Radical Roles for RAGE in the Pathogenesis of Oxidative Stress in Cardiovascular Diseases and Beyond

{kind=link}
Abstract
:1. RAGE—A Molecule of the Immunoglobulin Superfamily and Links to Oxidative Stress
2. RAGE and Oxidative Stress—First Evidence
3. RAGE and Oxidative Stress: Evidence in Multiple Cell Types
3.1. Cardiac Endothelial Cells
3.2. Vascular Smooth Muscle Cells (SMCs)
3.3. Inflammatory Cells
3.4. Endothelial Progenitor Cells (EPCs)
3.5. Cells of the Nervous System
3.6. Osteoblast Cells
3.7. Renal Cells
3.8. Pancreatic β Cells
4. RAGE and Oxidative Stress: Evidence from in Vivo Models
4.1. Cardiovascular System
4.2. Renal System
4.3. Central Nervous System
4.4. Liver
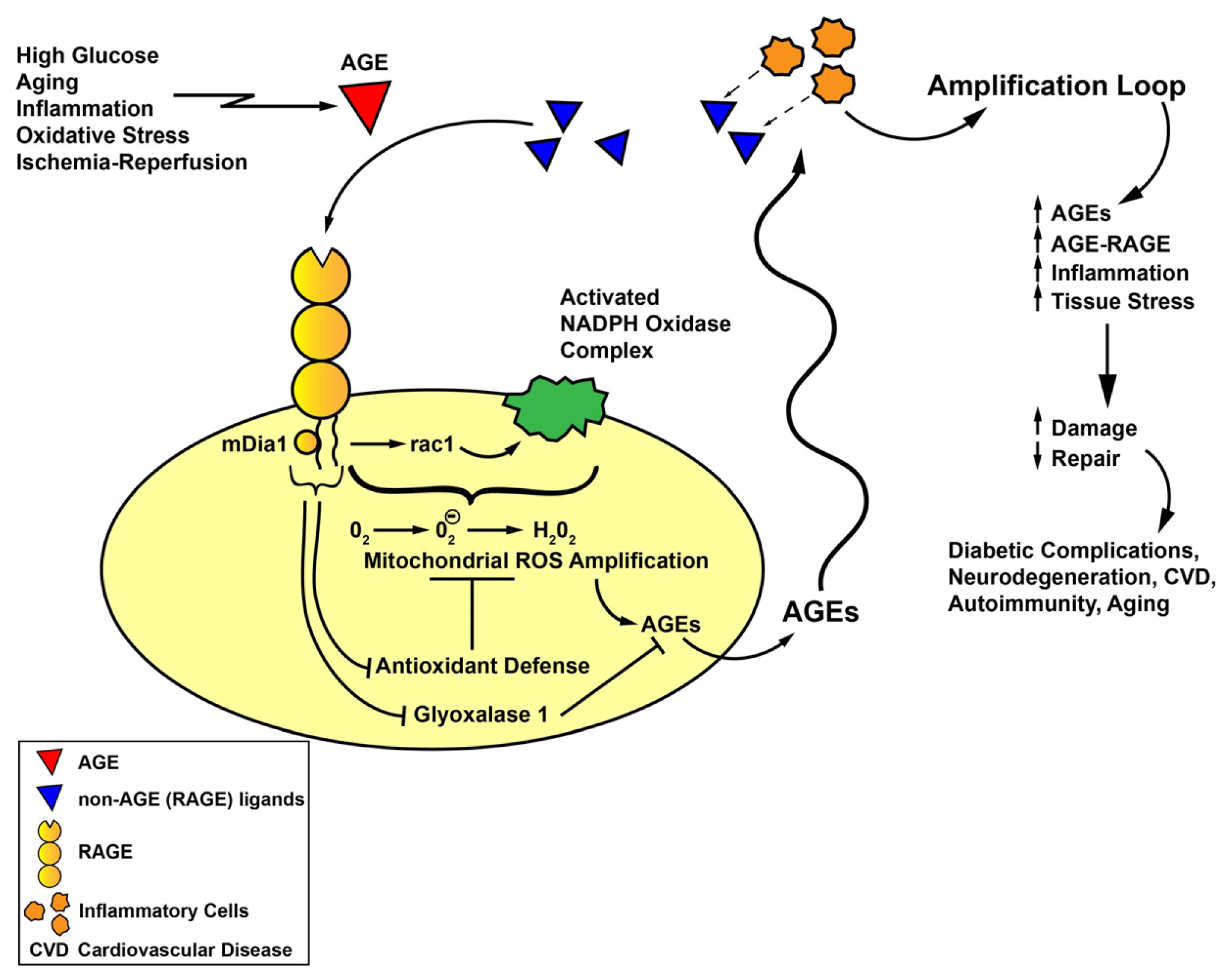
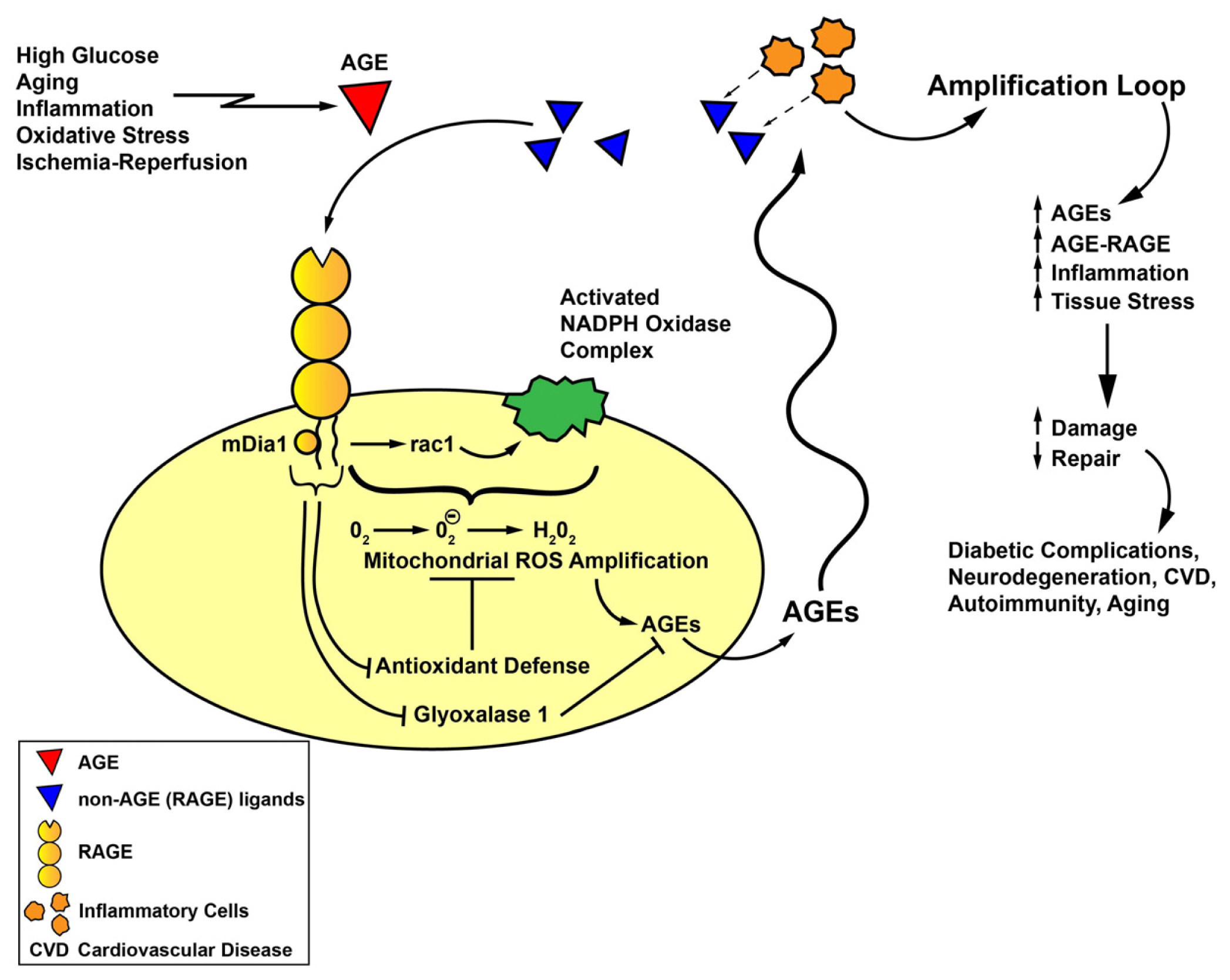
5. RAGE Signaling and Novel Roles for RAGE Cytoplasmic Domain Binding Partner, mDia1, in Oxidative Stress
6. AGE-RAGE and Oxidative Stress: Evidence from Human Subjects
6.1. Cardiometabolic and Renal Disease
6.2. Pulmonary Disease
6.3. Central Nervous System
6.4. Immune/Inflammatory Disorders: Rheumatoid Arthritis
6.5. Disorders of Reproduction: Preeclampsia
7. Summary and Perspectives
Acknowledgments
Conflicts of Interest
References
- Schmidt, A.M.; Vianna, M.; Gerlach, M.; Brett, J.; Ryan, J.; Kao, J.; Esposito, C.; Hegarty, H.; Hurley, W.; Clauss, M.; et al. Isolation and characterization of two binding proteins for advanced glycosylation end products from bovine lung, which are present on the endothelial cell surface. J. Biol. Chem 1992, 267, 14987–14997. [Google Scholar]
- Brett, J.; Schmidt, A.M.; Yan, S.D.; Zou, Y.S.; Weidman, E.; Pinsky, D.; Nowygrod, R.; Neeper, M.; Prysiecki, C.; Shaw, A.; et al. Survey of the distribution of a newly characterized receptor for advanced glycation endproducts of tissues. Am. J. Pathol 1993, 143, 1699–1712. [Google Scholar]
- Yan, S.F.; Ramasamy, R.; Schmidt, A.M. The RAGE axis: A fundamental mechanism signaling danger to the vulnerable vasculature. Circ. Res 2010, 106, 842–853. [Google Scholar]
- Hofmann, M.A.; Drury, S.; Fu, C.; Qu, W.; Taguchi, A.; Lu, Y.; Avila, C.; Kambham, N.; Bierhaus, A.; Nawroth, P.; et al. RAGE mediates a novel proinflammatory axis: A central cell surface receptor for S100/calgranulin polypeptides. Cell 1999, 25, 889–901. [Google Scholar]
- Taguchi, A.; Blood, D.C.; del Toro, G.; Canet, A.; Lee, D.C.; Qu, W.; Tanji, N.; Lu, Y.; Lalla, E.; Fu, C.; et al. Blockade of RAGE-amphoterin signaling suppresses tumor growth and metastases. Nature 2000, 405, 354–360. [Google Scholar]
- Yan, S.D.; Chen, X.; Fu, J.; Chen, M.; Zhu, H.; Roher, A.; Slattery, T.; Zhao, L.; Nagashima, M.; Morser, J.; et al. RAGE and amyloid beta peptide neurotoxicity in Alzheimer’s disease. Nature 1996, 382, 685–691. [Google Scholar]
- Rai, V.; Toure, F.; Chitayat, S.; Pei, R.; Song, F.; Li, Q.; Zhang, J.; Rosario, R.; Ramasamy, R.; Chazin, W.J.; et al. Lysophosphatidic acid targets vascular and oncogenic pathways via RAGE signaling. J. Exp. Med 2012, 209, 2339–2350. [Google Scholar]
- Chavakis, T.; Bierhaus, A.; Al-Fakhri, N.; Schneider, D.; Witte, S.; Linn, T.; Nagashima, M.; Morser, J.; Arnold, B.; Preissner, K.T.; et al. The pattern recognition receptor (RAGE) is a counterreceptor for leukocyte integrins: A novel pathway for inflammatory cell recruitment. J. Exp. Med 2003, 198, 1507–1515. [Google Scholar]
- Ma, W.; Rai, V.; Hudson, B.I.; Song, F.; Schmidt, A.M.; Barile, G.R. RAGE binds C1q and enhances C1q-mediated phagocytosis. Cell. Immunol 2012, 274, 72–82. [Google Scholar]
- Chang, J.S.; Wendt, T.; Qu, W.; Kong, L.; Zou, Y.S.; Schmidt, A.M.; Yan, S.F. Oxygen deprivation triggers upregulation of early growth response 1 by the receptor for advanced glycation end products. Circ. Res 2008, 102, 905–913. [Google Scholar]
- Sohal, R.S.; Allen, R.G. Oxidative stress as a causal factor in differentiation and aging: A unifying hypothesis. Exp. Gerontol 1990, 25, 499–522. [Google Scholar]
- Baynes, J.W. Role of oxidative stress in development of complications in diabetes. Diabetes 1991, 40, 405–412. [Google Scholar]
- Yan, S.D.; Schmidt, A.M.; Anderson, G.M.; Zhang, J.; Brett, J.; Zou, Y.S.; Pinsky, D.; Stern, D. Enhanced cellular oxidant stress by the interaction of advanced glycation end products with their receptors/binding proteins. J. Biol. Chem 1994, 269, 9889–9897. [Google Scholar]
- Wautier, M.P.; Chappey, O.; Corda, S.; Stern, D.M.; Schmidt, A.M.; Wautier, J.L. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am. J. Physiol. Endocrinol. Metab 2001, 280, E685–E694. [Google Scholar]
- Coughlan, M.T.; Thorburn, D.R.; Penfold, S.A.; Laskowski, A.; Harcourt, B.E.; Sourris, K.C.; Tan, A.L.Y.; Fukami, K.; Thallas-Bonke, V.; Nawroth, P.P.; et al. RAGE-induced cytosolic ROS promote mitochondrial superoxide generation in diabetes. J. Am. Soc. Nephrol 2009, 20, 742–752. [Google Scholar]
- Liu, Y.; Ma, Y.; Wang, R.; Xia, C.; Zhang, R.; Lian, K.; Luan, R.; Sun, L.; Yang, L.; Lau, W.B.; et al. Advanced glycation endproducts accelerate ischemia-reperfusion injury through receptor for advanced end product/nitrative thioredoxin inactivation in cardiac microvascular endothelial cells. Antioxid. Redox Signal 2011, 15, 1769–1778. [Google Scholar]
- Reddy, M.A.; Li, S.L.; Sahar, S.; Kim, Y.S.; Xu, Z.G.; Lanting, L.; Natarajan, R. Key role of Src kinase in S100B-induced activation of the receptor for advanced glycation endproducts in vascular smooth muscle cells. J. Biol. Chem 2006, 281, 13685–13693. [Google Scholar]
- Gawdzik, J.; Mathew, L.; Kim, G.; Puri, T.S.; Hofmann Bowman, M.A. Vascular remodeling and arterial calcification are directly mediated by S100A12 (EN-RAGE) in chronic kidney disease. Am. J. Nephrol 2011, 33, 250–259. [Google Scholar]
- Omori, K.; Ohira, T.; Uchida, Y.; Ayilavarapu, S.; Batista, E.L., Jr.; Yagi, M.; Iwata, T.; Liu, H.; Hasturk, H.; Kantarci, A.; et al. Priming of the neutrophil oxidative burst in diabetes requires preassembly of the NADPH oxidase. J. Leukoc. Biol 2008, 84, 292–301. [Google Scholar]
- Suzuki, Y.; Inoue, T.; Yoshimaru, T.; Ra, C. Galectin-3 but not galectin-1 induces mast cell death by oxidative stress and mitochondrial permeability transition. Biochim. Biophys. Acta 2008, 1783, 924–934. [Google Scholar]
- Ruiter, M.S.; van Golde, J.M.; Schaper, N.C.; Stehouwer, C.D.; Huijberts, M.S. Diabetes impairs arteriogenesis in the peripheral circulation: Review of molecular mechanisms. Clin. Sci. (Lond.) 2010, 119, 225–238. [Google Scholar]
- Felice, F.; Barsotti, M.C.; Poredos, P.; Balbarini, A.; di Stefano, R. Effect of aging on metabolic pathways in endothelial progenitor cells. Curr. Pharm. Des 2013, 19, 2351–2365. [Google Scholar]
- Chen, J.; Huang, L.; Song, M.; Yu, S.; Gao, P.; Jing, J. C-reactive protein upregulates receptor for advanced glycation endproducts expression and alters defenses in rat endothelial progenitor cells. J. Cardiovasc. Pharmacol 2009, 53, 359–367. [Google Scholar]
- Chen, J.; Song, M.; Yu, S.; Gao, P.; Yu, Y.; Wang, H.; Huang, L. Advanced glycation endproducts alter functions and promote apoptosis in endothelial progenitor cells through receptor for advanced glycation endproducts mediate overexpression of cell oxidant stress. Mol. Cell. Biochem 2010, 335, 137–146. [Google Scholar]
- Du Yan, S.; Zhu, H.; Fu, J.; Yan, S.F.; Roher, A.; Tourtellotte, W.W.; Rajavashisth, T.; Chen, X.; Godman, G.C.; Stern, D.; et al. Amyloid-beta peptide-receptor for advanced glycation endproduct interaction elicits neuronal expression of macrophage colony stimulating factor: A proinflammatory pathway in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 1997, 94, 5296–5301. [Google Scholar]
- Nitti, M.; d’Abramo, C.; Traverso, N.; Verzola, D.; Garibotto, G.; Poggi, A.; Odetti, P.; Cottalasso, D.; Marinari, U.M.; Pronzato, M.A.; et al. Central role of PKC delta in glycoxidation-dependent apoptosis of human neurons. Free Radic. Biol. Med 2005, 38, 846–856. [Google Scholar]
- Wang, Z.; Li, D.D.; Liang, Y.Y.; Wang, D.S.; Cai, N.S. Activation of astroyctes by advanced glycation endproducts: Cytokine induction and nitric oxide release. Acta Pharmacol. Sin 2002, 23, 974–980. [Google Scholar]
- Vincent, A.M.; Perrone, L.; Sullivan, K.A.; Backus, C.; Sastry, A.M.; Lastoskie, C.; Feldman, E.L. Receptor for advanced glycation endproducts activation injures primary sensory neurons via oxidative stress. Endocrinology 2007, 148, 548–558. [Google Scholar]
- Qin, J.; Goswami, R.; Dawson, S.; Dawson, G. Expression of the receptor for advanced glycation endproducts in oligodendrocytes in response to oxidative stress. J. Neurosci. Res 2008, 86, 2414–2422. [Google Scholar]
- Schurman, L.; McCarthy, A.D.; Sedlinsky, C.; Gangoiti, M.V.; Arnol, V.; Bruzzone, L.; Cortizo, A.M. Metformin reverts deleterious effects of advanced glycation endproducts (AGEs) on osteoblastic cells. Exp. Clin. Endocrinol. Diabetes 2008, 116, 333–340. [Google Scholar]
- Ide, Y.; Matsui, T.; Ishibashi, Y.; Takeuchi, M.; Yamagishi, S. Pigment epithelium derived factor inhibits advanced glycation endproduct elicited mesangial cell damage by blocking NF-κB activation. Microvasc. Res 2010, 80, 227–232. [Google Scholar]
- Chen, S.C.; Guh, J.Y.; Hwang, C.C.; Chiou, S.J.; Lin, T.D.; Ko, Y.M.; Huang, J.S.; Yang, Y.L.; Chuang, L.Y. Advanced glycation endproducts activate extracellular regulated kinase via the oxidative stress EGF receptor pathway in renal fibroblasts. J. Cell. Biochem 2010, 109, 38–48. [Google Scholar]
- Lee, B.W.; Chae, H.Y.; Kwon, S.J.; Park, S.Y.; Ihm, J.; Ihm, S.H. RAGE ligands induced apoptotic cell death of pancreatic-beta cells via oxidative stress. Int. J. Mol. Med 2010, 26, 813–818. [Google Scholar]
- Sun, L.; Ishida, T.; Yasuda, T.; Kojima, Y.; Honjo, T.; Yamamoto, Y.; Yamamoto, H.; Ishibashi, S.; Hirata, K.; Hayashi, Y. RAGE mediates oxidized LDL induced pro-inflammatory effects and atherosclerosis in non-diabetic LDL receptor deficient mice. Cardiovasc. Res 2009, 82, 371–381. [Google Scholar]
- Hofmann-Bowman, M.; Wilk, J.; Heydemann, A.; Kim, G.; Rehman, J.; Lodato, J.A.; Raman, J.; McNally, E.M. S100A12 mediates aortic wall remodeling and aortic aneurysm. Circ. Res 2010, 106, 145–154. [Google Scholar]
- Tikellis, C.; Thomas, M.C..; Harcourt, B.E.; Coughlan, M.T.; Pete, J.; Bialkowsi, K.; Tan, A.; Bierhaus, A.; Cooper, M.E.; Forbes, J.M. Cardiac inflammation associated with a Western diet is mediated via activation of RAGE by AGEs. Am. J. Physiol. Endocrinol. Metab 2008, 295, E323–E330. [Google Scholar]
- Bucciarelli, L.G.; Kaneko, M.; Ananthakrishnan, R.; Harja, E.; Lee, L.K.; Hwang, Y.; Lerner, S.; Bakr, S.; Li, Q.; Lu, Y.; et al. Receptor for advanced glycation endproducts: Key modulator of myocardial ischemic injury. Circulation 2006, 113, 1226–1234. [Google Scholar]
- Bucciarelli, L.G.; Ananthakrishnan, R.; Hwang, Y.C.; Kaneko, M.; Song, F.; Sell, D.R.; Strauch, C.; Monnier, V.M.; Yan, S.F.; Schmidt, A.M.; et al. RAGE and modulation of ischemic injury in the diabetic myocardium. Diabetes 2008, 57, 1941–1951. [Google Scholar]
- Loor, G.; Kondapalli, J.; Iwase, H.; Chandel, N.S.; Waypa, G.B.; Guzy, R.D.; vanden Hoek, T.L.; Schumacker, P.T. Mitochondrial oxidative stress triggers cell death in simulated ischemia-reperfusion. Biochim. Biophys. Acta 2011, 1813, 1382–1394. [Google Scholar]
- Di Lisa, F.; Menabo, R.; Canton, M.; Barile, M.; Bernardi, P. Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J. Biol. Chem 2001, 276, 2571–2575. [Google Scholar]
- Aleshin, A.; Ananthakrishnan, R.; Li, Q.; Rosario, R.; Lu, Y.; Qu, W.; Song, F.; Bakr, S.; Szabolcs, M.; D’Agati, V.; et al. RAGE modulates myocardial injury consequent to LAD infarction via impact on JNK and STAT signaling in a murine model. Am. J. Physiol. Heart Circ. Physiol 2008, 294, H1823–H1832. [Google Scholar]
- Tsoporis, J.N.; Izhar, S.; Leong-Poi, H.; Desjardins, J.F.; Huttunen, H.J.; Parker, T.G. S100B interaction with receptor for advanced glycation endproducts (RAGE): A novel receptor-mediated mechanism for myocyte apoptosis postinfarction. Circ. Res 2010, 106, 93–101. [Google Scholar]
- Inagi, R.; Yamamoto, Y.; Nangaku, M.; Usuda, N.; Okamato, H.; Kurokawa, K.; van ypersele de Strihou, C.; Yamamoto, H.; Miyata, T. A severe diabetic nephropathy model with early development of nodule-like lesions induced by megsin overexpression in RAGE/iNOS transgenic mice. Diabetes 2006, 55, 356–266. [Google Scholar]
- Tomino, Y.; Hagiwara, S.; Gohda, T. AGE-RAGE interaction and oxidative stress in obesity related renal dysfunction. Kidney Int 2011, 80, 133–135. [Google Scholar]
- Guo, J.; Ananthakrishnan, R.; Qu, W.; Lu, Y.; Reiniger, N.; Zeng, S.; Ma, W.; Rosario, R.; Yan, S.F.; Ramasamy, R.; et al. RAGE mediates podocyte injury in adriamycin-induced glomerulosclerosis. J. Am. Soc. Nephrol 2008, 19, 961–972. [Google Scholar]
- De Oliveira, M.R.; Oliveira, M.W.; Behr, G.A.; de Bittencourt Pasquali, M.A.; Moreira, J.C. Increased receptor for advanced glycation endproducts immunocontent in the cerebral cortex of vitamin A-treated rats. Neurochem. Res 2009, 34, 1410–1416. [Google Scholar]
- Kuhla, A.; Hettwer, C.; Menger, M.D.; Vollmar, B. Oxidative stress associated rise of hepatic protein glycation increases inflammatory liver injury in uncoupling protein 2 deficient mice. Lab. Invest 2010, 90, 1189–1198. [Google Scholar]
- Rabbani, N.; Thornalley, P.J. Glyoxalase in diabetes, obesity and related disorders. Semin. Cell Dev. Biol 2011, 22, 309–317. [Google Scholar]
- Ekong, U.; Zeng, S.; Dun, H.; Feirt, N.; Guo, J.; Ippagunta, N.; Guarrera, J.V.; Lu, Y.; Weinberg, A.; Qu, W.; et al. Blockade of the receptor for advanced glycation endproducts attenuates acetaminophen toxicity in mice. Gastroenterol. Hepatol 2006, 21, 682–688. [Google Scholar]
- Hudson, B.I.; Kalea, A.Z.; del Mar Arriero, M.; Harja, E.; Boulanger, E.; D’Agati, V.; Schmidt, A.M. Interaction of the RAGE cytoplasmic domain with diaphanous1 is required for ligand-stimulated cellular migration through activation of Rac1 and Cdc42. J. Biol. Chem 2008, 283, 34457–34468. [Google Scholar]
- Xu, Y.; Toure, F.; Qu, W.; Lin, L.; Song, F.; Shen, X.; Rosario, R.; Garcia, J.; Schmidt, A.M.; Yan, S.F. Advanced glycation endproduct (AGE) receptor for AGE (RAGE) signaling and upregulation of Egr-1 in hypoxic macrophages. J. Biol. Chem 2010, 285, 23233–23240. [Google Scholar]
- Toure, F.; Fritz, G.; Li, Q.; Rai, V.; Daffu, G.; Zou, Y.S.; Rosario, R.; Ramasamy, R.; Alberts, A.S.; Yan, S.F.; et al. Formin mDia1 mediates vascular remodeling via integration of oxidative and signal transduction pathways. Circ. Res 2012, 110, 1279–1293. [Google Scholar]
- Young, K.G.; Copeland, J.W. Formins and cell signaling. Biochim. Biophys. Acta 2010, 1803, 183–190. [Google Scholar]
- Rodriguez-Ayala, E.; Anderstam, B.; Suliman, M.E.; Seeberger, A.; Heimburger, O.; Lindholm, B.; Stenvinkel, P. Enhanced RAGE-mediated NFKB stimulation in inflamed hemodialysis patients. Atherosclerosis 2005, 180, 333–340. [Google Scholar]
- Raucci, A.; Cugusi, S.; Antonelli, A.; Barabino, S.M.; Monti, L.; Bierhaus, A.; Reiss, K.; Saftig, P.; Bianchi, M.E. A soluble form of the receptor for advanced glycation endproducts (RAGE) is produced by proteolytic cleavage of the membrane-bound form by the sheddase a disintegrin and metalloprotease 10 (ADAM10). FASEB J 2008, 22, 3716–3727. [Google Scholar]
- Zhang, L.; Bukulin, M.; Kojro, E.; Roth, A.; Metz, V.V.; Fahrenholz, F.; Nawroth, P.P.; Bierhaus, A.; Postina, R. Receptor for advanced glycation endproducts is subjected to protein ectodomain shedding by metalloproteinases. J. Biol. Chem 2008, 283, 35507–35516. [Google Scholar]
- Park, I.H.; Yeon, S.I.; Youn, J.H.; Choi, J.E.; Sasaki, N.; Choi, I.H.; Shin, J.S. Expression of a novel secreted splice variant of the receptor for advanced glycation end products (RAGE) in human brain astrocytes and peripheral blood mononuclear cells. Mol. Immunol 2004, 40, 1203–1211. [Google Scholar]
- Cheng, C.; Tsuneyama, K.; Kominami, R.; Shinohara, H.; Sakurai, S.; Yonekura, H.; Watanabe, T.; Takano, Y.; Yamamoto, H.; Yamamoto, Y. Expression profiling of endogenous secretory receptor for advanced glycation endproducts in human organs. Mod. Pathol 2005, 18, 1385–1396. [Google Scholar]
- Santilli, F.; Bucciarelli, L.; Noto, D.; Cefalu, A.B.; Davi, V.; Ferrante, E.; Pettinella, C.; Averna, M.R.; Ciabattoni, G.; Davi, G. Decreased plasma soluble RAGE in patients with hypercholesterolemia: Effects of statins. Free Radic. Biol. Med 2007, 43, 1255–1262. [Google Scholar]
- Devangelio, E.; Santilli, F.; Formoso, G.; Ferroni, P.; Bucciarelli, L.; Michetti, N.; Clissa, C.; Ciabattoni, G.; Consoli, A.; Davi, G. Soluble RAGE in type 2 diabetes: Association with oxidative stress. Free Radic. Biol. Med 2007, 43, 511–518. [Google Scholar]
- Rodino-Janeiro, B.K.; Salgado-Somoza, A.; Teijeira-Fernandez, E.; Gonzalez-Juanatey, J.R.; Alvarez, E.; Eiras, S. Receptor for advanced glycation endproducts expression in subcutaneous adipose tissue is related to coronary artery disease. Eur. J. Endocrinol 2011, 164, 529–537. [Google Scholar]
- Inghilleri, S.; Morbini, P.; Campo, I.; Zorzetto, M.; Oggionni, T.; Pozzi, E.; Luisetti, M. Factors influencing oxidative imbalance in pulmonary fibrosis: An immunohistochemical study. Pulm. Med 2011, 2011, 421409:1–421409:10. [Google Scholar]
- Dalfo, E.; Portero-Otin, M.; Ayala, V.; Martinez, A.; Pamplona, R.; Ferrer, I. Evidence of oxidative stress in the neocortex in incidental Lewy body disease. J. Neuropathol. Exp. Neurol 2005, 64, 816–830. [Google Scholar]
- Freixes, M.; Rodriguez, A.; Dalfo, E.; Ferrer, I. Oxidation, glycoxidation, lipoxidation, nitration and responses to oxidative stress in the cerebral cortex in Creutzfeld-Jakob disease. Neurobiol. Aging 2006, 27, 1807–1815. [Google Scholar]
- Ferrante, E.; Vazzana, N.; Santilli, F.; di Cicco, M.; Lauriti, C.; di Battista, L.; Ciabattoni, G.; di Matteo, L.; Davi, G. Determinants of thromboxane biosynthesis in rheumatoid arthritis: Role of RAGE and oxidant stress. Free Radic. Biol. Med 2010, 49, 857–864. [Google Scholar]
- Chekir, C.; Nakatsuka, M.; Noguchi, S.; Konishi, H.; Kamada, Y.; Sasaki, A.; Hao, L.; Hiramatsu, Y. Accumulation of advanced glycation endproducts in women with preeclampsia: Possible involvement of placental oxidative stress and nitrative stress. Placenta 2006, 27, 225–233. [Google Scholar]
- Anderson, M.M.; Heinecke, J.W. Production of Nepsilon-(carboxymethyl)lysine is impaired in mice deficient in NADPH oxidase: A role for phagocyte derived oxidants in the formation of advanced glycation endproducts during inflammation. Diabetes 2003, 52, 2137–2143. [Google Scholar]
- Reiniger, N.; Lau, K.; McCalla, D.; Eby, B.; Cheng, B.; Lu, Y.; Qu, W.; Quadri, N.; Ananthakrishnan, R.; Furmansky, M.; et al. Deletion of the receptor for advanced glycation endproducts reduces glomerulosclerosis and preserves renal function in the diabetic OVE26 mice. Diabetes 2010, 59, 2043–2054. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Daffu, G.; Del Pozo, C.H.; O'Shea, K.M.; Ananthakrishnan, R.; Ramasamy, R.; Schmidt, A.M. Radical Roles for RAGE in the Pathogenesis of Oxidative Stress in Cardiovascular Diseases and Beyond. Int. J. Mol. Sci. 2013, 14, 19891-19910. https://doi.org/10.3390/ijms141019891
Daffu G, Del Pozo CH, O'Shea KM, Ananthakrishnan R, Ramasamy R, Schmidt AM. Radical Roles for RAGE in the Pathogenesis of Oxidative Stress in Cardiovascular Diseases and Beyond. International Journal of Molecular Sciences. 2013; 14(10):19891-19910. https://doi.org/10.3390/ijms141019891
Chicago/Turabian StyleDaffu, Gurdip, Carmen Hurtado Del Pozo, Karen M. O'Shea, Radha Ananthakrishnan, Ravichandran Ramasamy, and Ann Marie Schmidt. 2013. "Radical Roles for RAGE in the Pathogenesis of Oxidative Stress in Cardiovascular Diseases and Beyond" International Journal of Molecular Sciences 14, no. 10: 19891-19910. https://doi.org/10.3390/ijms141019891