Proteomic Analysis Identifies an NADPH Oxidase 1 (Nox1)-Mediated Role for Actin-Related Protein 2/3 Complex Subunit 2 (ARPC2) in Promoting Smooth Muscle Cell Migration

Abstract
:1. Introduction
2. Results and Discussion
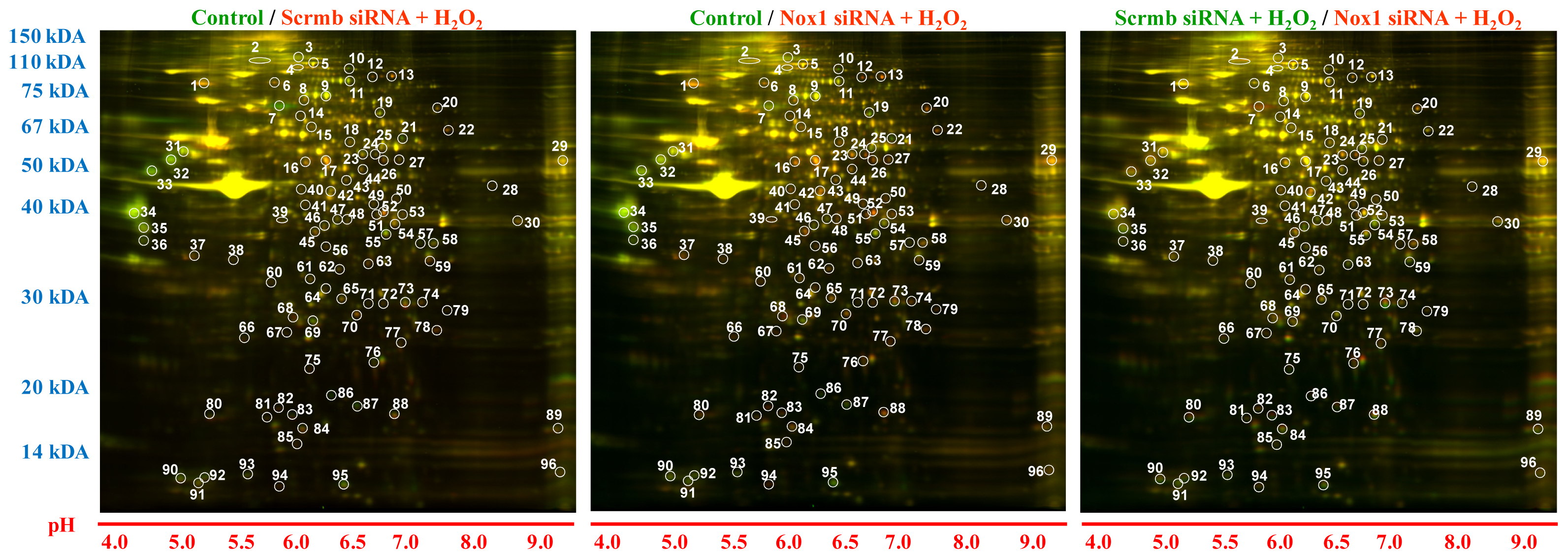
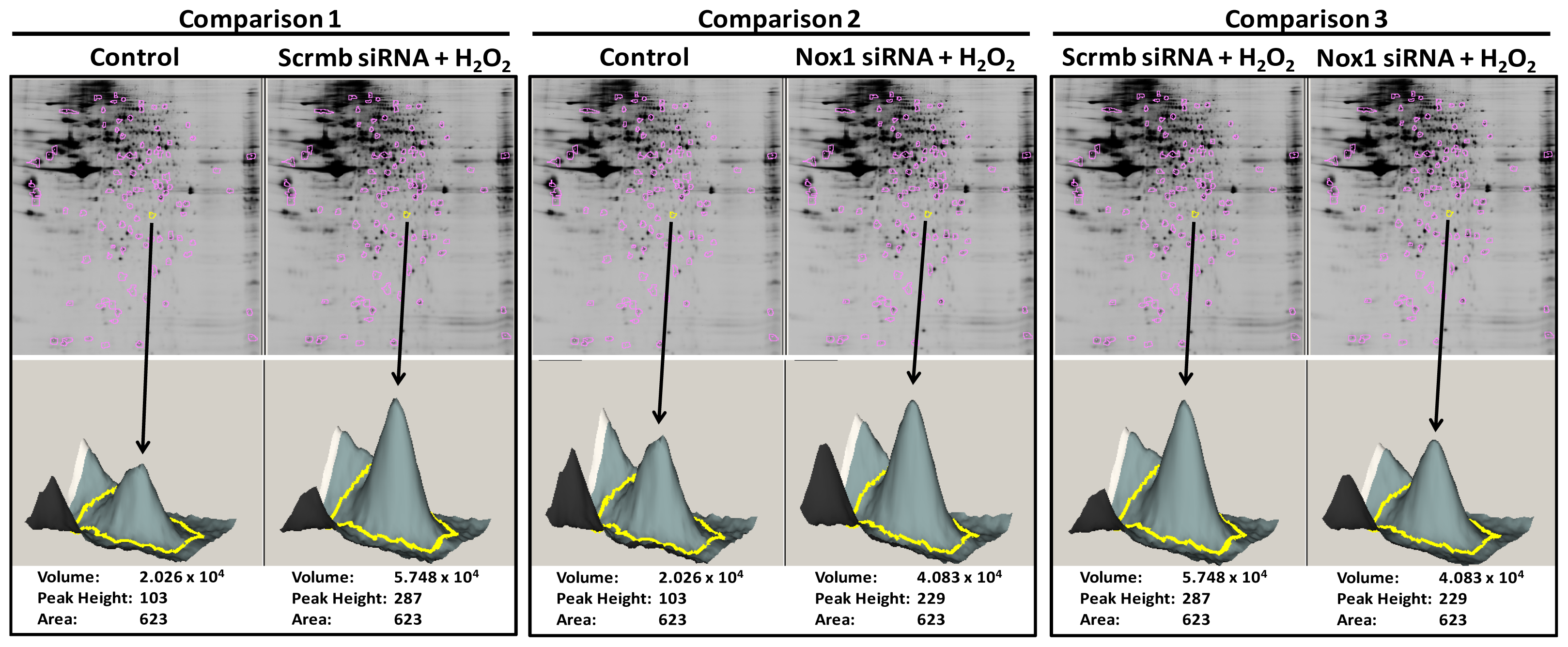
2.1. 2D-Gel Proteomic Analysis Identifies Novel Proteins Downstream of Nox1 Activation in VSMC
- (a)
- Comparison 1: “Scrmb siRNA + H2O2”/“control” ≥ 1.3-fold or ≤ 0.7-fold
- (b)
- Comparisons 1 & 2: (“Scrmb siRNA + H2O2”/“control”) − (“Nox1 siRNA + H2O2”/“control”) ≥ 0.3-fold or ≤ −0.3-fold
- (c)
- Comparison 3: “Scrmb siRNA + H2O2”/“Nox1 siRNA + H2O2” ≥ 0.2 fold. Control connotes vehicle treatment in untransfected VSMC.
2.2. Upregulation of ARPC2 Protein Expression in VSMCs via Nox1
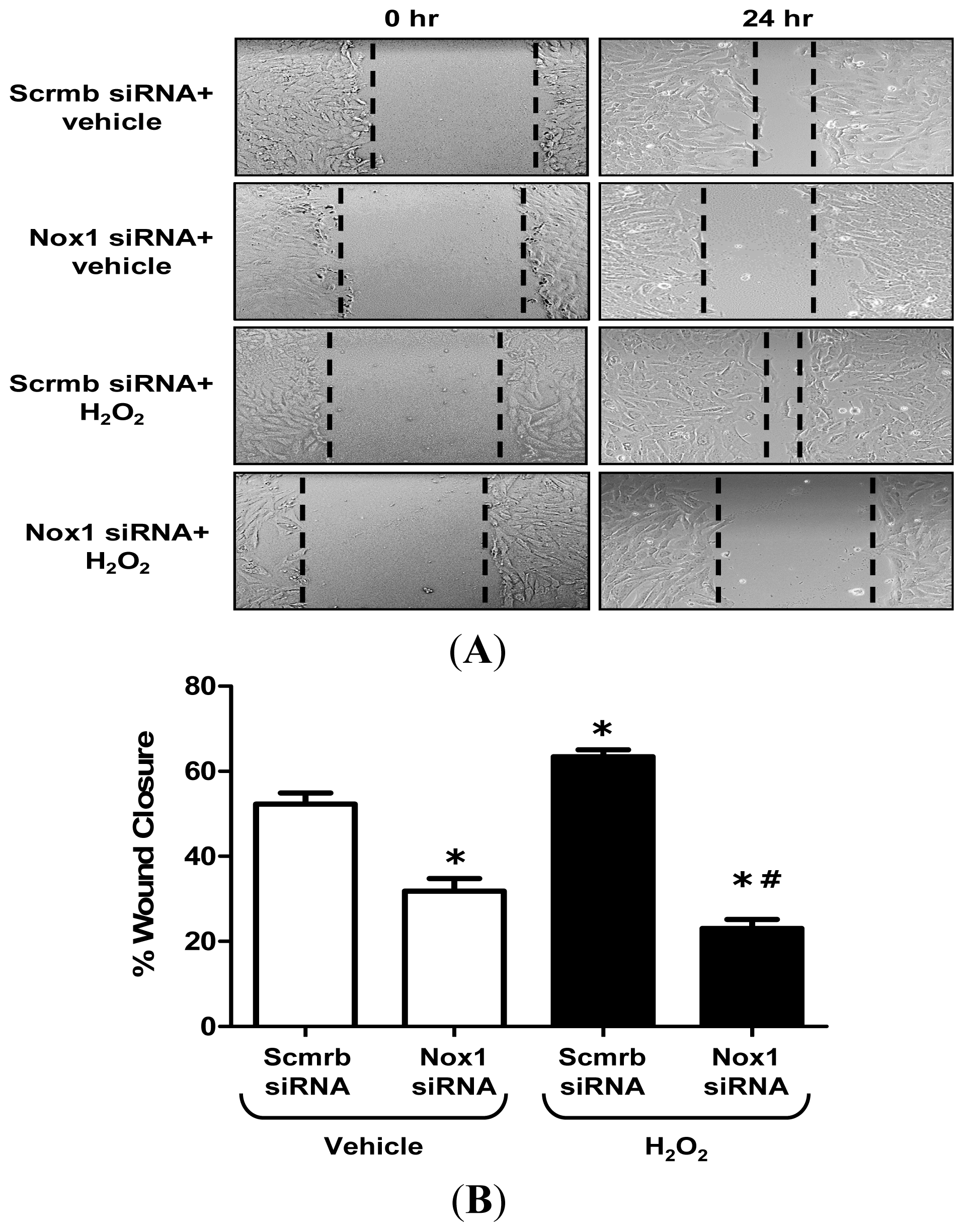
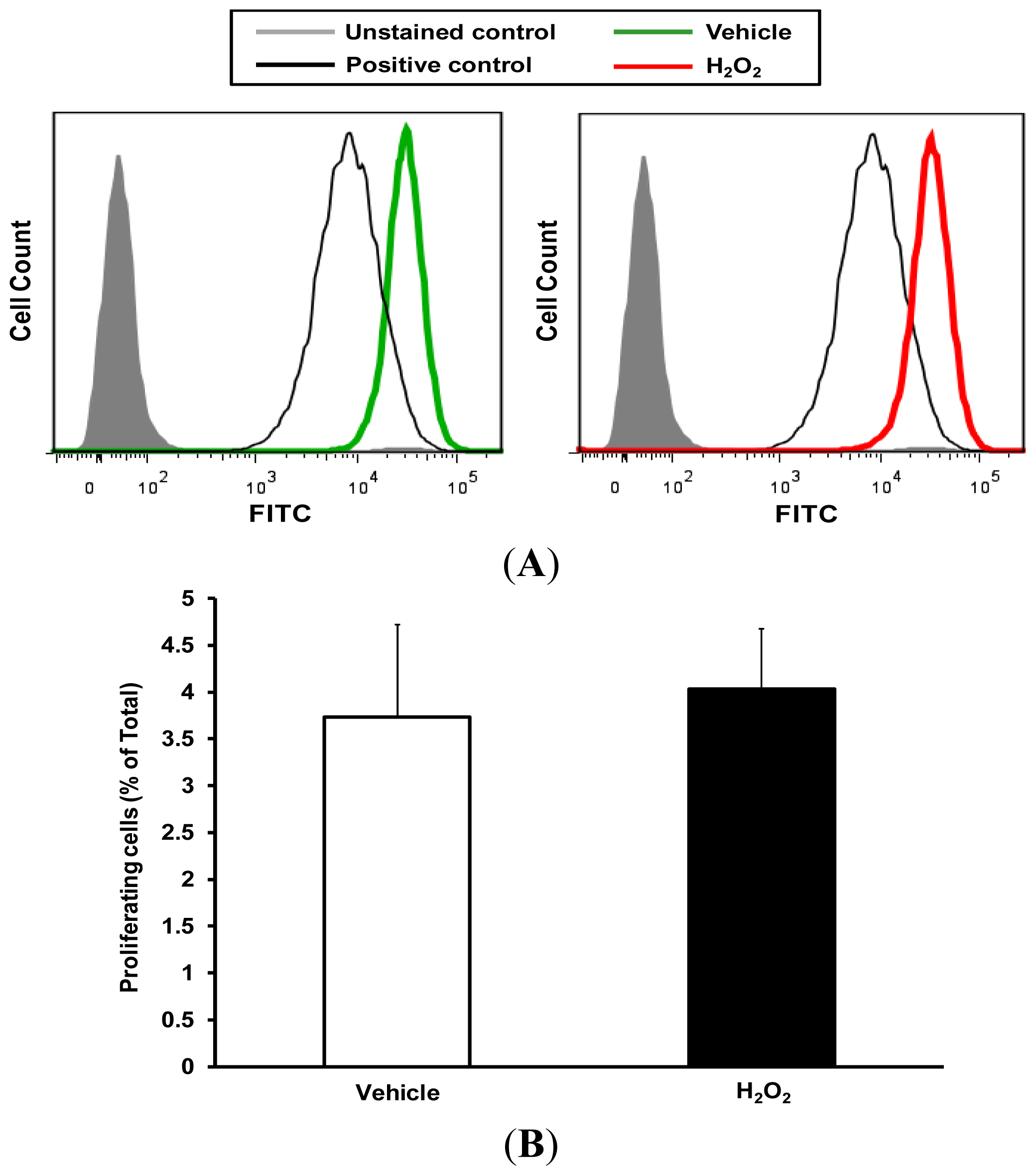
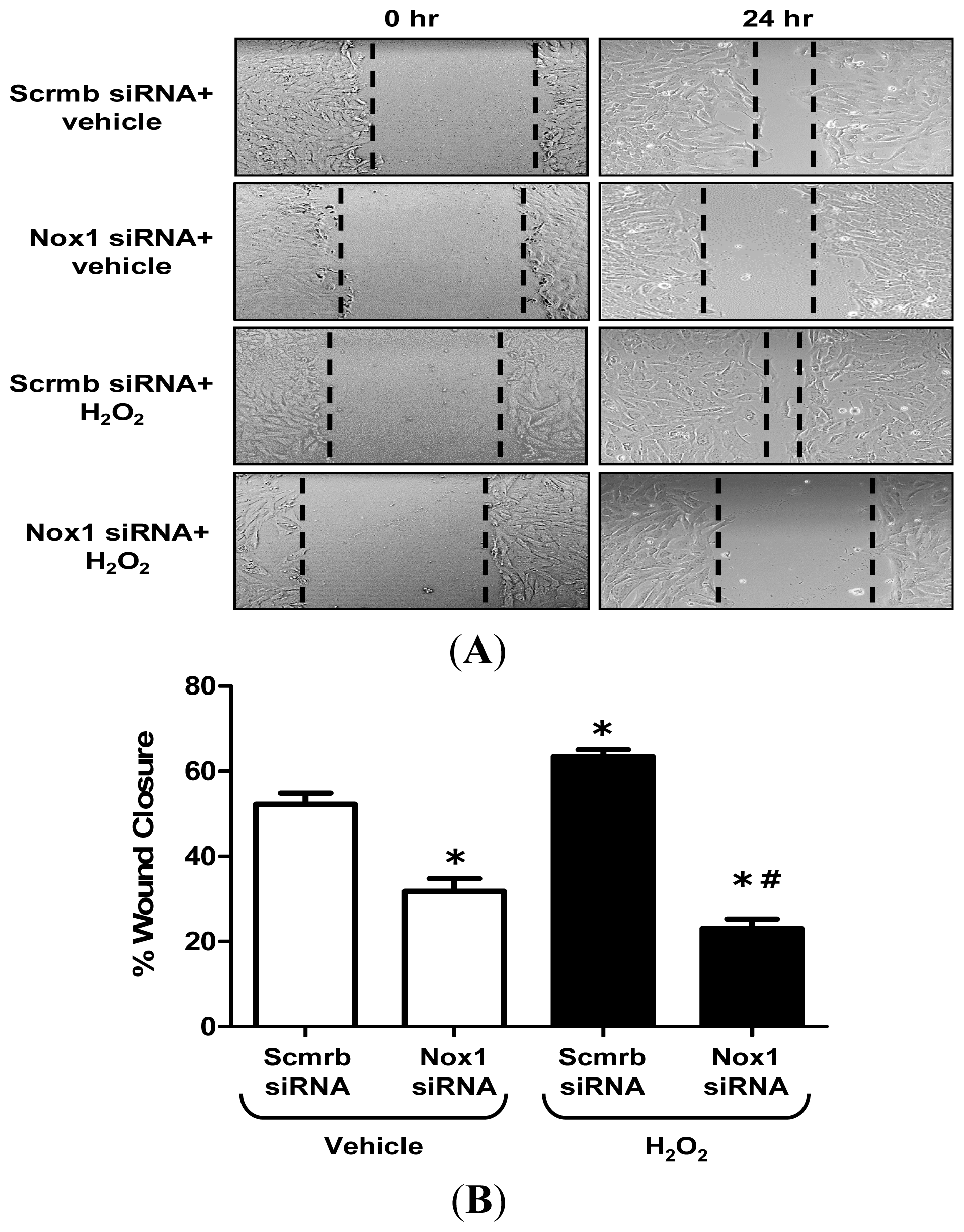
2.3. H2O2 Stimulates VSMC Migration via Nox1
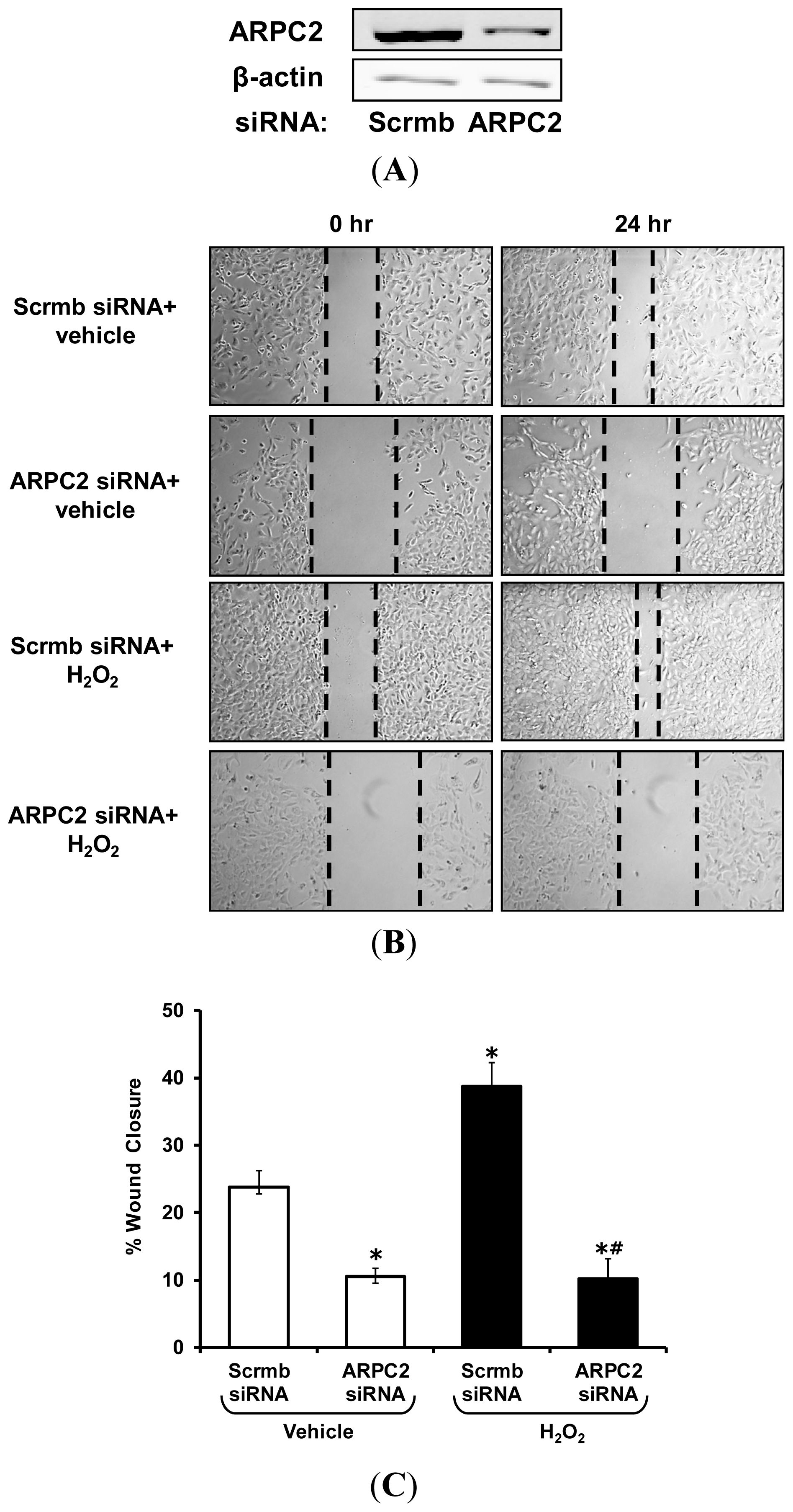
2.4. Gene Silencing of ARPC2 Attenuates VSMC Migration
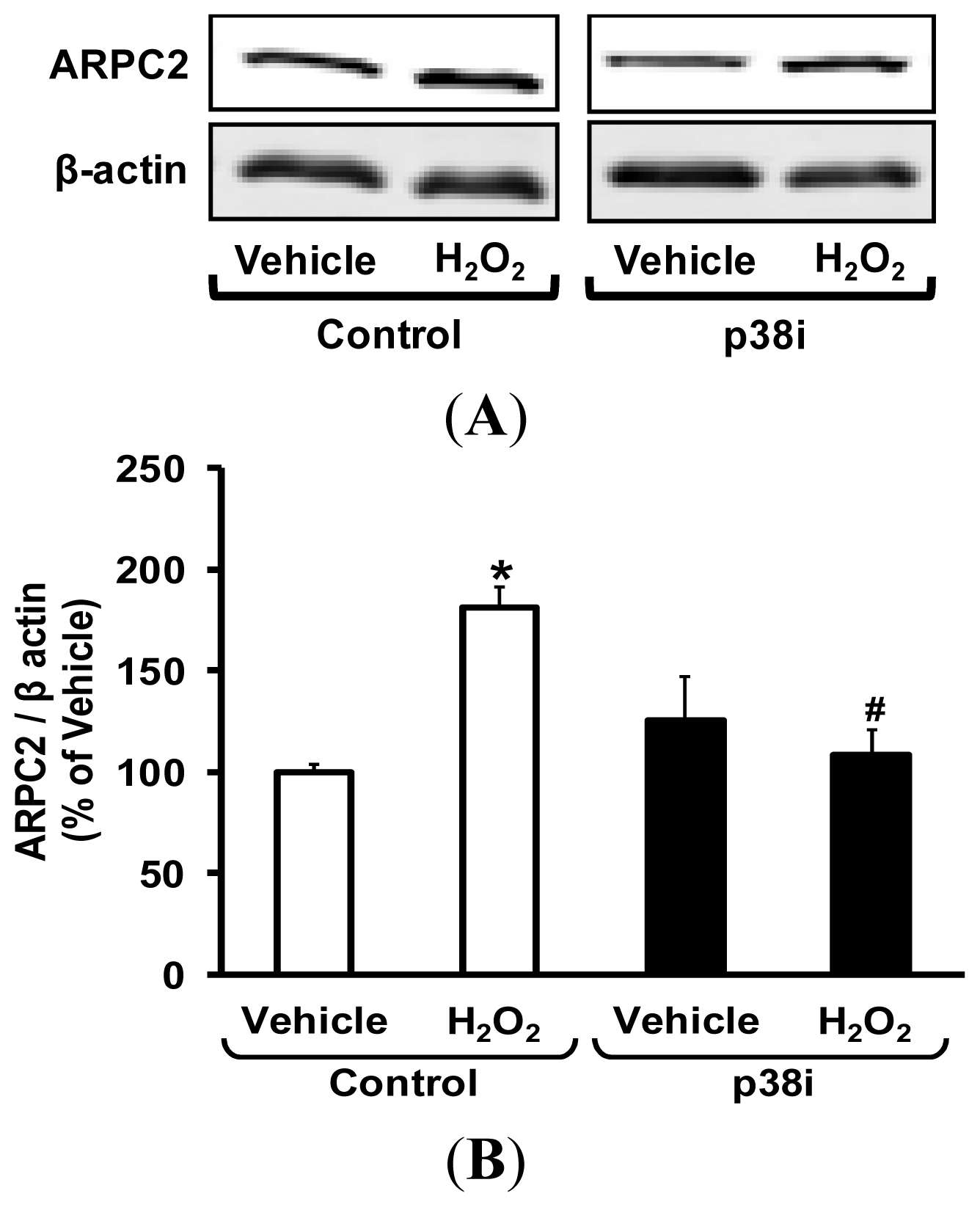
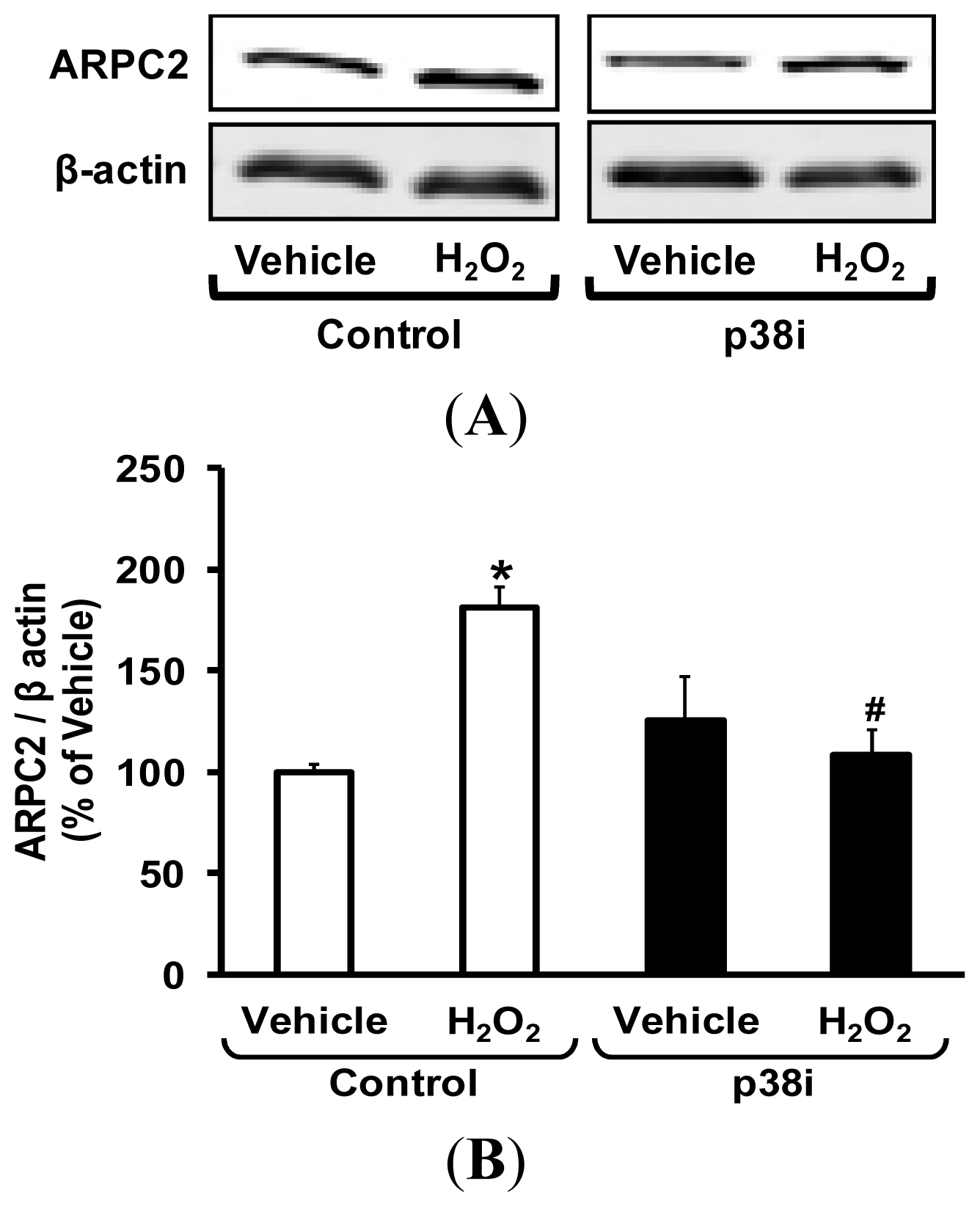
2.5. Pharmacological Inhibition of p38 MAPK Attenuates H2O2-Induced ARPC2 Expression
3. Experimental Section
3.1. Cell Culture
3.2. Gene Silencing
3.3. Western Blot
3.4. Proteomic Analysis Using Two-Dimensional Differential In-Gel Electrophoresis (2D-DIGE) Combined with Mass Spectroscopy (MS)
3.4.1. 2D-DIGE
3.4.2. Mass Spectrometry
3.5. Wound Migration “Scratch” Assay
3.6. Carboxyfluorescein Succinimidyl Ester (CFSE) Quantification
3.7. Statistical Analysis
4. Conclusions


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comparison | Treatment conditions | |
|---|---|---|
| 1 | Control | Scrmb siRNA + H2O2 |
| 2 | Control | Nox1 siRNA + H2O2 |
| 3 | Scrmb siRNA + H2O2 | Nox1 siRNA + H2O2 |
| Spot number | Protein name | Up/Down-regulation | Accession No. | Protein Score | Molecular weight (Da) | Protein Score C. I.%/Total Ion C. I.% | Major function |
|---|---|---|---|---|---|---|---|
| 2 | Fibronectin 1, isoform CRA_d | ↓ | gi|149015981 | 411 | 262,586 | 100/100 | Cell adhesion and differentiation |
| 4 | Heterogeneous nuclear ribonucleoprotein U | ↑ | gi|148747541 | 601 | 87,678 | 100/100 | Regulation of mRNA metabolism and transport |
| 7 | Fibronectin 3 | ↓ | gi|204158 | 111 | 74,876 | 100/100 | Wound healing, cell adhesion and differentation |
| 32 | Vimentin, isoform CRA_b | ↓ | gi|149021114 | 1,250 | 53,668 | 100/100 | Type III intermediate filament, maintains cellular integrity |
| 33 | Vimentin | ↓ | gi|14389299 | 1350 | 53,700 | 100/100 | Type III intermediate filament, maintains cellular integrity |
| 57 | Insulin-like growth factor (IGF)-binding protein 2 precursor | ↓ | gi|148747421 | 584 | 32,833 | 100/100 | Regulates bioavailability of IGF |
| 59 | 36 kda voltage dependent anion channel | ↑ | gi|299036 | 355 | 31,700 | 100/100 | Voltage-dependent anion channel protein |
| 63 | ARPC2 | ↑ | gi|205686193 | 823 | 34,369 | 100/100 | Regulation of actin cytoskeleton |
| 86 | Cofilin-1 | ↓ | gi|8393101 | 148 | 18,520 | 100/100 | Regulation of actin cytoskeleton dynamics |
| 87 | Destrin | ↓ | gi|62665569 | 241 | 18,507 | 100/100 | Actin-depolymerizing protein |
Acknowledgments
Conflicts of Interest
References
- Madhur, M.S.; Funt, S.A.; Li, L.; Vinh, A.; Chen, W.; Lob, H.E.; Iwakura, Y.; Blinder, Y.; Rahman, A.; Quyyumi, A.A.; et al. Role of interleukin 17 in inflammation, atherosclerosis, and vascular function in apolipoprotein e-deficient mice. Arterioscler. Thromb. Vasc. Biol 2011, 31, 1565–1572. [Google Scholar]
- Sparks, M.A.; Makhanova, N.A.; Griffiths, R.C.; Snouwaert, J.N.; Koller, B.H.; Coffman, T.M. Thromboxane receptors in smooth muscle promote hypertension, vascular remodeling, and sudden death. Hypertension 2013, 61, 166–173. [Google Scholar]
- Johnson, J.L.; Dwivedi, A.; Somerville, M.; George, S.J.; Newby, A.C. Matrix metalloproteinase (MMP)-3 activates MMP-9 mediated vascular smooth muscle cell migration and neointima formation in mice. Arterioscler. Thromb. Vasc. Biol 2011, 31, e35–e44. [Google Scholar]
- Al Ghouleh, I.; Khoo, N.K.; Knaus, U.G.; Griendling, K.K.; Touyz, R.M.; Thannickal, V.J.; Barchowsky, A.; Nauseef, W.M.; Kelley, E.E.; Bauer, P.M.; et al. Oxidases and peroxidases in cardiovascular and lung disease: New concepts in reactive oxygen species signaling. Free Radic. Biol. Med 2011, 51, 1271–1288. [Google Scholar]
- Csanyi, G.; Taylor, W.R.; Pagano, P.J. NOX and inflammation in the vascular adventitia. Free Radic. Biol. Med 2009, 47, 1254–1266. [Google Scholar]
- Szocs, K.; Lassegue, B.; Sorescu, D.; Hilenski, L.L.; Valppu, L.; Couse, T.L.; Wilcox, J.N.; Quinn, M.T.; Lambeth, J.D.; Griendling, K.K. Upregulation of Nox-based NAD(P)H oxidases in restenosis after carotid injury. Arterioscler. Thromb. Vasc. Biol 2002, 22, 21–27. [Google Scholar]
- Griendling, K.K. Novel NAD(P)H oxidases in the cardiovascular system. Heart 2004, 90, 491–493. [Google Scholar]
- Csanyi, G.; Cifuentes-Pagano, E.; Al Ghouleh, I.; Ranayhossaini, D.J.; Egana, L.; Lopes, L.R.; Jackson, H.M.; Kelley, E.E.; Pagano, P.J. Nox2 B-loop peptide, Nox2ds, specifically inhibits the NADPH oxidase Nox2. Free Radic. Biol. Med 2011, 51, 1116–1125. [Google Scholar]
- Dikalov, S.I.; Dikalova, A.E.; Bikineyeva, A.T.; Schmidt, H.H.; Harrison, D.G.; Griendling, K.K. Distinct roles of Nox1 and Nox4 in basal and angiotensin II-stimulated superoxide and hydrogen peroxide production. Free Radic. Biol. Med 2008, 45, 1340–1351. [Google Scholar]
- Al Ghouleh, I.; Frazziano, G.; Rodriguez, A.I.; Csanyi, G.; Maniar, S.; St Croix, C.M.; Kelley, E.E.; Egana, L.A.; Song, G.J.; Bisello, A.; et al. Aquaporin 1, Nox1, and Ask1 mediate oxidant-induced smooth muscle cell hypertrophy. Cardiovasc. Res 2013, 97, 134–142. [Google Scholar]
- Brown, D.I.; Griendling, K.K. Nox proteins in signal transduction. Free Radic. Biol. Med 2009, 47, 1239–1253. [Google Scholar]
- Sung, H.J.; Eskin, S.G.; Sakurai, Y.; Yee, A.; Kataoka, N.; McIntire, L.V. Oxidative stress produced with cell migration increases synthetic phenotype of vascular smooth muscle cells. Ann. Biomed. Eng 2005, 33, 1546–1554. [Google Scholar]
- Goley, E.D.; Rammohan, A.; Znameroski, E.A.; Firat-Karalar, E.N.; Sept, D.; Welch, M.D. An actin-filament-binding interface on the Arp2/3 complex is critical for nucleation and branch stability. Proc. Natl. Acad. Sci. USA 2010, 107, 8159–8164. [Google Scholar]
- Goley, E.D.; Welch, M.D. The ARP2/3 complex: An actin nucleator comes of age. Nat. Rev. Mol. Cell Biol 2006, 7, 713–726. [Google Scholar]
- Liao, G.; Simone, B.; Liu, G. Mis-localization of Arp2 mRNA impairs persistence of directional cell migration. Exp. Cell Res 2011, 317, 812–822. [Google Scholar]
- Rauhala, H.E.; Teppo, S.; Niemela, S.; Kallioniemi, A. Silencing of the ARP2/3 complex disturbs pancreatic cancer cell migration. Anticancer Res 2013, 33, 45–52. [Google Scholar]
- Suraneni, P.; Rubinstein, B.; Unruh, J.R.; Durnin, M.; Hanein, D.; Li, R. The Arp2/3 complex is required for lamellipodia extension and directional fibroblast cell migration. J. Cell Biol 2012, 197, 239–251. [Google Scholar]
- Cascino, T.; Csanyi, G.; Al Ghouleh, I.; Montezano, A.C.; Touyz, R.M.; Haurani, M.J.; Pagano, P.J. Adventitia-derived hydrogen peroxide impairs relaxation of the rat carotid artery via smooth muscle cell p38 mitogen-activated protein kinase. Antioxid. Redox. Signal 2011, 15, 1507–1515. [Google Scholar]
- Di Lisa, F.; Kaludercic, N.; Paolocci, N. Beta(2)-Adrenoceptors, NADPH oxidase, ROS and p38 MAPK: Another ‘radical’ road to heart failure? Br. J. Pharmacol 2011, 162, 1009–1011. [Google Scholar]
- Ushio-Fukai, M.; Alexander, R.W.; Akers, M.; Griendling, K.K. p38 Mitogen-activated protein kinase is a critical component of the redox-sensitive signaling pathways activated by angiotensin II. Role in vascular smooth muscle cell hypertrophy. J. Biol. Chem 1998, 273, 15022–15029. [Google Scholar]
- Viedt, C.; Soto, U.; Krieger-Brauer, H.I.; Fei, J.; Elsing, C.; Kubler, W.; Kreuzer, J. Differential activation of mitogen-activated protein kinases in smooth muscle cells by angiotensin II: Involvement of p22phox and reactive oxygen species. Arterioscler. Thromb. Vasc. Biol 2000, 20, 940–948. [Google Scholar]
- Lo, H.M.; Tsai, Y.J.; Du, W.Y.; Tsou, C.J.; Wu, W.B. A naturally occurring carotenoid, lutein, reduces PDGF and H2O2 signaling and compromised migration in cultured vascular smooth muscle cells. J. Biomed. Sci 2012, 19, 18. [Google Scholar]
- Schindeler, A.; Lavulo, L.; Harvey, R.P. Muscle costameric protein, Chisel/Smpx, associates with focal adhesion complexes and modulates cell spreading in vitro via a Rac1/p38 pathway. Exp. Cell Res 2005, 307, 367–380. [Google Scholar]
- Singh, S.; Powell, D.W.; Rane, M.J.; Millard, T.H.; Trent, J.O.; Pierce, W.M.; Klein, J.B.; Machesky, L.M.; McLeish, K.R. Identification of the p16-Arc subunit of the Arp 2/3 complex as a substrate of MAPK-activated protein kinase 2 by proteomic analysis. J. Biol. Chem 2003, 278, 36410–36417. [Google Scholar]
- Huang, X.; Zhang, J.; Liu, J.; Sun, L.; Zhao, H.; Lu, Y.; Wang, J.; Li, J. C-reactive protein promotes adhesion of monocytes to endothelial cells via NADPH oxidase-mediated oxidative stress. J. Cell Biochem 2012, 113, 857–867. [Google Scholar]
- Piao, Y.J.; Seo, Y.H.; Hong, F.; Kim, J.H.; Kim, Y.J.; Kang, M.H.; Kim, B.S.; Jo, S.A.; Jo, I.; Jue, D.M.; et al. Nox 2 stimulates muscle differentiation via NF-kappaB/iNOS pathway. Free Radic. Biol. Med 2005, 38, 989–1001. [Google Scholar]
- Lim, W.S.; Ng, D.L.; Kor, S.B.; Wong, H.K.; Tengku-Muhammad, T.S.; Choo, Q.C.; Chew, C.H. Tumour necrosis factor alpha down-regulates the expression of peroxisome proliferator activated receptor alpha (PPARalpha) in human hepatocarcinoma HepG2 cells by activation of NF-kappaB pathway. Cytokine 2013, 61, 266–274. [Google Scholar]
- Maalouf, S.W.; Talhouk, R.S.; Schanbacher, F.L. Inflammatory responses in epithelia: Endotoxin-induced IL-6 secretion and iNOS/NO production are differentially regulated in mouse mammary epithelial cells. J. Inflamm 2010, 7, 58. [Google Scholar]
- Oliveira-Marques, V.; Marinho, H.S.; Cyrne, L.; Antunes, F. Role of hydrogen peroxide in NF-kappaB activation: From inducer to modulator. Antioxid. Redox. Signal 2009, 11, 2223–2243. [Google Scholar]
- Al Ghouleh, I.; Magder, S. NADPH oxidase-derived superoxide destabilizes lipopolysaccharide-induced interleukin 8 mRNA via p38, extracellular signal-regulated kinase mitogen-activated protein kinase, and the destabilizing factor tristetraprolin. Shock 2012, 37, 433–440. [Google Scholar]
- Shimizu, H.; Hirose, Y.; Nishijima, F.; Tsubakihara, Y.; Miyazaki, H. ROS and PDGF-beta [corrected] receptors are critically involved in indoxyl sulfate actions that promote vascular smooth muscle cell proliferation and migration. Am. J. Physiol. Cell Physiol 2009, 297, C389–C396. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ghouleh, I.A.; Rodríguez, A.; Pagano, P.J.; Csányi, G. Proteomic Analysis Identifies an NADPH Oxidase 1 (Nox1)-Mediated Role for Actin-Related Protein 2/3 Complex Subunit 2 (ARPC2) in Promoting Smooth Muscle Cell Migration. Int. J. Mol. Sci. 2013, 14, 20220-20235. https://doi.org/10.3390/ijms141020220
Ghouleh IA, Rodríguez A, Pagano PJ, Csányi G. Proteomic Analysis Identifies an NADPH Oxidase 1 (Nox1)-Mediated Role for Actin-Related Protein 2/3 Complex Subunit 2 (ARPC2) in Promoting Smooth Muscle Cell Migration. International Journal of Molecular Sciences. 2013; 14(10):20220-20235. https://doi.org/10.3390/ijms141020220
Chicago/Turabian StyleGhouleh, Imad Al, Andrés Rodríguez, Patrick J. Pagano, and Gábor Csányi. 2013. "Proteomic Analysis Identifies an NADPH Oxidase 1 (Nox1)-Mediated Role for Actin-Related Protein 2/3 Complex Subunit 2 (ARPC2) in Promoting Smooth Muscle Cell Migration" International Journal of Molecular Sciences 14, no. 10: 20220-20235. https://doi.org/10.3390/ijms141020220