The Heterochromatic Barrier to DNA Double Strand Break Repair: How to Get the Entry Visa

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Overview of the DNA Damage Response and Chromatin Changes that Arise at DNA Double Strand Breaks
3. Evidence that Heterochromatin Influences the Signal Transduction Response at a DSB
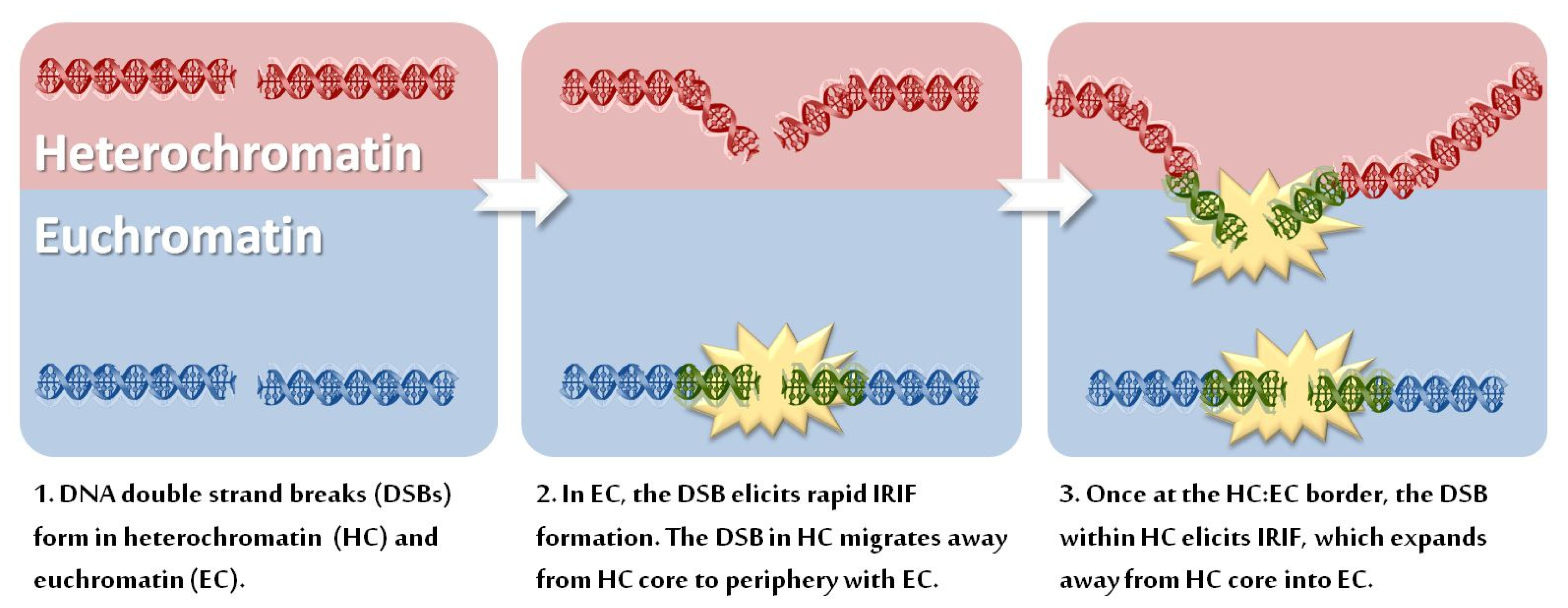
3.1. Movement of DSB-Containing DNA to the Periphery of Chromocentres
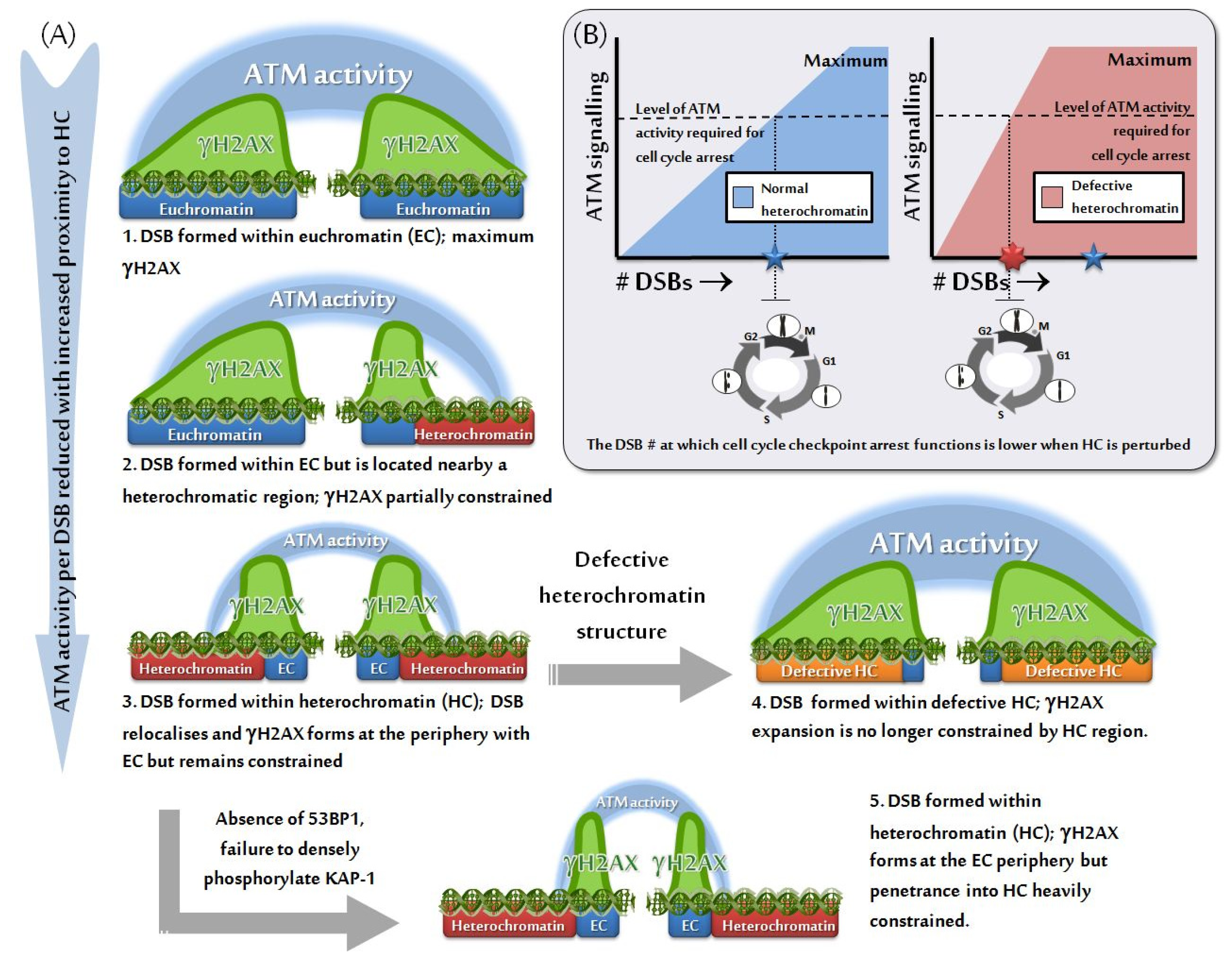
3.2. HC Is also a Barrier to IRIF Expansion
3.3. Can IRIF Formation Be Initiated in HC Regions?
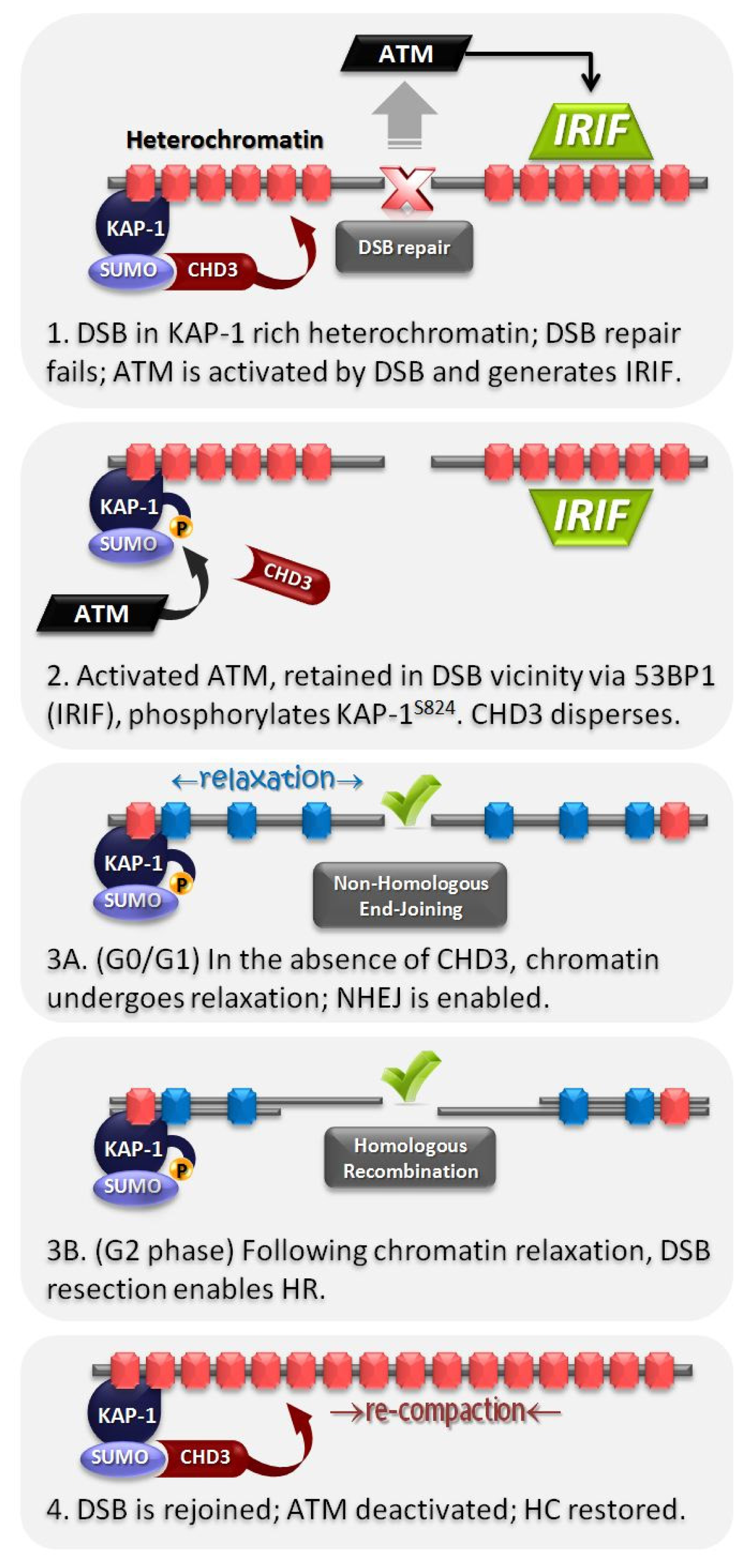
4. HC Is a Barrier to DSB Repair and Is Relieved by ATM-Dependent Signaling to KAP-1
5. Fast and Slow DSB Repair Processes
6. Role of the Mediator Protein, 53BP1, for pKAP-1 Foci Formation and HC-DSB Repair
7. pKAP-1 Foci Formation Promotes Dispersal of the ATP-Dependent Chromatin Remodeling Enzyme, CHD3, from DSB Sites
8. Impact of HC on the Efficacy of Checkpoint Arrest
9. Summary and Future Perspectives
Acknowledgements
References
- Baldeyron, C.; Soria, G.; Roche, D.; Cook, A. J.; Almouzni, G. HP1α recruitment to DNA damage by p150CAF-1 promotes homologous recombination repair. J. Cell Biol 2011, 193, 81–95. [Google Scholar]
- Ayoub, N.; Jeyasekharan, A.D.; Bernal, J.A.; Venkitaraman, A.R. HP1-β mobilization promotes chromatin changes that initiate the DNA damage response. Nature 2008, 453, 682–686. [Google Scholar]
- Ayoub, N.; Jeyasekharan, A.D.; Venkitaraman, A.R. Mobilization and recruitment of HP1: A bimodal response to DNA breakage. Cell Cycle 2009, 8, 2945–2950. [Google Scholar]
- Miller, K.M.; Tjeertes, J.V.; Coates, J.; Legube, G.; Polo, S.E.; Britton, S.; Jackson, S.P. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat. Struct. Mol. Biol 2010, 17, 1144–1151. [Google Scholar]
- Kasparek, T.R.; Humphrey, T.C. DNA double-strand break repair pathways, chromosomal rearrangements and cancer. Semin. Cell Dev. Biol 2011, 22, 886–897. [Google Scholar]
- Wyman, C.; Kanaar, R. DNA double-strand break repair: All’s well that ends well. Annu. Rev. Genet 2006, 40, 363–383. [Google Scholar]
- Lavin, M.F. Ataxia-telangiectasia: From a rare disorder to a paradigm for cell signalling and cancer. Nat. Rev. Mol. Cell Biol 2008, 9, 759–769. [Google Scholar]
- Uziel, T.; Lerenthal, Y.; Moyal, L.; Andegeko, Y.; Mittelman, L.; Shiloh, Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J 2003, 22, 5612–5621. [Google Scholar]
- Al-Hakim, A.; Escribano-Diaz, C.; Landry, M.C.; O’Donnell, L.; Panier, S.; Szilard, R.K.; Durocher, D. The ubiquitous role of ubiquitin in the DNA damage response. DNA Repair 2010, 9, 1229–1240. [Google Scholar]
- Poulsen, M.; Lukas, C.; Lukas, J.; Bekker-Jensen, S.; Mailand, N. Human RNF169 is a negative regulator of the ubiquitin-dependent response to DNA double-strand breaks. J. Cell Biol 2012, 197, 189–199. [Google Scholar]
- Danielsen, J.R.; Povlsen, L.K.; Villumsen, B.H.; Streicher, W.; Nilsson, J.; Wikstrom, M.; Bekker-Jensen, S.; Mailand, N. DNA damage-inducible SUMOylation of HERC2 promotes RNF8 binding via a novel SUMO-binding Zinc finger. J. Cell Biol 2012, 197, 179–187. [Google Scholar]
- Shiloh, Y.; Shema, E.; Moyal, L.; Oren, M. RNF20-RNF40: A ubiquitin-driven link between gene expression and the DNA damage response. FEBS Lett 2011, 585, 2795–2802. [Google Scholar]
- Moyal, L.; Lerenthal, Y.; Gana-Weisz, M.; Mass, G.; So, S.; Wang, S.Y.; Eppink, B.; Chung, Y.M.; Shalev, G.; Shema, E.; et al. Requirement of ATM-dependent monoubiquitylation of histone H2B for timely repair of DNA double-strand breaks. Mol. Cell 2011, 41, 529–542. [Google Scholar]
- Yin, Y.; Seifert, A.; Chua, J.S.; Maure, J.F.; Golebiowski, F.; Hay, R.T. SUMO-targeted ubiquitin E3 ligase RNF4 is required for the response of human cells to DNA damage. Genes Dev 2012, 26, 1196–1208. [Google Scholar]
- Galanty, Y.; Belotserkovskaya, R.; Coates, J.; Jackson, S.P. RNF4, a SUMO-targeted ubiquitin E3 ligase, promotes DNA double-strand break repair. Genes Dev 2012, 26, 1179–1195. [Google Scholar]
- Galanty, Y.; Belotserkovskaya, R.; Coates, J.; Polo, S.; Miller, K.M.; Jackson, S.P. Mammalian SUMO E3-ligases PIAS1 and PIAS4 promote responses to DNA double-strand breaks. Nature 2009, 462, 935–939. [Google Scholar]
- Smits, V.A.; Warmerdam, D.O.; Martin, Y.; Freire, R. Mechanisms of ATR-mediated checkpoint signalling. Front. Biosci 2010, 15, 840–853. [Google Scholar]
- Miller, K.M.; Jackson, S.P. Histone marks: Repairing DNA breaks within the context of chromatin. Biochem. Soc. Trans 2012, 40, 370–376. [Google Scholar]
- Polo, S.E.; Jackson, S.P. Dynamics of DNA damage response proteins at DNA breaks: A focus on protein modifications. Genes Dev 2011, 25, 409–433. [Google Scholar]
- Luijsterburg, M.S.; van Attikum, H. Chromatin and the DNA damage response: The cancer connection. Mol. Oncol 2011, 5, 349–367. [Google Scholar]
- Kruhlak, M.J.; Celeste, A.; Nussenzweig, A. Spatio-temporal dynamics of chromatin containing DNA breaks. Cell Cycle 2006, 5, 1910–1912. [Google Scholar]
- Kruhlak, M.J.; Celeste, A.; Dellaire, G.; Fernandez-Capetillo, O.; Muller, W.G.; McNally, J.G.; Bazett-Jones, D.P.; Nussenzweig, A. Changes in chromatin structure and mobility in living cells at sites of DNA double-strand breaks. J.Cell Biol 2006, 172, 823–834. [Google Scholar]
- Falk, M.; Lukasova, E.; Gabrielova, B.; Ondrej, V.; Kozubek, S. Chromatin dynamics during DSB repair. Biochim. Biophys. Acta 2007, 1773, 1534–1545. [Google Scholar]
- Kim, J.A.; Kruhlak, M.; Dotiwala, F.; Nussenzweig, A.; Haber, J.E. Heterochromatin is refractory to γ-H2AX modification in yeast and mammals. J. Cell Biol 2007, 178, 209–218. [Google Scholar]
- Karagiannis, T.C.; Harikrishnan, K.N.; El-Osta, A. Disparity of histone deacetylase inhibition on repair of radiation-induced DNA damage on euchromatin and constitutive heterochromatin compartments. Oncogene 2007, 26, 3963–3971. [Google Scholar]
- Cowell, I.G.; Sunter, N.J.; Singh, P.B.; Austin, C.A.; Durkacz, B.W.; Tilby, M.J. γH2AX foci form preferentially in euchromatin after ionising-radiation. PLoS ONE 2007, 2, e1057. [Google Scholar]
- Goodarzi, A.A.; Noon, A.T.; Deckbar, D.; Ziv, Y.; Shiloh, Y.; Lobrich, M.; Jeggo, P.A. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol. Cell 2008, 31, 167–177. [Google Scholar]
- Noon, A.T.; Shibata, A.; Rief, N.; Lobrich, M.; Stewart, G.S.; Jeggo, P.A.; Goodarzi, A.A. 53BP1-dependent robust localized KAP-1 phosphorylation is essential for heterochromatic DNA double-strand break repair. Nat. Cell Biol 2010, 12, 177–184. [Google Scholar]
- Jakob, B.; Splinter, J.; Conrad, S.; Voss, K.O.; Zink, D.; Durante, M.; Lobrich, M.; Taucher-Scholz, G. DNA double-strand breaks in heterochromatin elicit fast repair protein recruitment, histone H2AX phosphorylation and relocation to euchromatin. Nucleic Acids Res 2011, 39, 6489–6499. [Google Scholar]
- Chiolo, I.; Minoda, A.; Colmenares, S.U.; Polyzos, A.; Costes, S.V.; Karpen, G.H. Double-strand breaks in heterochromatin move outside of a dynamic HP1α domain to complete recombinational repair. Cell 2011, 144, 732–744. [Google Scholar]
- Falk, M.; Lukasova, E.; Kozubek, S. Chromatin structure influences the sensitivity of DNA to gamma-radiation. Biochim. Biophys. Acta 2008, 1783, 2398–2414. [Google Scholar]
- Falk, M.; Lukasova, E.; Kozubek, S. Higher-order chromatin structure in DSB induction, repair and misrepair. Mutat. Res 2010, 704, 88–100. [Google Scholar]
- Brunton, H.; Goodarzi, A.A.; Noon, A.T.; Shrikhande, A.; Hansen, R.S.; Jeggo, P.A.; Shibata, A. Analysis of human syndromes with disordered chromatin reveals the impact of heterochromatin on the efficacy of ATM-dependent G2/M checkpoint arrest. Mol. Cell Biol 2011, 31, 4022–4035. [Google Scholar]
- Zhang, Y.; Adachi, M.; Zou, H.; Hareyama, M.; Imai, K.; Shinomura, Y. Histone deacetylase inhibitors enhance phosphorylation of histone H2AX after ionizing radiation. Int. J. Radiat. Oncol. Biol. Phys 2006, 65, 859–866. [Google Scholar]
- Murga, M.; Jaco, I.; Fan, Y.; Soria, R.; Martinez-Pastor, B.; Cuadrado, M.; Yang, S.M.; Blasco, M.A.; Skoultchi, A.I.; Fernandez-Capetillo, O. Global chromatin compaction limits the strength of the DNA damage response. J. Cell Biol 2007, 178, 1101–1108. [Google Scholar]
- Costes, S.V.; Chiolo, I.; Pluth, J.M.; Barcellos-Hoff, M.H.; Jakob, B. Spatiotemporal characterization of ionizing radiation induced DNA damage foci and their relation to chromatin organization. Mutat. Res 2010, 704, 78–87. [Google Scholar]
- Riballo, E.; Kuhne, M.; Rief, N.; Doherty, A.; Smith, G.C.; Recio, M.J.; Reis, C.; Dahm, K.; Fricke, A.; Krempler, A.; et al. A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to gamma-H2AX foci. Mol. Cell 2004, 16, 715–724. [Google Scholar]
- Ziv, Y.; Bielopolski, D.; Galanty, Y.; Lukas, C.; Taya, Y.; Schultz, D.C.; Lukas, J.; Bekker-Jensen, S.; Bartek, J.; Shiloh, Y. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat. Cell Biol 2006, 8, 870–876. [Google Scholar]
- White, D.E.; Negorev, D.; Peng, H.; Ivanov, A.V.; Maul, G.G.; Rauscher, F.J., III. KAP1, a novel substrate for PIKK family members, colocalizes with numerous damage response factors at DNA lesions. Cancer Res. 2006, 66, 11594–11599. [Google Scholar]
- Blasius, M.; Forment, J.V.; Thakkar, N.; Wagner, S.A.; Choudhary, C.; Jackson, S.P. A phospho-proteomic screen identifies substrates of the checkpoint kinase Chk1. Genome Biol 2011, 12, R78. [Google Scholar]
- White, D.; Rafalska-Metcalf, I.U.; Ivanov, A.V.; Corsinotti, A.; Peng, H.; Lee, S.C.; Trono, D.; Janicki, S.M.; Rauscher, F.J., III. The ATM substrate KAP1 controls DNA repair in heterochromatin: regulation by HP1 proteins and serine 473/824 phosphorylation. Mol. Cancer Res. 2012, 10, 401–414. [Google Scholar]
- Hu, C.; Zhang, S.; Gao, X.; Xu, X.; Lv, Y.; Zhang, Y.; Zhu, Z.; Zhang, C.; Li, Q.; Wong, J.; et al. Roles of kruppel-associated box (KRAB)-associated co-repressor KAP1 Ser-473 phosphorylation in DNA damage response. J. Biol. Chem 2012, 287, 18937–18952. [Google Scholar]
- Lee, D.H.; Goodarzi, A.A.; Adelmant, G.O.; Pan, Y.; Jeggo, P.A.; Marto, J.A.; Chowdhury, D. Phosphoproteomic analysis reveals that PP4 dephosphorylates KAP-1 impacting the DNA damage response. EMBO J 2012, 31, 2403–2415. [Google Scholar]
- Woodbine, L.; Brunton, H.; Goodarzi, A.A.; Shibata, A.; Jeggo, P.A. Endogenously induced DNA double strand breaks arise in heterochromatic DNA regions and require ataxia telangiectasia mutated and Artemis for their repair. Nucleic Acids Res 2011, 39, 6986–6997. [Google Scholar]
- Beucher, A.; Birraux, J.; Tchouandong, L.; Barton, O.; Shibata, A.; Conrad, S.; Goodarzi, A.A.; Krempler, A.; Jeggo, P. A.; Lobrich, M. ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. EMBO J 2009, 28, 3413–3427. [Google Scholar]
- Shibata, A.; Conrad, S.; Birraux, J.; Geuting, V.; Barton, O.; Ismail, A.; Kakarougkas, A.; Meek, K.; Taucher-Scholz, G.; Lobrich, M.; et al. Factors determining DNA double-strand break repair pathway choice in G2 phase. EMBO J 2011, 30, 1079–1092. [Google Scholar]
- Mallette, F.A.; Mattiroli, F.; Cui, G.; Young, L.C.; Hendzel, M.J.; Mer, G.; Sixma, T.K.; Richard, S. RNF8- and RNF168-dependent degradation of KDM4A/JMJD2A triggers 53BP1 recruitment to DNA damage sites. EMBO J 2012, 31, 1865–1878. [Google Scholar]
- Panier, S.; Durocher, D. Regulatory ubiquitylation in response to DNA double-strand breaks. DNA Repair 2009, 8, 436–443. [Google Scholar]
- Lee, J.H.; Goodarzi, A.A.; Jeggo, P.A.; Paull, T.T. 53BP1 promotes ATM activity through direct interactions with the MRN complex. EMBO J 2010, 29, 574–585. [Google Scholar]
- Sun, Y.; Jiang, X.; Xu, Y.; Ayrapetov, M.K.; Moreau, L.A.; Whetstine, J.R.; Price, B.D. Histone H3 methylation links DNA damage detection to activation of the tumour suppressor Tip60. Nat. Cell Biol 2009, 11, 1376–1382. [Google Scholar]
- Xu, Y.; Sun, Y.; Jiang, X.; Ayrapetov, M.K.; Moskwa, P.; Yang, S.; Weinstock, D.M.; Price, B.D. The p400 ATPase regulates nucleosome stability and chromatin ubiquitination during DNA repair. J. Cell Biol 2010, 191, 31–43. [Google Scholar]
- Jha, S.; Shibata, E.; Dutta, A. Human Rvb1/Tip49 is required for the histone acetyltransferase activity of Tip60/NuA4 and for the downregulation of phosphorylation on H2AX after DNA damage. Mol. Cell Biol. 2008, 28, 2690–2700. [Google Scholar]
- Ivanov, A.V.; Peng, H.; Yurchenko, V.; Yap, K.L.; Negorev, D.G.; Schultz, D.C.; Psulkowski, E.; Fredericks, W.J.; White, D.E.; Maul, G.G.; et al. PHD domain-mediated E3 ligase activity directs intramolecular sumoylation of an adjacent bromodomain required for gene silencing. Mol. Cell 2007, 28, 823–837. [Google Scholar]
- Schultz, D.C.; Friedman, J.R.; Rauscher, F.J., III. Targeting histone deacetylase complexes via KRAB-zinc finger proteins: the PHD and bromodomains of KAP-1 form a cooperative unit that recruits a novel isoform of the Mi-2α subunit of NuRD. Genes Dev. 2001, 15, 428–443. [Google Scholar]
- Li, X.; Lee, Y.K.; Jeng, J.C.; Yen, Y.; Schultz, D.C.; Shih, H.M.; Ann, D.K. Role for KAP1 serine 824 phosphorylation and sumoylation/desumoylation switch in regulating KAP1-mediated transcriptional repression. J. Biol. Chem 2007, 282, 36177–36189. [Google Scholar]
- Goodarzi, A.A.; Kurka, T.; Jeggo, P.A. KAP-1 phosphorylation regulates CHD3 nucleosome remodeling during the DNA double-strand break response. Nat. Struct. Mol. Biol 2011, 18, 831–839. [Google Scholar]
- Deckbar, D.; Birraux, J.; Krempler, A.; Tchouandong, L.; Beucher, A.; Walker, S.; Stiff, T.; Jeggo, P.A.; Lobrich, M. Chromosome breakage after G2 checkpoint release. J. Cell Biol 2007, 176, 748–755. [Google Scholar]
- Fernet, M.; Megnin-Chanet, F.; Hall, J.; Favaudon, V. Control of the G2/M checkpoints after exposure to low doses of ionising radiation: Implications for hyper-radiosensitivity. DNA Repair 2010, 9, 48–57. [Google Scholar]


© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Goodarzi, A.A.; Jeggo, P.A. The Heterochromatic Barrier to DNA Double Strand Break Repair: How to Get the Entry Visa. Int. J. Mol. Sci. 2012, 13, 11844-11860. https://doi.org/10.3390/ijms130911844
Goodarzi AA, Jeggo PA. The Heterochromatic Barrier to DNA Double Strand Break Repair: How to Get the Entry Visa. International Journal of Molecular Sciences. 2012; 13(9):11844-11860. https://doi.org/10.3390/ijms130911844
Chicago/Turabian StyleGoodarzi, Aaron A., and Penny A. Jeggo. 2012. "The Heterochromatic Barrier to DNA Double Strand Break Repair: How to Get the Entry Visa" International Journal of Molecular Sciences 13, no. 9: 11844-11860. https://doi.org/10.3390/ijms130911844