Atorvastatin Attenuates Bleomycin-Induced Pulmonary Fibrosis via Suppressing iNOS Expression and the CTGF (CCN2)/ERK Signaling Pathway

Abstract
:1. Introduction
2. Results
2.1. Protective Effects of Atorvastatin against Bleomycin Modulated Body Weight and Lung Indices
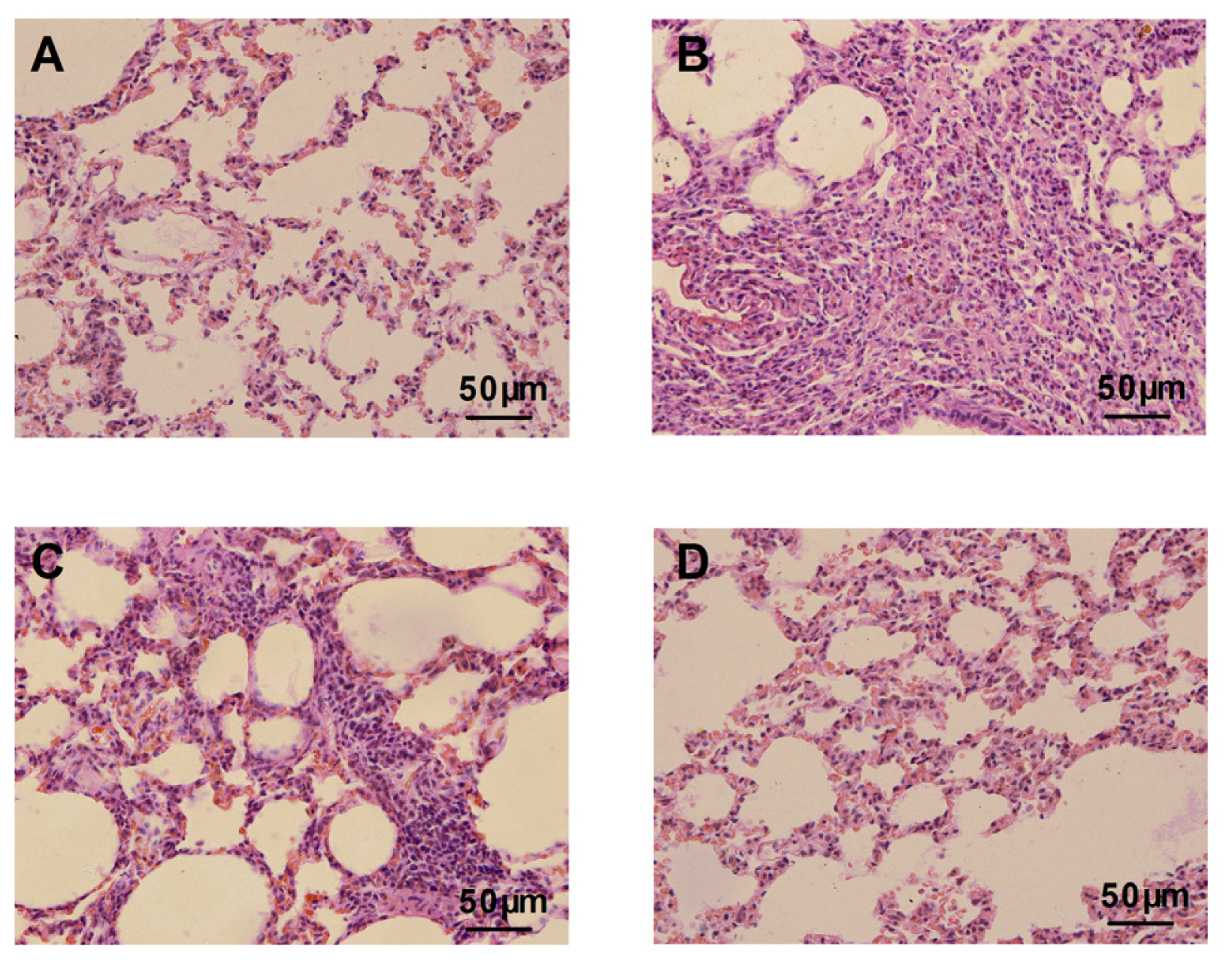
2.2. Atorvastatin Attenuated Bleomycin Mediated Histological Changes
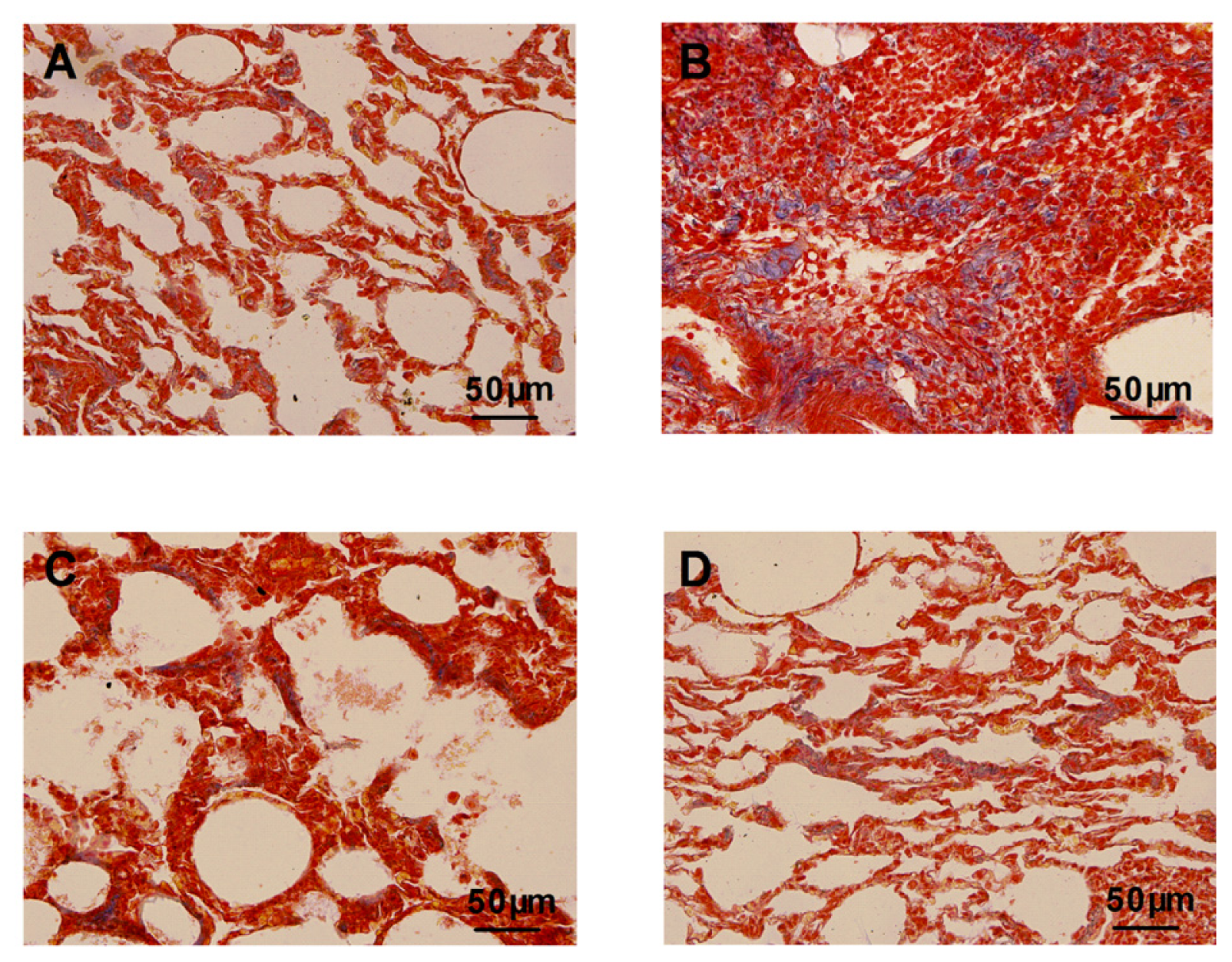
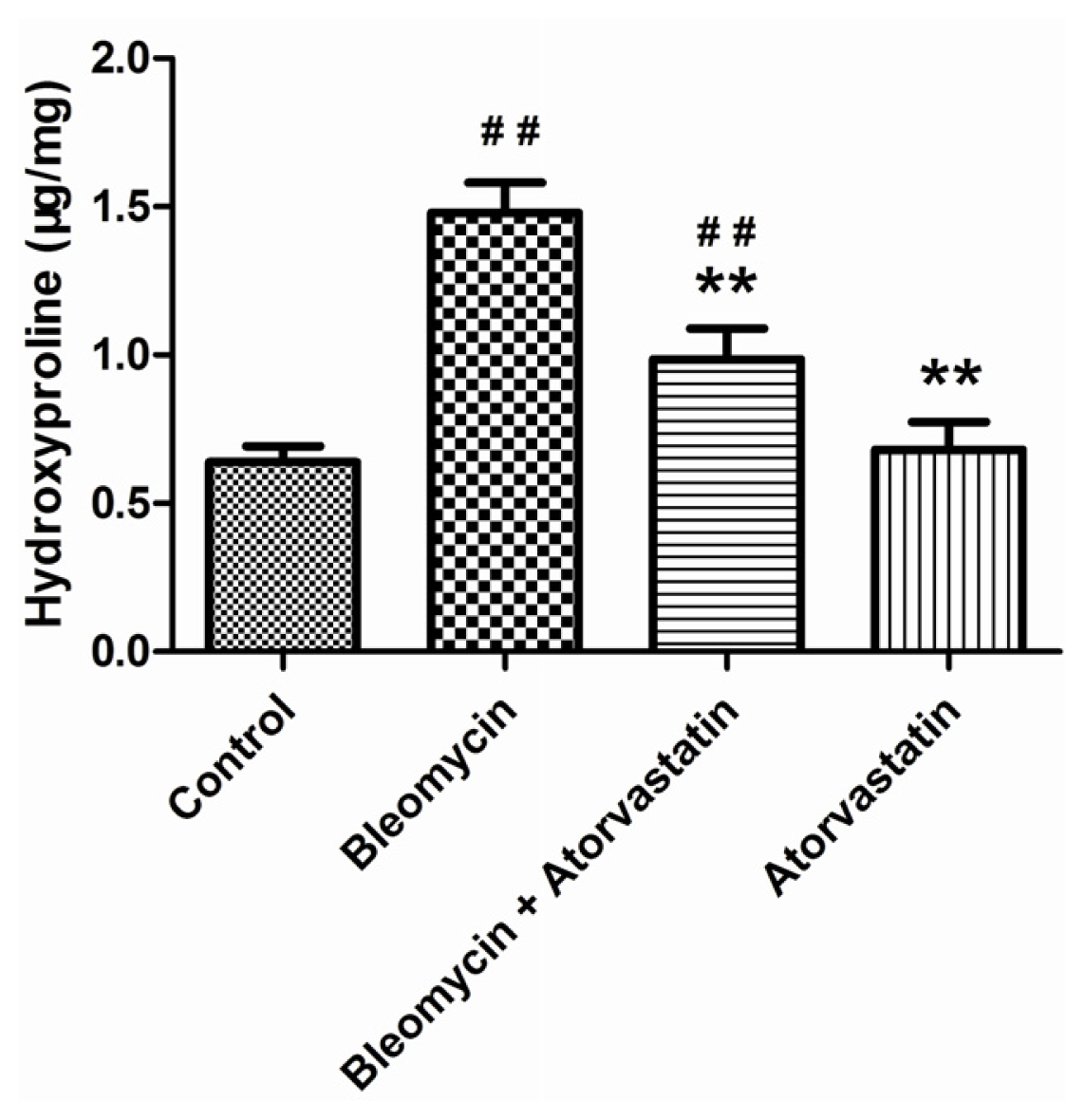
2.3. Inhibition Effects of Atorvastatin on Collagen Depositions and Hydroxyproline Content
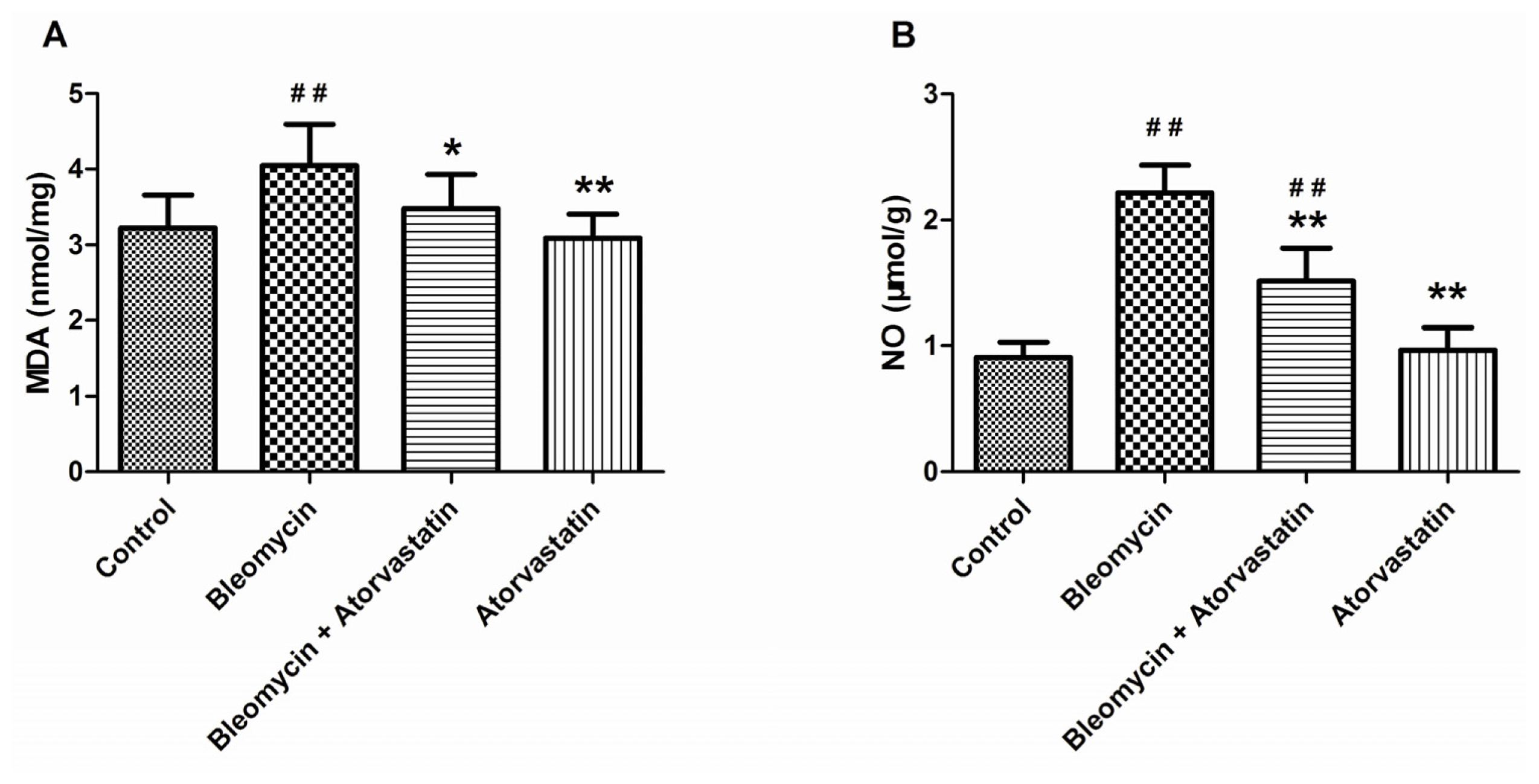
2.4. Suppressive Effects of Atorvastatin on Oxidative Stress Markers
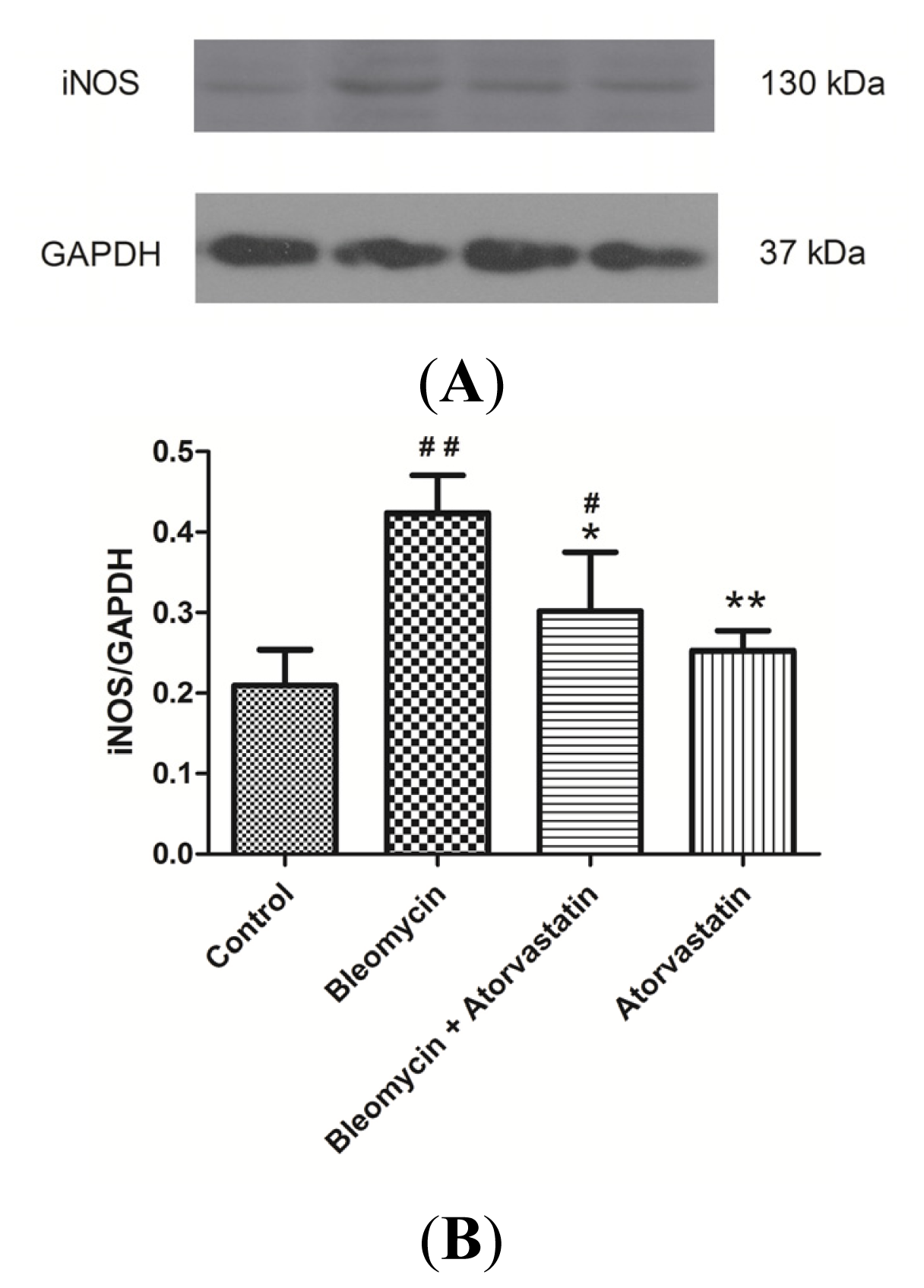
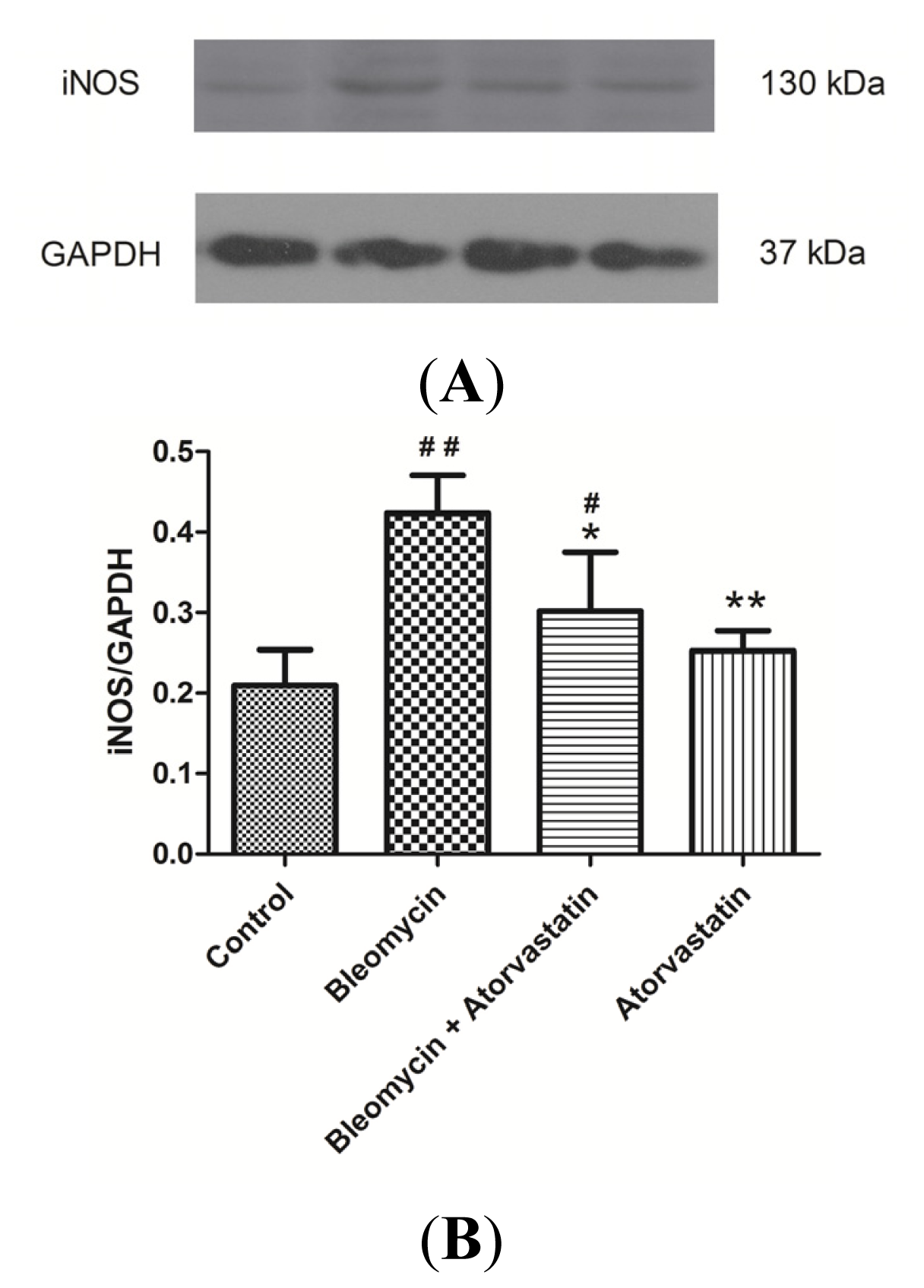
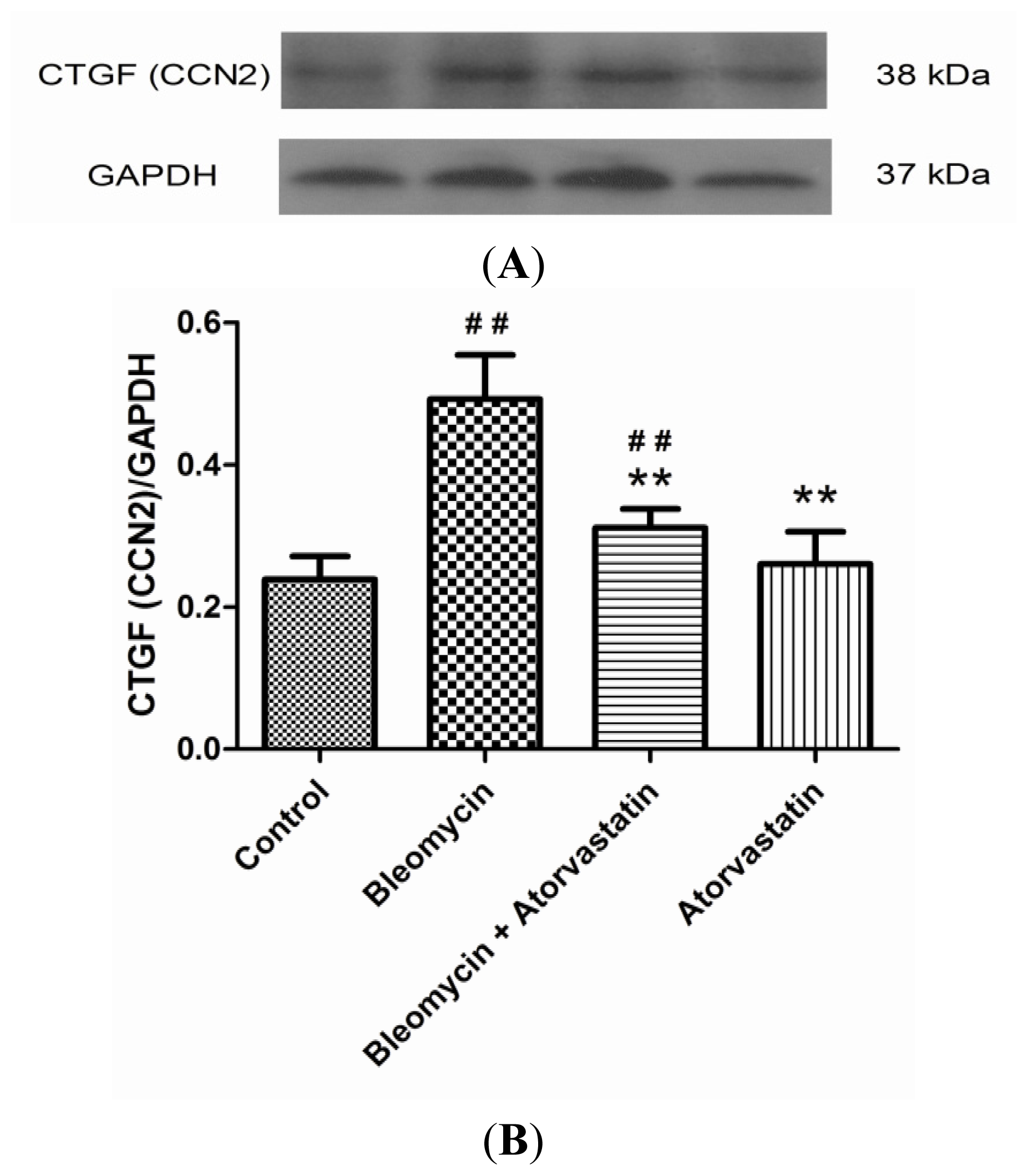
2.5. Down-Regulation of iNOS and CTGF (CCN2) Expression by Atorvastatin Treatment
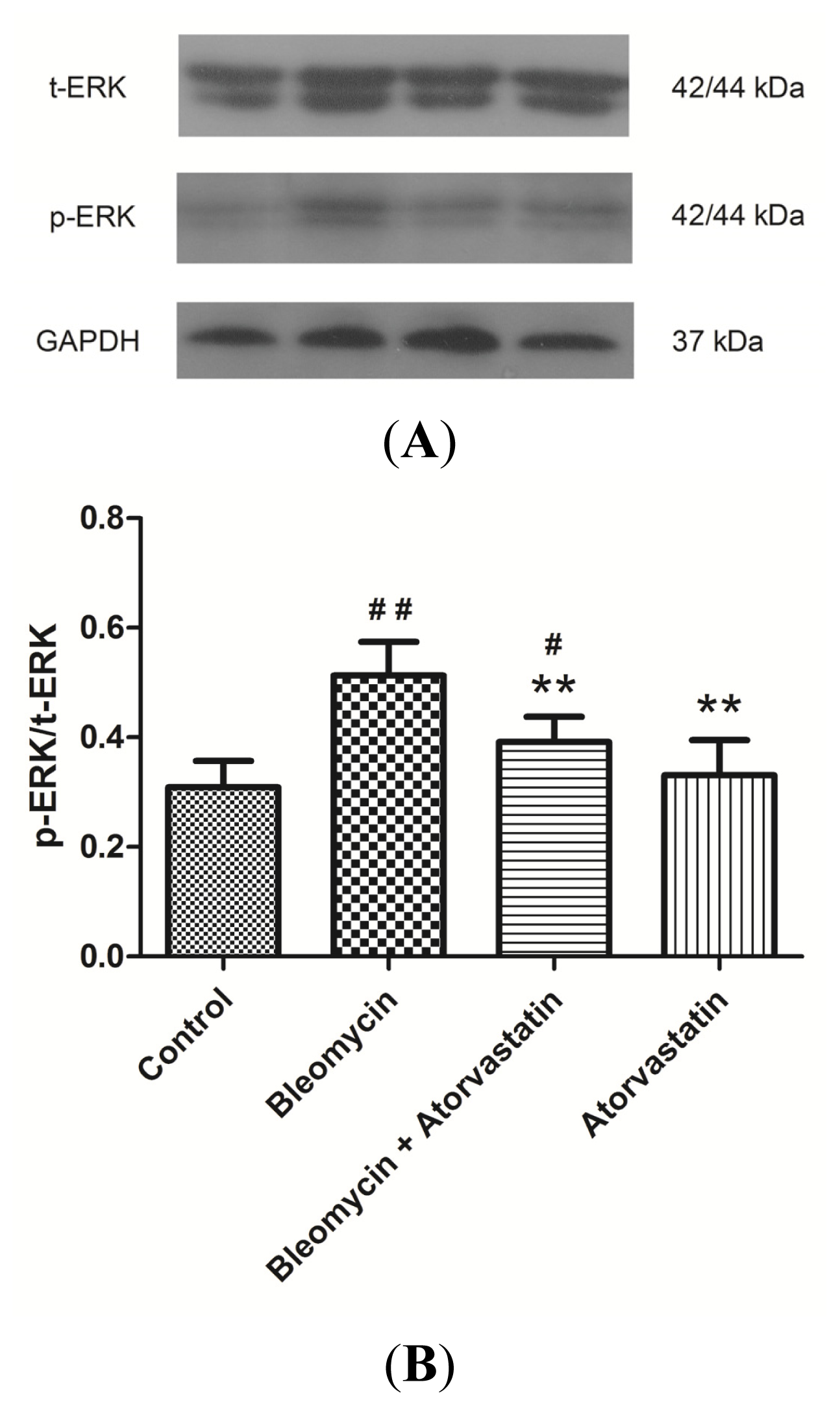
2.6. Down-Regulation of Phosphorylation of ERK by Atorvastatin Treatment
3. Discussion
4. Experimental Section
4.1. Animal and Maintenance
4.2. Experimental Protocol
4.3. Histological Examination and Masson’s Trichrome Staining
4.4. Estimation of Hydroxyproline (HYP)
4.5. Measurements of Malondialdehyde (MDA) and Nitric Oxide (NO) Level
4.6. Western Blot Analysis
4.7. Statistical Analysis
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Selman, M.; Thannickal, V.J.; Pardo, A.; Zisman, D.A.; Martinez, F.J.; Lynch, J.P., 3rd. Idiopathic pulmonary fibrosis: Pathogenesis and therapeutic approaches. Drugs 2004, 64, 405–430. [Google Scholar]
- Kim, D.S. Acute exacerbations in patients with idiopathic pulmonary fibrosis. Respir. Res 2013, 14, 86. [Google Scholar]
- Cottin, V. Changing the idiopathic pulmonary fibrosis treatment approach and improving patient outcomes. Eur. Respir. Rev 2012, 21, 161–167. [Google Scholar]
- Raghu, G.; Chang, J. Idiopathic pulmonary fibrosis: Current trends in management. Clin. Chest Med 2004, 25, 621–636. [Google Scholar]
- Maher, T.M. Idiopathic pulmonary fibrosis: Pathobiology of novel approaches to treatment. Clin. Chest Med 2012, 33, 69–83. [Google Scholar]
- Antoniou, K.M.; Margaritopoulos, G.A.; Siafakas, N.M. Pharmacological treatment of idiopathic pulmonary fibrosis: From the past to the future. Eur. Respir. Rev 2013, 22, 281–291. [Google Scholar]
- Rahman, I.; Skwarska, E.; Henry, M.; Davis, M.; O’Connor, C.M.; FitzGerald, M.X.; Greening, A.; MacNee, W. Systemic and pulmonary oxidative stress in idiopathic pulmonary fibrosis. Free Radic. Biol. Med 1999, 27, 60–68. [Google Scholar]
- Kinnula, V.L.; Fattman, C.L.; Tan, R.J.; Oury, T.D. Oxidative stress in pulmonary fibrosis: A possible role for redox modulatory therapy. Am. J. Respir. Crit. Care Med 2005, 172, 417–422. [Google Scholar]
- Daniil, Z.D.; Papageorgiou, E.; Koutsokera, A.; Kostikas, K.; Kiropoulos, T.; Papaioannou, A.I.; Gourgoulianis, K.I. Serum levels of oxidative stress as a marker of disease severity in idiopathic pulmonary fibrosis. Pulm. Pharmacol. Ther 2008, 21, 26–31. [Google Scholar]
- Kliment, C.R.; Englert, J.M.; Gochuico, B.R.; Yu, G.; Kaminski, N.; Rosas, I.; Oury, T.D. Oxidative stress alters syndecan-1 distribution in lungs with pulmonary fibrosis. J. Biol. Chem 2009, 284, 3537–3545. [Google Scholar]
- Chitra, P.; Saiprasad, G.; Manikandan, R.; Sudhandiran, G. Berberine attenuates bleomycin induced pulmonary toxicity and fibrosis via suppressing NF-κB dependant TGF-β activation: A biphasic experimental study. Toxicol. Lett 2013, 219, 178–193. [Google Scholar]
- Fubini, B.; Hubbard, A. Reactive oxygen species (ROS) and reactive nitrogen species (RNS) generation by silica in inflammation and fibrosis. Free Radic. Biol. Med 2003, 34, 1507–1516. [Google Scholar]
- Bargagli, E.; Olivieri, C.; Bennett, D.; Prasse, A.; Muller-Quernheim, J.; Rottoli, P. Oxidative stress in the pathogenesis of diffuse lung diseases: A review. Respir. Med 2009, 103, 1245–1256. [Google Scholar]
- Paredi, P.; Kharitonov, S.A.; Barnes, P.J. Analysis of expired air for oxidation products. Am. J. Respir. Crit. Care Med 2002, 166, S31–S37. [Google Scholar]
- Barnes, P.J.; Hansel, T.T. Prospects for new drugs for chronic obstructive pulmonary disease. Lancet 2004, 364, 985–996. [Google Scholar]
- Kalayarasan, S.; Sriram, N.; Sudhandiran, G. Diallyl sulfide attenuates bleomycin-induced pulmonary fibrosis: Critical role of iNOS, NF-kappaB, TNF-alpha and IL-1beta. Life Sci 2008, 82, 1142–1153. [Google Scholar]
- Minder, C.M.; Blaha, M.J.; Horne, A.; Michos, E.D.; Kaul, S.; Blumenthal, R.S. Evidence-based use of statins for primary prevention of cardiovascular disease. Am. J. Med 2012, 125, 440–446. [Google Scholar]
- Davignon, J. Pleiotropic effects of pitavastatin. Br. J. Clin. Pharmacol 2012, 73, 518–535. [Google Scholar]
- Athyros, V.G.; Kakafika, A.I.; Tziomalos, K.; Karagiannis, A.; Mikhailidis, D.P. Pleiotropic effects of statins-clinical evidence. Curr. Pharm. Des 2009, 15, 479–489. [Google Scholar]
- Mikael, L.G.; Rozen, R. Homocysteine modulates the effect of simvastatin on expression of ApoA-I and NF-kappaB/iNOS. Cardiovasc. Res 2008, 80, 151–158. [Google Scholar]
- Araújo, F.A.; Rocha, M.A.; Mendes, J.B.; Andrade, S.P. Atorvastatin inhibits inflammatory angiogenesis in mice through down regulation of VEGF, TNF-alpha and TGF-beta1. Biomed. Pharmacother 2010, 64, 29–34. [Google Scholar]
- Shyamsundar, M.; McKeown, S.T.; O’Kane, C.M.; Craig, T.R.; Brown, V.; Thickett, D.R.; Matthay, M.A.; Taggart, C.C.; Backman, J.T.; Elborn, J.S.; et al. Simvastatin decreases lipopolysaccharide-induced pulmonary inflammation in healthy volunteers. Am. J. Respir. Crit. Care Med 2009, 179, 1107–1114. [Google Scholar]
- Massaro, M.; Zampolli, A.; Scoditti, E.; Carluccio, M.A.; Storelli, C.; Distante, A.; de Caterina, R. Statins inhibit cyclooxygenase-2 and matrix metalloproteinase-9 in human endothelial cells: Anti-angiogenic actions possibly contributing to plaque stability. Cardiovasc. Res 2010, 86, 311–320. [Google Scholar]
- Jiang, C.; Huang, H.; Liu, J.; Wang, Y.; Lu, Z.; Xu, Z. Fasudil, a rho-kinase inhibitor, attenuates bleomycin-induced pulmonary fibrosis in mice. Int. J. Mol. Sci 2012, 13, 8293–8307. [Google Scholar]
- Brigstock, D.R.; Goldschmeding, R.; Katsube, K.I.; Lam, S.C.; Lau, L.F.; Lyons, K.; Naus, C.; Perbal, B.; Riser, B.; Takigawa, M.; et al. Proposal for a unified CCN nomenclature. Mol. Pathol 2003, 56, 127–128. [Google Scholar]
- Leask, A. Possible strategies for anti-fibrotic drug intervention in scleroderma. J. Cell Commun. Signal 2011, 5, 125–129. [Google Scholar]
- Mason, R.M. Fell-Muir lecture: Connective tissue growth factor (CCN2)—A pernicious and pleiotropic player in the development of kidney fibrosis. Int. J. Exp. Pathol 2013, 94, 1–16. [Google Scholar]
- White, E.S.; Lazar, M.H.; Thannickal, V.J. Pathogenetic mechanisms in usual interstitial pneumonia/idiopathic pulmonary fibrosis. J. Pathol 2003, 201, 343–354. [Google Scholar]
- Gothelf, A.; Mir, L.M.; Gehl, J. Electrochemotherapy: Results of cancer treatment using enhanced delivery of bleomycin by electroporation. Cancer Treat Rev 2003, 29, 371–387. [Google Scholar]
- Azambuja, E.; Fleck, J.F.; Batista, R.G.; Menna Barreto, S.S. Bleomycin lung toxicity: Who are the patients with increased risk? Pulm. Pharmacol. Ther 2005, 18, 363–366. [Google Scholar]
- Moeller, A.; Ask, K.; Warburton, D.; Gauldie, J.; Kolb, M. The bleomycin animal model: A useful tool to investigate treatment options for idiopathic pulmonary fibrosis? Int. J. Biochem. Cell Biol 2008, 40, 362–382. [Google Scholar]
- Wan, Y.Y.; Tian, G.Y.; Guo, H.S.; Kang, Y.M.; Yao, Z.H.; Li, X.L.; Liu, Q.H.; Lin, D.J. Endostatin, an angiogenesis inhibitor, ameliorates bleomycin-induced pulmonary fibrosis in rats. Respir. Res 2013, 14, 56. [Google Scholar]
- Chaudhary, N.I.; Schnapp, A.; Park, J.E. Pharmacologic differentiation of inflammation and fibrosis in the rat bleomycin model. Am. J. Respir. Crit. Care Med 2006, 173, 769–776. [Google Scholar]
- Mouratis, M.A.; Aidinis, V. Modeling pulmonary fibrosis with bleomycin. Curr. Opin. Pulm. Med 2011, 17, 355–361. [Google Scholar]
- Cheresh, P.; Kim, S.J.; Tulasiram, S.; Kamp, D.W. Oxidative stress and pulmonary fibrosis. Biochim. Biophys. Acta 2013, 1832, 1028–1040. [Google Scholar]
- Tsutsui, M.; Shimokawa, H.; Otsuji, Y.; Yanagihara, N. Pathophysiological relevance of NO signaling in the cardiovascular system: novel insight from mice lacking all NO synthases. Pharmacol. Ther 2010, 128, 499–508. [Google Scholar]
- Scotton, C.J.; Chambers, R.C. Molecular targets in pulmonary fibrosis: The myofibroblast in focus. Chest 2007, 132, 1311–1121. [Google Scholar]
- Phanish, M.K.; Winn, S.K.; Dockrell, M.E. Connective tissue growth factor-(CTGF,CCN2)—A marker, mediator and therapeutic target for renal fibrosis. Nephron Exp. Nephrol 2010, 114, e83–e92. [Google Scholar]
- Gressner, O.A.; Gressner, A.M. Connective tissue growth factor: A fibrogenic master switch in fibrotic liver diseases. Liver Int 2008, 28, 1065–1079. [Google Scholar]
- Ponticos, M.; Holmes, A.M.; Shi-Wen, X.; Leoni, P.; Khan, K.; Rajkumar, V.S.; Hoyles, R.K.; Bou-Gharios, G.; Black, C.M.; Denton, C.P.; et al. Pivotal role of connective tissue growth factor in lung fibrosis: MAPK-dependent transcriptional activation of type I collagen. Arthritis Rheum 2009, 60, 2142–2155. [Google Scholar]
- Tannheimer, S.L.; Wright, C.D.; Salmon, M. Combination of roflumilast with a beta-2 adrenergic receptor agonist inhibits proinflammatory and profibrotic mediator release from human lung fibroblasts. Respir. Res 2012, 13, 28. [Google Scholar]
- Rodrigues-Díez, R.; Lavoz, C.; Rayego-Mateos, S.; Civantos, E.; Rodríguez-Vita, J.; Mezzano, S.; Ortiz, A.; Egido, J.; Ruiz-Ortega, M. Statins inhibit angiotensin II/Smad pathway and related vascular fibrosis, by a TGF-β-independent process. PLoS One 2010, 5, e14145. [Google Scholar]
- Galuppo, M.; Esposito, E.; Mazzon, E.; di Paola, R.; Paterniti, I.; Impellizzeri, D.; Cuzzocrea, S. MEK inhibition suppresses the development of lung fibrosis in the bleomycin model. Naunyn-Schmiedebergs Arch. Pharmacol 2011, 384, 21–37. [Google Scholar]
- Antoniou, K.M.; Margaritopoulos, G.A.; Soufla, G.; Symvoulakis, E.; Vassalou, E.; Lymbouridou, R.; Samara, K.D.; Kappou, D.; Spandidos, D.A.; Siafakas, N.M. Expression analysis of Akt and MAPK signaling pathways in lung tissue of patients with idiopathic pulmonary fibrosis (IPF). J. Recept. Signal Transduct. Res 2010, 30, 262–269. [Google Scholar]
- Adamali, H.I.; Maher, T.M. Current and novel drug therapies for idiopathic pulmonary fibrosis. Drug Des. Devel. Ther 2012, 6, 261–272. [Google Scholar]
- Whelan, T.P. Lung transplantation for interstitial lung disease. Clin. Chest Med 2012, 33, 179–189. [Google Scholar]
- McShane, P.J.; Garrity, E.R., Jr. Minimization of immunosuppression after lung transplantation: Current trends. Transpl. Int 2009, 22, 90–95. [Google Scholar]
- Strieter, R.M.; Mehrad, B. New mechanisms of pulmonary fibrosis. Chest 2009, 136, 1364–1370. [Google Scholar]
- Richeldi, L. Assessing the treatment effect from multiple trials in idiopathic pulmonary fibrosis. Eur. Respir. Rev 2012, 21, 147–151. [Google Scholar]
- Nadrous, H.F.; Ryu, J.H.; Douglas, W.W.; Decker, P.A.; Olson, E.J. Impact of angiotensin-converting enzyme inhibitors and statins on survival in idiopathic pulmonary fibrosis. Chest 2004, 126, 438–446. [Google Scholar]
- Alexeeff, S.E.; Litonjua, A.A.; Sparrow, D.; Vokonas, P.S.; Schwartz, J. Statin use reduces decline in lung function: VA Normative Aging Study. Am. J. Respir. Crit. Care Med 2007, 176, 742–747. [Google Scholar]
- Yang, J.; Song, T.B.; Zhao, Z.H.; Qiu, S.D.; Hu, X.D.; Chang, L. Vasoactive intestinal peptide protects against ischemic brain damage induced by focal cerebral ischemia in rats. Brain Res 2011, 1398, 94–101. [Google Scholar]
- Punithavathi, D.; Venkatesan, N.; Babu, M. Curcumin inhibition of bleomycin-induced pulmonary fibrosis in rats. Br. J. Pharmacol 2000, 131, 169–172. [Google Scholar]
- Edwards, C.A.; O’Brien, W.D., Jr. Modified assay for determination of hydroxyproline in a tissue hydrolyzate. Clin. Chim. Acta 1980, 104, 161–167. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Initial Body weight (g) | Final Body Weight (g) | Lung Weight (mg) | Lung Indices (mg/kg) |
|---|---|---|---|---|
| Control | 208.9 ± 7.8 | 304.9 ± 15.1 | 1,393.8 ± 129.9 | 4,565.5 ± 257.6 |
| Bleomycin | 208.0 ± 9.7 | 283.8 ± 13.6 # | 2,190.0 ± 336.2 ## | 7,714.1 ± 1,104.0 ## |
| Bleomycin + Atorvastatin | 208.8 ± 9.5 | 290.5 ± 17.3 | 1,930.0 ± 200.1 ##,* | 6,633.7 ± 402.2 ##,* |
| Atorvastatin | 208.3 ± 10.0 | 292.5 ± 12.1 | 1,366.3 ± 116.6 ** | 4,664.5 ± 231.1 ** |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhu, B.; Ma, A.-Q.; Yang, L.; Dang, X.-M. Atorvastatin Attenuates Bleomycin-Induced Pulmonary Fibrosis via Suppressing iNOS Expression and the CTGF (CCN2)/ERK Signaling Pathway. Int. J. Mol. Sci. 2013, 14, 24476-24491. https://doi.org/10.3390/ijms141224476
Zhu B, Ma A-Q, Yang L, Dang X-M. Atorvastatin Attenuates Bleomycin-Induced Pulmonary Fibrosis via Suppressing iNOS Expression and the CTGF (CCN2)/ERK Signaling Pathway. International Journal of Molecular Sciences. 2013; 14(12):24476-24491. https://doi.org/10.3390/ijms141224476
Chicago/Turabian StyleZhu, Bo, Ai-Qun Ma, Lan Yang, and Xiao-Min Dang. 2013. "Atorvastatin Attenuates Bleomycin-Induced Pulmonary Fibrosis via Suppressing iNOS Expression and the CTGF (CCN2)/ERK Signaling Pathway" International Journal of Molecular Sciences 14, no. 12: 24476-24491. https://doi.org/10.3390/ijms141224476