scholar]
- Trachootham, D.; Zhou, Y.; Zhang, H.; Demizu, Y.; Chen, Z.; Pelicano, H.; Chiao, P.J.; Achanta, G.; Arlinghaus, R.B.; Liu, J.; et al. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell 2006, 10, 241–252. [Google Scholar]
- Wang, L.G.; Chiao, J.W. Prostate cancer chemopreventive activity of phenethyl isothiocyanate through epigenetic regulation (review). Int. J. Oncol 2010, 37, 533–539. [Google Scholar]


© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Roy, N.; Bagchi, S.; Raychaudhuri, P. Damaged DNA Binding Protein 2 in Reactive Oxygen Species (ROS) Regulation and Premature Senescence. Int. J. Mol. Sci. 2012, 13, 11012-11026. https://doi.org/10.3390/ijms130911012
Roy N, Bagchi S, Raychaudhuri P. Damaged DNA Binding Protein 2 in Reactive Oxygen Species (ROS) Regulation and Premature Senescence. International Journal of Molecular Sciences. 2012; 13(9):11012-11026. https://doi.org/10.3390/ijms130911012
Chicago/Turabian StyleRoy, Nilotpal, Srilata Bagchi, and Pradip Raychaudhuri. 2012. "Damaged DNA Binding Protein 2 in Reactive Oxygen Species (ROS) Regulation and Premature Senescence" International Journal of Molecular Sciences 13, no. 9: 11012-11026. https://doi.org/10.3390/ijms130911012