The Yin-Yang of DNA Damage Response: Roles in Tumorigenesis and Cellular Senescence


 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
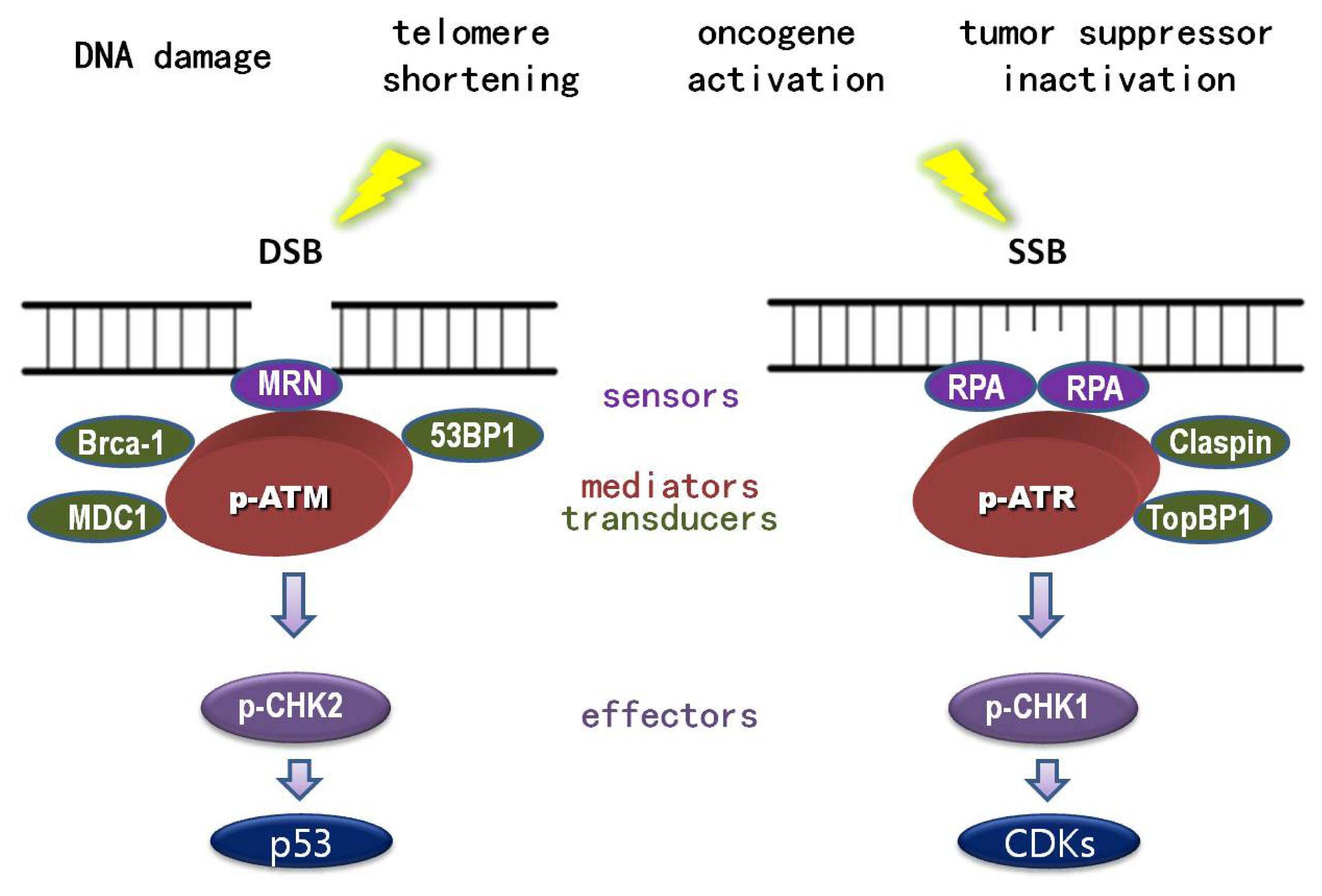
1.1. DNA Damage
1.2. DNA Damage Response (DDR)
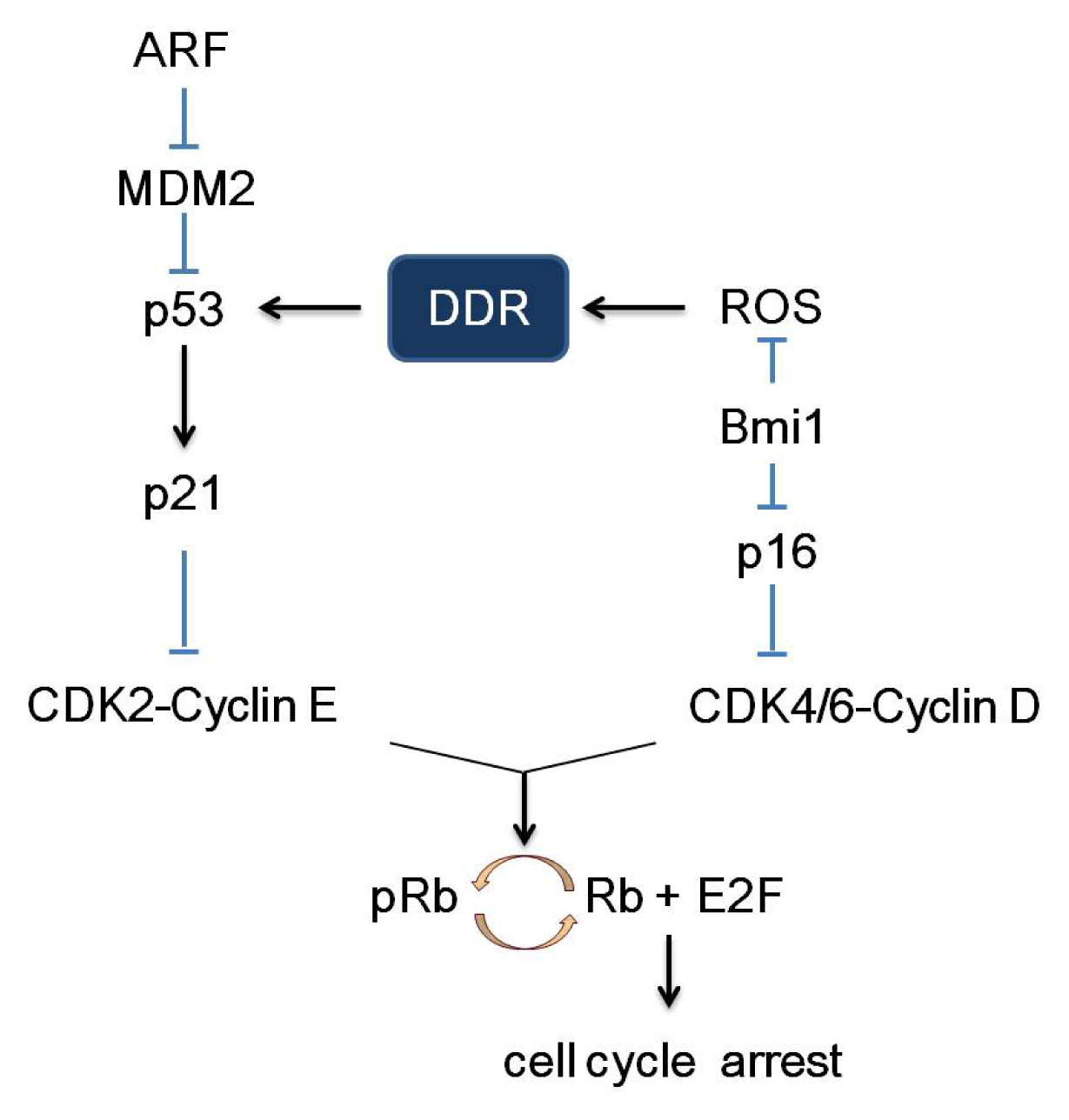
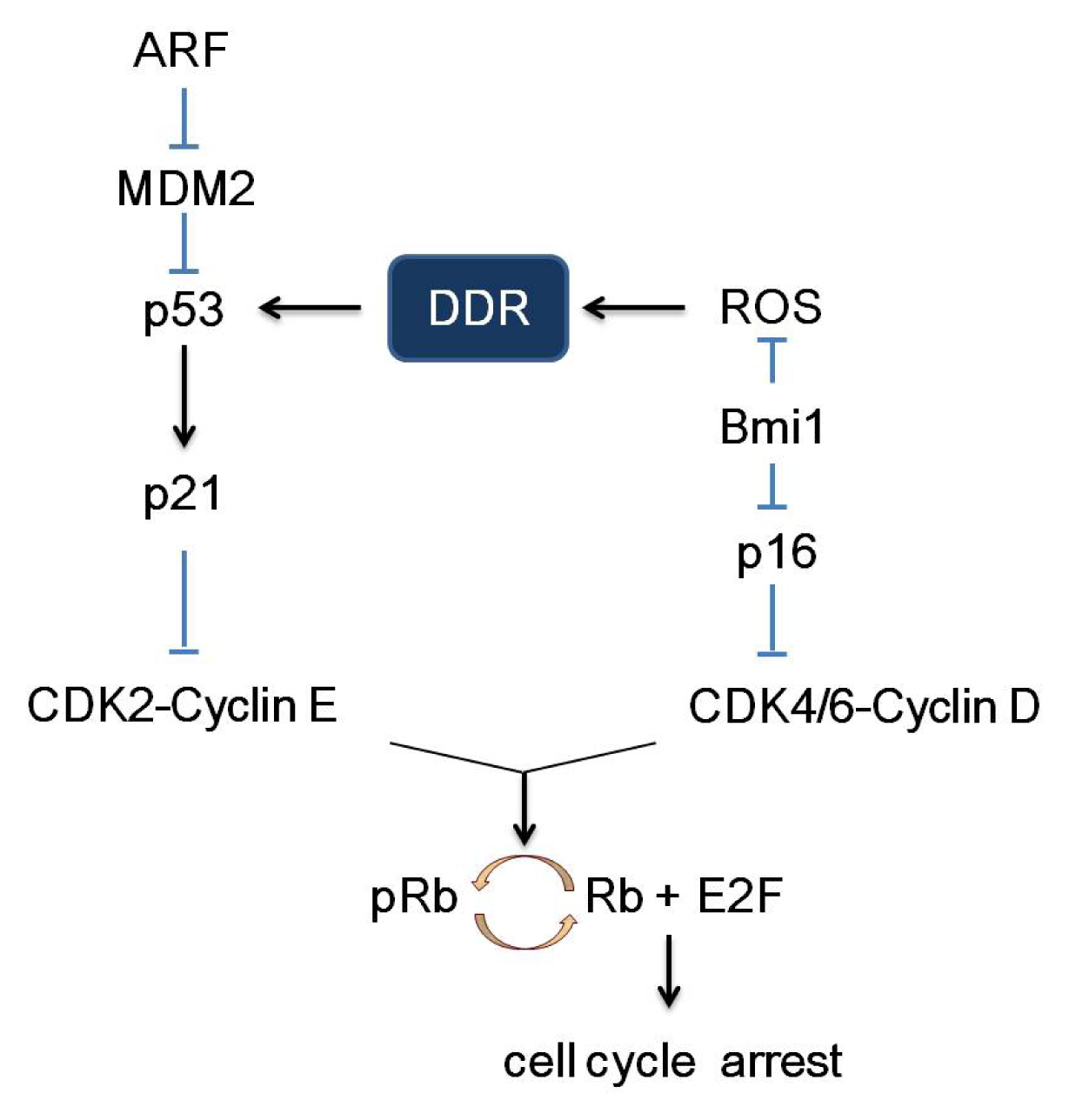
2. Pathways of Senescence-Associated Cell Cycle Arrest
3. DDR Is the Common Link between Tumorigenesis and Senescence
3.1. p53
3.2. p21 and p27
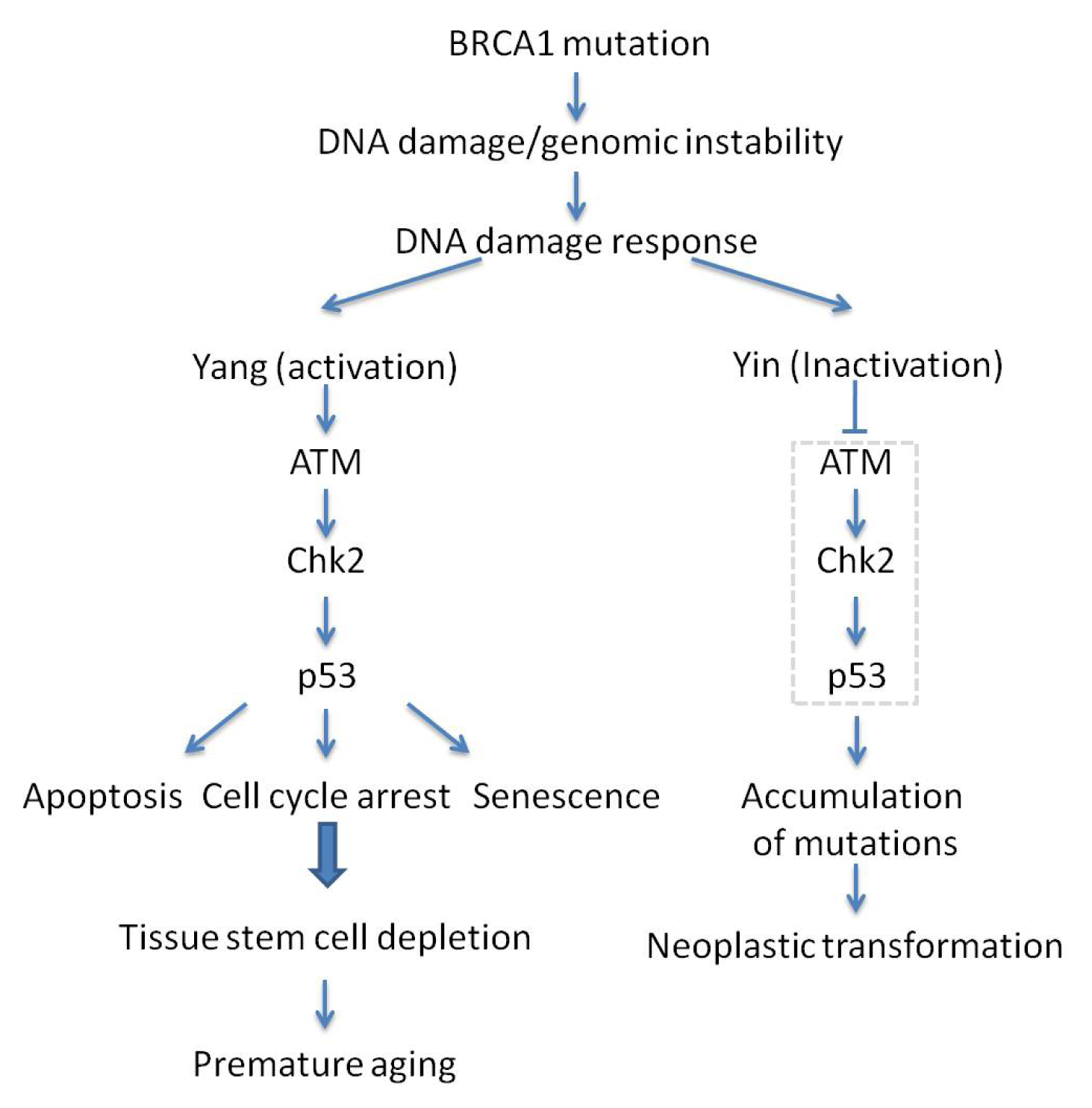
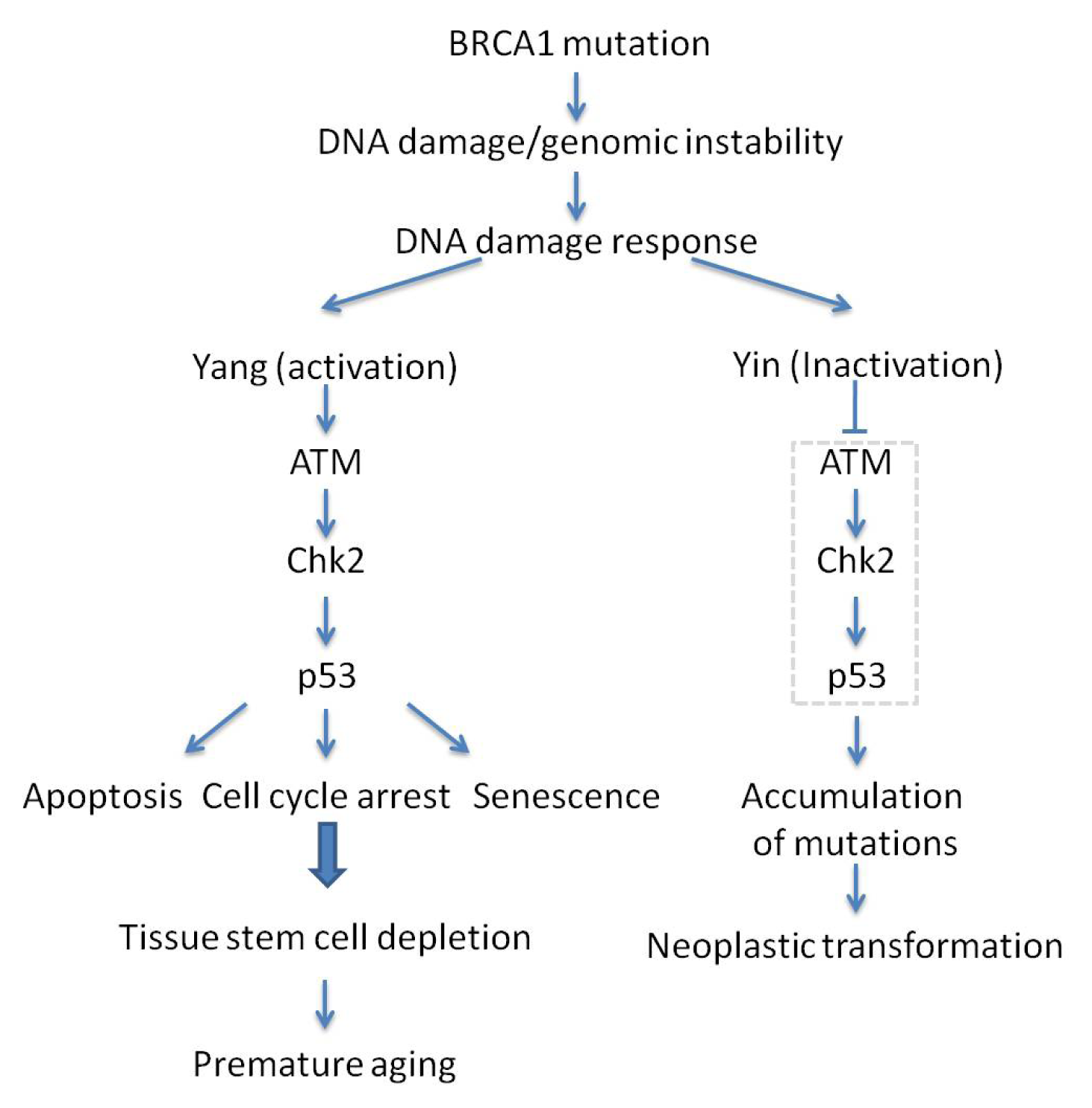
3.3. BRCA1
3.4. p16 and Bmi1
4. Will the Yin-Yang of DDR Be Beneficial for Clinical Treatment of Cancer?
5. Conclusions
Acknowledgments
Conflict of Interest
References
- Thompson, L.H. Recognition, signaling, and repair of DNA double-strand breaks produced by ionizing radiation in mammalian cells: The molecular choreography. Mutat. Res 2012, 751, 158–246. [Google Scholar]
- Lopez-Girona, A.; Tanaka, K.; Chen, X.B.; Baber, B.A.; McGowan, C.H.; Russell, P. Serine-345 is required for Rad3-dependent phosphorylation and function of checkpoint kinase Chk1 in fission yeast. Proc. Natl. Acad. Sci. USA 2001, 98, 11289–11294. [Google Scholar]
- Reinhardt, H.C.; Yaffe, M.B. Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Curr. Opin. Cell Biol 2009, 21, 245–255. [Google Scholar]
- Guo, Z.; Kumagai, A.; Wang, S.X.; Dunphy, W.G. Requirement for Atr in phosphorylation of Chk1 and cell cycle regulation in response to DNA replication blocks and UV-damaged DNA in Xenopus egg extracts. Genes Dev 2000, 14, 2745–2756. [Google Scholar]
- Zhao, H.; Piwnica-Worms, H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol. Cell Biol 2001, 21, 4129–4139. [Google Scholar]
- Lee, J.H.; Paull, T.T. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 2005, 308, 551–554. [Google Scholar]
- Suzuki, K.; Kodama, S.; Watanabe, M. Recruitment of ATM protein to double strand DNA irradiated with ionizing radiation. J. Biol. Chem 1999, 274, 25571–25575. [Google Scholar]
- Lee, J.H.; Paull, T.T. Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene 2007, 26, 7741–7748. [Google Scholar]
- Lee, J.H.; Paull, T.T. Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science 2004, 304, 93–96. [Google Scholar]
- Lisby, M.; Barlow, J.H.; Burgess, R.C.; Rothstein, R. Choreography of the DNA damage response: Spatiotemporal relationships among checkpoint and repair proteins. Cell 2004, 118, 699–713. [Google Scholar]
- Kim, J.S.; Krasieva, T.B.; Kurumizaka, H.; Chen, D.J.; Taylor, A.M.; Yokomori, K. Independent and sequential recruitment of NHEJ and HR factors to DNA damage sites in mammalian cells. J. Cell Biol 2005, 170, 341–347. [Google Scholar]
- Uziel, T.; Lerenthal, Y.; Moyal, L.; Andegeko, Y.; Mittelman, L.; Shiloh, Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J 2003, 22, 5612–5621. [Google Scholar]
- Dart, D.A.; Adams, K.E.; Akerman, I.; Lakin, N.D. Recruitment of the cell cycle checkpoint kinase ATR to chromatin during S-phase. J. Biol. Chem 2004, 279, 16433–16440. [Google Scholar]
- Lupardus, P.J.; Byun, T.; Yee, M.C.; Hekmat-Nejad, M.; Cimprich, K.A. A requirement for replication in activation of the ATR-dependent DNA damage checkpoint. Genes Dev 2002, 16, 2327–2332. [Google Scholar]
- Zou, L.; Elledge, S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548. [Google Scholar]
- Smith, J.; Tho, L.M.; Xu, N.; Gillespie, D.A. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res 2010, 108, 73–112. [Google Scholar]
- Cortez, D.; Guntuku, S.; Qin, J.; Elledge, S.J. ATR and ATRIP: Partners in checkpoint signaling. Science 2001, 294, 1713–1716. [Google Scholar]
- Noon, A.T.; Goodarzi, A.A. 53BP1-mediated DNA double strand break repair: Insert bad pun here. DNA Repair 2011, 10, 1071–1076. [Google Scholar]
- Polo, S.E.; Jackson, S.P. Dynamics of DNA damage response proteins at DNA breaks: A focus on protein modifications. Genes Dev 2011, 25, 409–433. [Google Scholar]
- Banin, S.; Moyal, L.; Shieh, S.; Taya, Y.; Anderson, C.W.; Chessa, L.; Smorodinsky, N.I.; Prives, C.; Reiss, Y.; Shiloh, Y.; Ziv, Y. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 1998, 281, 1674–1677. [Google Scholar]
- Canman, C.E.; Lim, D.S.; Cimprich, K.A.; Taya, Y.; Tamai, K.; Sakaguchi, K.; Appella, E.; Kastan, M.B.; Siliciano, J.D. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science 1998, 281, 1677–1679. [Google Scholar]
- Khanna, K.K.; Keating, K.E.; Kozlov, S.; Scott, S.; Gatei, M.; Hobson, K.; Taya, Y.; Gabrielli, B.; Chan, D.; Lees-Miller, S.P.; Lavin, M.F. ATM associates with and phosphorylates p53: Mapping the region of interaction. Nat. Genet 1998, 20, 398–400. [Google Scholar]
- Lakin, N.D.; Hann, B.C.; Jackson, S.P. The ataxia-telangiectasia related protein ATR mediates DNA-dependent phosphorylation of p53. Oncogene 1999, 18, 3989–3995. [Google Scholar]
- Meek, D.W. Tumour suppression by p53: A role for the DNA damage response? Nat. Rev. Cancer 2009, 9, 714–723. [Google Scholar]
- Meulmeester, E.; Maurice, M.M.; Boutell, C.; Teunisse, A.F.; Ovaa, H.; Abraham, T.E.; Dirks, R.W.; Jochemsen, A.G. Loss of HAUSP-mediated deubiquitination contributes to DNA damage-induced destabilization of Hdmx and Hdm2. Mol. Cell 2005, 18, 565–576. [Google Scholar]
- Meulmeester, E.; Pereg, Y.; Shiloh, Y.; Jochemsen, A.G. ATM-mediated phosphorylations inhibit Mdmx/Mdm2 stabilization by HAUSP in favor of p53 activation. Cell Cycle 2005, 4, 1166–1170. [Google Scholar]
- Stommel, J.M.; Wahl, G.M. Accelerated MDM2 auto-degradation induced by DNA-damage kinases is required for p53 activation. EMBO J 2004, 23, 1547–1556. [Google Scholar]
- Oren, M. Decision making by p53: Life, death and cancer. Cell Death Differ 2003, 10, 431–442. [Google Scholar]
- Sionov, R.V.; Haupt, Y. The cellular response to p53: The decision between life and death. Oncogene 1999, 18, 6145–6157. [Google Scholar]
- Kuerbitz, S.J.; Plunkett, B.S.; Walsh, W.V.; Kastan, M.B. Wild-type p53 is a cell cycle checkpoint determinant following irradiation. Proc. Natl. Acad. Sci. USA 1992, 89, 7491–7495. [Google Scholar]
- Dulic, V.; Kaufmann, W.K.; Wilson, S.J.; Tlsty, T.D.; Lees, E.; Harper, J.W.; Elledge, S.J.; Reed, S.I. p53-dependent inhibition of cyclin-dependent kinase activities in human fibroblasts during radiation-induced G1 arrest. Cell 1994, 76, 1013–1023. [Google Scholar]
- Reed, S.I.; Bailly, E.; Dulic, V.; Hengst, L.; Resnitzky, D.; Slingerland, J. G1 control in mammalian cells. J. Cell Sci. Suppl 1994, 18, 69–73. [Google Scholar]
- Liu, J.; Cao, L.; Chen, J.; Song, S.; Lee, I.H.; Quijano, C.; Liu, H.; Keyvanfar, K.; Chen, H.; Cao, L.Y.; et al. Bmi1 regulates mitochondrial function and the DNA damage response pathway. Nature 2009, 459, 387–392. [Google Scholar]
- Hayflick, L. The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res 1965, 37, 614–636. [Google Scholar]
- Herbig, U.; Jobling, W.A.; Chen, B.P.; Chen, D.J.; Sedivy, J.M. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a). Mol. Cell 2004, 14, 501–513. [Google Scholar]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar]
- Bartek, J.; Lukas, J.; Bartkova, J. DNA damage response as an anti-cancer barrier: Damage threshold and the concept of ‘conditional haploinsufficiency’. Cell Cycle 2007, 6, 2344–2347. [Google Scholar]
- Campisi, J. Cellular senescence as a tumor-suppressor mechanism. Trends Cell Biol 2001, 11, S27–S31. [Google Scholar]
- Shay, J.W.; Roninson, I.B. Hallmarks of senescence in carcinogenesis and cancer therapy. Oncogene 2004, 23, 2919–2933. [Google Scholar]
- Campisi, J. Senescent cells, tumor suppression, and organismal aging: Good citizens, bad neighbors. Cell 2005, 120, 513–522. [Google Scholar]
- Lundberg, A.S.; Hahn, W.C.; Gupta, P.; Weinberg, R.A. Genes involved in senescence and immortalization. Curr. Opin. Cell Biol 2000, 12, 705–709. [Google Scholar]
- Jacobs, J.J.; Kieboom, K.; Marino, S.; DePinho, R.A.; van Lohuizen, M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature 1999, 397, 164–168. [Google Scholar]
- Carnero, A.; Hudson, J.D.; Price, C.M.; Beach, D.H. p16INK4A and p19ARF act in overlapping pathways in cellular immortalization. Nat. Cell Biol 2000, 2, 148–155. [Google Scholar]
- Sharpless, N.E. Ink4a/Arf links senescence and aging. Exp. Gerontol 2004, 39, 1751–1759. [Google Scholar]
- Brown, J.P.; Wei, W.; Sedivy, J.M. Bypass of senescence after disruption of p21CIP1/WAF1 gene in normal diploid human fibroblasts. Science 1997, 277, 831–834. [Google Scholar]
- Zilfou, J.T.; Lowe, S.W. Tumor suppressive functions of p53. Cold Spring Harb. Perspect. Biol 2009, 1, a001883. [Google Scholar]
- Bos, J.L. The ras gene family and human carcinogenesis. Mutat. Res 1988, 195, 255–271. [Google Scholar]
- Braig, M.; Schmitt, C.A. Oncogene-induced senescence: Putting the brakes on tumor development. Cancer Res 2006, 66, 2881–2884. [Google Scholar]
- Lowe, S.W.; Cepero, E.; Evan, G. Intrinsic tumour suppression. Nature 2004, 432, 307–315. [Google Scholar]
- Bartkova, J.; Rezaei, N.; Liontos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.V.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C.; et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006, 444, 633–637. [Google Scholar]
- Di Micco, R.; Fumagalli, M.; Cicalese, A.; Piccinin, S.; Gasparini, P.; Luise, C.; Schurra, C.; Garre, M.; Nuciforo, P.G.; Bensimon, A.; et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006, 444, 638–642. [Google Scholar]
- Collado, M.; Serrano, M. Senescence in tumours: Evidence from mice and humans. Nat. Rev. Cancer 2010, 10, 51–57. [Google Scholar]
- Bartkova, J.; Horejsi, Z.; Koed, K.; Kramer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar]
- Gorgoulis, V.G.; Vassiliou, L.V.; Karakaidos, P.; Zacharatos, P.; Kotsinas, A.; Liloglou, T.; Venere, M.; Ditullio, R.A., Jr.; Kastrinakis, N.G.; Levy, B.; et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005, 434, 907–913. [Google Scholar]
- DiTullio, R.A., Jr; Mochan, T.A.; Venere, M.; Bartkova, J.; Sehested, M.; Bartek, J.; Halazonetis, T.D. 53BP1 functions in an ATM-dependent checkpoint pathway that is constitutively activated in human cancer. Nat. Cell Biol. 2002, 4, 998–1002. [Google Scholar]
- Bartkova, J.; Bakkenist, C.J.; Rajpert-De Meyts, E.; Skakkebaek, N.E.; Sehested, M.; Lukas, J.; Kastan, M.B.; Bartek, J. ATM activation in normal human tissues and testicular cancer. Cell Cycle 2005, 4, 838–845. [Google Scholar]
- Nuciforo, P.G.; Luise, C.; Capra, M.; Pelosi, G.; d’Adda di Fagagna, F. Complex engagement of DNA damage response pathways in human cancer and in lung tumor progression. Carcinogenesis 2007, 28, 2082–2088. [Google Scholar]
- Fan, C.; Quan, R.; Feng, X.; Gillis, A.; He, L.; Matsumoto, E.D.; Salama, S.; Cutz, J.C.; Kapoor, A.; Tang, D. ATM activation is accompanied with earlier stages of prostate tumorigenesis. Biochim. Biophys. Acta 2006, 1763, 1090–1097. [Google Scholar]
- Chen, Z.; Trotman, L.C.; Shaffer, D.; Lin, H.K.; Dotan, Z.A.; Niki, M.; Koutcher, J.A.; Scher, H.I.; Ludwig, T.; Gerald, W.; et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005, 436, 725–730. [Google Scholar]
- Braig, M.; Lee, S.; Loddenkemper, C.; Rudolph, C.; Peters, A.H.; Schlegelberger, B.; Stein, H.; Dorken, B.; Jenuwein, T.; Schmitt, C.A. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature 2005, 436, 660–665. [Google Scholar]
- Schmitt, C.A.; Fridman, J.S.; Yang, M.; Lee, S.; Baranov, E.; Hoffman, R.M.; Lowe, S.W. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell 2002, 109, 335–346. [Google Scholar]
- Michaloglou, C.; Vredeveld, L.C.; Soengas, M.S.; Denoyelle, C.; Kuilman, T.; van der Horst, C.M.; Majoor, D.M.; Shay, J.W.; Mooi, W.J.; Peeper, D.S. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2005, 436, 720–724. [Google Scholar]
- Lazzerini Denchi, E.; Attwooll, C.; Pasini, D.; Helin, K. Deregulated E2F activity induces hyperplasia and senescence-like features in the mouse pituitary gland. Mol. Cell Biol 2005, 25, 2660–2672. [Google Scholar]
- Collado, M.; Gil, J.; Efeyan, A.; Guerra, C.; Schuhmacher, A.J.; Barradas, M.; Benguria, A.; Zaballos, A.; Flores, J.M.; Barbacid, M.; et al. Tumour biology: Senescence in premalignant tumours. Nature 2005, 436, 642. [Google Scholar]
- Mallette, F.A.; Gaumont-Leclerc, M.F.; Ferbeyre, G. The DNA damage signaling pathway is a critical mediator of oncogene-induced senescence. Genes Dev 2007, 21, 43–48. [Google Scholar]
- Ohtani, N.; Takahashi, A.; Mann, D.J.; Hara, E. Cellular senescence: A double-edged sword in the fight against cancer. Exp. Dermatol 2012, 21, 1–4. [Google Scholar]
- Giaimo, S.; d’Adda di Fagagna, F. Is cellular senescence an example of antagonistic pleiotropy? Aging Cell 2012, 11, 378–383. [Google Scholar]
- Larsson, L.G. Oncogene- and tumor suppressor gene-mediated suppression of cellular senescence. Semin. Cancer Biol 2011, 21, 367–376. [Google Scholar]
- Tyner, S.D.; Venkatachalam, S.; Choi, J.; Jones, S.; Ghebranious, N.; Igelmann, H.; Lu, X.; Soron, G.; Cooper, B.; Brayton, C.; et al. p53 mutant mice that display early ageing-associated phenotypes. Nature 2002, 415, 45–53. [Google Scholar]
- Dumble, M.; Moore, L.; Chambers, S.M.; Geiger, H.; van Zant, G.; Goodell, M.A.; Donehower, L.A. The impact of altered p53 dosage on hematopoietic stem cell dynamics during aging. Blood 2007, 109, 1736–1742. [Google Scholar]
- Donehower, L.A.; Harvey, M.; Slagle, B.L.; McArthur, M.J.; Montgomery, C.A., Jr; Butel, J.S.; Bradley, A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992, 356, 215–221. [Google Scholar]
- Jacks, T.; Remington, L.; Williams, B.O.; Schmitt, E.M.; Halachmi, S.; Bronson, R.T.; Weinberg, R.A. Tumor spectrum analysis in p53-mutant mice. Curr. Biol 1994, 4, 1–7. [Google Scholar]
- Liu, G.; Chen, X. Regulation of the p53 transcriptional activity. J. Cell Biochem 2006, 97, 448–458. [Google Scholar]
- Soussi, T.; Wiman, K.G. Shaping genetic alterations in human cancer: The p53 mutation paradigm. Cancer Cell 2007, 12, 303–312. [Google Scholar]
- Rodier, F.; Campisi, J.; Bhaumik, D. Two faces of p53: Aging and tumor suppression. Nucleic Acids Res 2007, 35, 7475–7484. [Google Scholar]
- Garcia-Cao, I.; Garcia-Cao, M.; Martin-Caballero, J.; Criado, L.M.; Klatt, P.; Flores, J.M.; Weill, J.C.; Blasco, M.A.; Serrano, M. “Super p53” mice exhibit enhanced DNA damage response, are tumor resistant and age normally. EMBO J 2002, 21, 6225–6235. [Google Scholar]
- Serrano, M.; Blasco, M.A. Cancer and ageing: Convergent and divergent mechanisms. Nat. Rev. Mol. Cell Biol 2007, 8, 715–722. [Google Scholar]
- Benson, E.K.; Zhao, B.; Sassoon, D.A.; Lee, S.W.; Aaronson, S.A. Effects of p21 deletion in mouse models of premature aging. Cell Cycle 2009, 8, 2002–2004. [Google Scholar]
- Zhao, B.; Benson, E.K.; Qiao, R.; Wang, X.; Kim, S.; Manfredi, J.J.; Lee, S.W.; Aaronson, S.A. Cellular senescence and organismal ageing in the absence of p21(CIP1/WAF1) in ku80(−/−) mice. EMBO Rep 2009, 10, 71–78. [Google Scholar]
- Davis, T.; Singhrao, S.K.; Wyllie, F.S.; Haughton, M.F.; Smith, P.J.; Wiltshire, M.; Wynford-Thomas, D.; Jones, C.J.; Faragher, R.G.; Kipling, D. Telomere-based proliferative lifespan barriers in Werner-syndrome fibroblasts involve both p53-dependent and p53-independent mechanisms. J. Cell Sci 2003, 116, 1349–1357. [Google Scholar]
- Stoyanova, T.; Roy, N.; Bhattacharjee, S.; Kopanja, D.; Valli, T.; Bagchi, S.; Raychaudhuri, P. p21 cooperates with DDB2 protein in suppression of ultraviolet ray-induced skin malignancies. J. Biol. Chem 2012, 287, 3019–3028. [Google Scholar]
- Choudhury, A.R.; Ju, Z.; Djojosubroto, M.W.; Schienke, A.; Lechel, A.; Schaetzlein, S.; Jiang, H.; Stepczynska, A.; Wang, C.; Buer, J.; et al. Cdkn1a deletion improves stem cell function and lifespan of mice with dysfunctional telomeres without accelerating cancer formation. Nat. Genet. 2007, 39, 99–105. [Google Scholar]
- Rudolph, K.L.; Chang, S.; Lee, H.W.; Blasco, M.; Gottlieb, G.J.; Greider, C.; DePinho, R.A. Longevity, stress response, and cancer in aging telomerase-deficient mice. Cell 1999, 96, 701–712. [Google Scholar]
- Chin, L.; Artandi, S.E.; Shen, Q.; Tam, A.; Lee, S.L.; Gottlieb, G.J.; Greider, C.W.; DePinho, R.A. p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell 1999, 97, 527–638. [Google Scholar]
- Artandi, S.E.; Chang, S.; Lee, S.L.; Alson, S.; Gottlieb, G.J.; Chin, L.; DePinho, R.A. Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature 2000, 406, 641–645. [Google Scholar]
- Martin-Caballero, J.; Flores, J.M.; Garcia-Palencia, P.; Serrano, M. Tumor susceptibility of p21(Waf1/Cip1)-deficient mice. Cancer Res 2001, 61, 6234–6238. [Google Scholar]
- Stivala, L.A.; Cazzalini, O.; Prosperi, E. The cyclin-dependent kinase inhibitor p21CDKN1A as a target of anti-cancer drugs. Curr. Cancer Drug Targets 2012, 12, 85–96. [Google Scholar]
- Roy, N.; Stoyanova, T.; Dominguez-Brauer, C.; Park, H.J.; Bagchi, S.; Raychaudhuri, P. DDB2, an essential mediator of premature senescence. Mol. Cell Biol 2010, 30, 2681–2692. [Google Scholar]
- Itoh, T.; Cado, D.; Kamide, R.; Linn, S. DDB2 gene disruption leads to skin tumors and resistance to apoptosis after exposure to ultraviolet light but not a chemical carcinogen. Proc. Natl. Acad. Sci. USA 2004, 101, 2052–2057. [Google Scholar]
- Yoon, T.; Chakrabortty, A.; Franks, R.; Valli, T.; Kiyokawa, H.; Raychaudhuri, P. Tumor-prone phenotype of the DDB2-deficient mice. Oncogene 2005, 24, 469–478. [Google Scholar]
- Stoyanova, T.; Roy, N.; Kopanja, D.; Bagchi, S.; Raychaudhuri, P. DDB2 decides cell fate following DNA damage. Proc. Natl. Acad. Sci. USA 2009, 106, 10690–10695. [Google Scholar]
- Stoyanova, T.; Yoon, T.; Kopanja, D.; Mokyr, M.B.; Raychaudhuri, P. The xeroderma pigmentosum group E gene product DDB2 activates nucleotide excision repair by regulating the level of p21Waf1/Cip1. Mol. Cell Biol 2008, 28, 177–187. [Google Scholar]
- Abbas, T.; Dutta, A. p21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar]
- Cazzalini, O.; Scovassi, A.I.; Savio, M.; Stivala, L.A.; Prosperi, E. Multiple roles of the cell cycle inhibitor p21(CDKN1A) in the DNA damage response. Mutat. Res 2010, 704, 12–20. [Google Scholar]
- Fotedar, R.; Bendjennat, M.; Fotedar, A. Role of p21WAF1 in the cellular response to UV. Cell Cycle 2004, 3, 134–137. [Google Scholar]
- Roninson, I.B. Oncogenic functions of tumour suppressor p21(Waf1/Cip1/Sdi1): Association with cell senescence and tumour-promoting activities of stromal fibroblasts. Cancer Lett 2002, 179, 1–14. [Google Scholar]
- Gartel, A.L. Is p21 an oncogene? Mol. Cancer Ther 2006, 5, 1385–1386. [Google Scholar]
- Nickeleit, I.; Zender, S.; Kossatz, U.; Malek, N.P. p27kip1: A target for tumor therapies? Cell Div 2007, 2, 13. [Google Scholar]
- Slingerland, J.; Pagano, M. Regulation of the cdk inhibitor p27 and its deregulation in cancer. J. Cell Physiol 2000, 183, 10–17. [Google Scholar]
- Loda, M.; Cukor, B.; Tam, S.W.; Lavin, P.; Fiorentino, M.; Draetta, G.F.; Jessup, J.M.; Pagano, M. Increased proteasome-dependent degradation of the cyclin-dependent kinase inhibitor p27 in aggressive colorectal carcinomas. Nat. Med 1997, 3, 231–234. [Google Scholar]
- Porter, P.L.; Malone, K.E.; Heagerty, P.J.; Alexander, G.M.; Gatti, L.A.; Firpo, E.J.; Daling, J.R.; Roberts, J.M. Expression of cell-cycle regulators p27Kip1 and cyclin E, alone and in combination, correlate with survival in young breast cancer patients. Nat. Med 1997, 3, 222–225. [Google Scholar]
- Yang, R.M.; Naitoh, J.; Murphy, M.; Wang, H.J.; Phillipson, J.; deKernion, J.B.; Loda, M.; Reiter, R.E. Low p27 expression predicts poor disease-free survival in patients with prostate cancer. J. Urol 1998, 159, 941–945. [Google Scholar]
- Chu, I.M.; Hengst, L.; Slingerland, J.M. The Cdk inhibitor p27 in human cancer: Prognostic potential and relevance to anticancer therapy. Nat. Rev. Cancer 2008, 8, 253–267. [Google Scholar]
- Fero, M.L.; Rivkin, M.; Tasch, M.; Porter, P.; Carow, C.E.; Firpo, E.; Polyak, K.; Tsai, L.H.; Broudy, V.; Perlmutter, R.M.; Kaushansky, K.; Roberts, J.M. A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27(Kip1)-deficient mice. Cell 1996, 85, 733–744. [Google Scholar]
- Muraoka, R.S.; Lenferink, A.E.; Law, B.; Hamilton, E.; Brantley, D.M.; Roebuck, L.R.; Arteaga, C.L. ErbB2/Neu-induced, cyclin D1-dependent transformation is accelerated in p27-haploinsufficient mammary epithelial cells but impaired in p27-null cells. Mol. Cell Biol 2002, 22, 2204–2219. [Google Scholar]
- Besson, A.; Gurian-West, M.; Chen, X.; Kelly-Spratt, K.S.; Kemp, C.J.; Roberts, J.M. A pathway in quiescent cells that controls p27Kip1 stability, subcellular localization, and tumor suppression. Genes Dev 2006, 20, 47–64. [Google Scholar]
- Cordon-Cardo, C.; Koff, A.; Drobnjak, M.; Capodieci, P.; Osman, I.; Millard, S.S.; Gaudin, P.B.; Fazzari, M.; Zhang, Z.F.; Massague, J.; Scher, H.I. Distinct altered patterns of p27KIP1 gene expression in benign prostatic hyperplasia and prostatic carcinoma. J. Natl. Cancer Inst 1998, 90, 1284–1291. [Google Scholar]
- Fero, M.L.; Randel, E.; Gurley, K.E.; Roberts, J.M.; Kemp, C.J. The murine gene p27Kip1 is haplo-insufficient for tumour suppression. Nature 1998, 396, 177–180. [Google Scholar]
- Miki, Y.; Swensen, J.; Shattuck-Eidens, D.; Futreal, P.A.; Harshman, K.; Tavtigian, S.; Liu, Q.; Cochran, C.; Bennett, L.M.; Ding, W.; et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994, 266, 66–71. [Google Scholar]
- Alberg, A.J.; Helzlsouer, K.J. Epidemiology, prevention, and early detection of breast cancer. Curr. Opin. Oncol 1997, 9, 505–511. [Google Scholar]
- Brody, L.C.; Biesecker, B.B. Breast cancer susceptibility genes. BRCA1 and BRCA2. Medicine 1998, 77, 208–226. [Google Scholar]
- Paterson, J.W. BRCA1: A review of structure and putative functions. Dis. Markers 1998, 13, 261–274. [Google Scholar]
- Rahman, N.; Stratton, M.R. The genetics of breast cancer susceptibility. Annu. Rev. Genet 1998, 32, 95–121. [Google Scholar]
- Scully, R.; Livingston, D.M. In search of the tumour-suppressor functions of BRCA1 and BRCA2. Nature 2000, 408, 429–432. [Google Scholar]
- Zhang, J.; Powell, S.N. The role of the BRCA1 tumor suppressor in DNA double-strand break repair. Mol. Cancer Res 2005, 3, 531–539. [Google Scholar]
- Deng, C.X.; Scott, F. Role of the tumor suppressor gene BRCA1 in genetic stability and mammary gland tumor formation. Oncogene 2000, 19, 1059–1064. [Google Scholar]
- Zheng, L.; Li, S.; Boyer, T.G.; Lee, W.H. Lessons learned from BRCA1 and BRCA2. Oncogene 2000, 19, 6159–6175. [Google Scholar]
- Venkitaraman, A.R. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell 2002, 108, 171–182. [Google Scholar]
- Cao, L.; Li, W.; Kim, S.; Brodie, S.G.; Deng, C.X. Senescence, aging, and malignant transformation mediated by p53 in mice lacking the BRCA1 full-length isoform. Genes Dev 2003, 17, 201–213. [Google Scholar]
- Cao, L.; Kim, S.; Xiao, C.; Wang, R.H.; Coumoul, X.; Wang, X.; Li, W.M.; Xu, X.L.; de Soto, J.A.; Takai, H.; et al. ATM-Chk2-p53 activation prevents tumorigenesis at an expense of organ homeostasis upon BRCA1 deficiency. EMBO J. 2006, 25, 2167–2177. [Google Scholar]
- Hakem, R.; de la Pompa, J.L.; Elia, A.; Potter, J.; Mak, T.W. Partial rescue of BRCA1 (5–6) early embryonic lethality by p53 or p21 null mutation. Nat. Genet 1997, 16, 298–302. [Google Scholar]
- Xu, X.; Qiao, W.; Linke, S.P.; Cao, L.; Li, W.M.; Furth, P.A.; Harris, C.C.; Deng, C.X. Genetic interactions between tumor suppressors BRCA1 and p53 in apoptosis, cell cycle and tumorigenesis. Nat. Genet 2001, 28, 266–271. [Google Scholar]
- Cao, L.; Xu, X.; Bunting, S.F.; Liu, J.; Wang, R.H.; Cao, L.L.; Wu, J.J.; Peng, T.N.; Chen, J.; Nussenzweig, A.; Deng, C.X.; Finkel, T. A selective requirement for 53BP1 in the biological response to genomic instability induced by BRCA1 deficiency. Mol. Cell 2009, 35, 534–541. [Google Scholar]
- Wang, W.; Wu, J.; Zhang, Z.; Tong, T. Characterization of regulatory elements on the promoter region of p16(INK4a) that contribute to overexpression of p16 in senescent fibroblasts. J. Biol. Chem 2001, 276, 48655–48661. [Google Scholar]
- Palmero, I.; McConnell, B.; Parry, D.; Brookes, S.; Hara, E.; Bates, S.; Jat, P.; Peters, G. Accumulation of p16INK4a in mouse fibroblasts as a function of replicative senescence and not of retinoblastoma gene status. Oncogene 1997, 15, 495–503. [Google Scholar]
- Tsutsui, T.; Kumakura, S.; Yamamoto, A.; Kanai, H.; Tamura, Y.; Kato, T.; Anpo, M.; Tahara, H.; Barrett, J.C. Association of p16(INK4a) and pRb inactivation with immortalization of human cells. Carcinogenesis 2002, 23, 2111–2117. [Google Scholar]
- Melk, A.; Kittikowit, W.; Sandhu, I.; Halloran, K.M.; Grimm, P.; Schmidt, B.M.; Halloran, P.F. Cell senescence in rat kidneys in vivo increases with growth and age despite lack of telomere shortening. Kidney Int 2003, 63, 2134–2143. [Google Scholar]
- Nielsen, G.P.; Stemmer-Rachamimov, A.O.; Shaw, J.; Roy, J.E.; Koh, J.; Louis, D.N. Immunohistochemical survey of p16INK4A expression in normal human adult and infant tissues. Lab Invest 1999, 79, 1137–1143. [Google Scholar]
- Zindy, F.; Quelle, D.E.; Roussel, M.F.; Sherr, C.J. Expression of the p16INK4a tumor suppressor versus other INK4 family members during mouse development and aging. Oncogene 1997, 15, 203–211. [Google Scholar]
- Krimpenfort, P.; Quon, K.C.; Mooi, W.J.; Loonstra, A.; Berns, A. Loss of p16Ink4a confers susceptibility to metastatic melanoma in mice. Nature 2001, 413, 83–86. [Google Scholar]
- Sharpless, N.E.; Bardeesy, N.; Lee, K.H.; Carrasco, D.; Castrillon, D.H.; Aguirre, A.J.; Wu, E.A.; Horner, J.W.; DePinho, R.A. Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature 2001, 413, 86–91. [Google Scholar]
- Gil, J.; Peters, G. Regulation of the INK4b-ARF-INK4a tumour suppressor locus: All for one or one for all. Nat. Rev. Mol. Cell Biol 2006, 7, 667–677. [Google Scholar]
- Kim, W.Y.; Sharpless, N.E. The regulation of INK4/ARF in cancer and aging. Cell 2006, 127, 265–275. [Google Scholar]
- Van der Lugt, N.M.; Domen, J.; Linders, K.; van Roon, M.; Robanus-Maandag, E.; te Riele, H.; van der Valk, M.; Deschamps, J.; Sofroniew, M.; van Lohuizen, M.; et al. Posterior transformation, neurological abnormalities, and severe hematopoietic defects in mice with a targeted deletion of the bmi-1 proto-oncogene. Genes Dev. 1994, 8, 757–769. [Google Scholar]
- Haupt, Y.; Alexander, W.S.; Barri, G.; Klinken, S.P.; Adams, J.M. Novel zinc finger gene implicated as myc collaborator by retrovirally accelerated lymphomagenesis in E mu-myc transgenic mice. Cell 1991, 65, 753–763. [Google Scholar]
- Van Lohuizen, M.; Verbeek, S.; Scheijen, B.; Wientjens, E.; van der Gulden, H.; Berns, A. Identification of cooperating oncogenes in E mu-myc transgenic mice by provirus tagging. Cell 1991, 65, 737–752. [Google Scholar]
- Itahana, K.; Zou, Y.; Itahana, Y.; Martinez, J.L.; Beausejour, C.; Jacobs, J.J.; van Lohuizen, M.; Band, V.; Campisi, J.; Dimri, G.P. Control of the replicative life span of human fibroblasts by p16 and the polycomb protein Bmi-1. Mol. Cell Biol 2003, 23, 389–401. [Google Scholar]
- Takeuchi, S.; Takahashi, A.; Motoi, N.; Yoshimoto, S.; Tajima, T.; Yamakoshi, K.; Hirao, A.; Yanagi, S.; Fukami, K.; Ishikawa, Y.; et al. Intrinsic cooperation between p16INK4a and p21Waf1/Cip1 in the onset of cellular senescence and tumor suppression. in vivo. Cancer Res. 2010, 70, 9381–9390. [Google Scholar]
- Carbone, C.J.; Grana, X.; Reddy, E.P.; Haines, D.S. p21 loss cooperates with INK4 inactivation facilitating immortalization and Bcl-2-mediated anchorage-independent growth of oncogene-transduced primary mouse fibroblasts. Cancer Res 2007, 67, 4130–4137. [Google Scholar]
- Roninson, I.B. Tumor cell senescence in cancer treatment. Cancer Res 2003, 63, 2705–2715. [Google Scholar]
- Roberson, R.S.; Kussick, S.J.; Vallieres, E.; Chen, S.Y.; Wu, D.Y. Escape from therapy-induced accelerated cellular senescence in p53-null lung cancer cells and in human lung cancers. Cancer Res 2005, 65, 2795–2803. [Google Scholar]
- Sidi, R.; Pasello, G.; Opitz, I.; Soltermann, A.; Tutic, M.; Rehrauer, H.; Weder, W.; Stahel, R.A.; Felley-Bosco, E. Induction of senescence markers after neo-adjuvant chemotherapy of malignant pleural mesothelioma and association with clinical outcome: An exploratory analysis. Eur. J. Cancer 2011, 47, 326–332. [Google Scholar]
- Kastan, M.B.; Bartek, J. Cell-cycle checkpoints and cancer. Nature 2004, 432, 316–323. [Google Scholar]
- Chang, B.D.; Swift, M.E.; Shen, M.; Fang, J.; Broude, E.V.; Roninson, I.B. Molecular determinants of terminal growth arrest induced in tumor cells by a chemotherapeutic agent. Proc. Natl. Acad. Sci. USA 2002, 99, 389–394. [Google Scholar]
- Krtolica, A.; Parrinello, S.; Lockett, S.; Desprez, P.Y.; Campisi, J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: A link between cancer and aging. Proc. Natl. Acad. Sci. USA 2001, 98, 12072–12077. [Google Scholar]
- Schmitt, C.A. Cellular senescence and cancer treatment. Biochim. Biophys. Acta 2007, 1775, 5–20. [Google Scholar]
- Coppe, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Munoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 2008, 6, 2853–2868. [Google Scholar]
- Rodier, F.; Campisi, J. Four faces of cellular senescence. J. Cell Biol 2011, 192, 547–556. [Google Scholar]
- Sparmann, A.; Bar-Sagi, D. Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell 2004, 6, 447–458. [Google Scholar]
- Ancrile, B.; Lim, K.H.; Counter, C.M. Oncogenic Ras-induced secretion of IL6 is required for tumorigenesis. Genes Dev 2007, 21, 1714–1719. [Google Scholar]
- Park, E.J.; Lee, J.H.; Yu, G.Y.; He, G.; Ali, S.R.; Holzer, R.G.; Osterreicher, C.H.; Takahashi, H.; Karin, M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 2010, 140, 197–208. [Google Scholar]
- Acosta, J.C.; O’Loghlen, A.; Banito, A.; Guijarro, M.V.; Augert, A.; Raguz, S.; Fumagalli, M.; Da Costa, M.; Brown, C.; Popov, N.; et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 2008, 133, 1006–1018. [Google Scholar]
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.; Douma, S.; van Doorn, R.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 2008, 133, 1019–1031. [Google Scholar]
- Dilley, T.K.; Bowden, G.T.; Chen, Q.M. Novel mechanisms of sublethal oxidant toxicity: Induction of premature senescence in human fibroblasts confers tumor promoter activity. Exp. Cell Res 2003, 290, 38–48. [Google Scholar]
© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Li, X.; Xu, H.; Xu, C.; Lin, M.; Song, X.; Yi, F.; Feng, Y.; Coughlan, K.A.; Cho, W.C.-s.; Kim, S.S.; et al. The Yin-Yang of DNA Damage Response: Roles in Tumorigenesis and Cellular Senescence. Int. J. Mol. Sci. 2013, 14, 2431-2448. https://doi.org/10.3390/ijms14022431
Li X, Xu H, Xu C, Lin M, Song X, Yi F, Feng Y, Coughlan KA, Cho WC-s, Kim SS, et al. The Yin-Yang of DNA Damage Response: Roles in Tumorigenesis and Cellular Senescence. International Journal of Molecular Sciences. 2013; 14(2):2431-2448. https://doi.org/10.3390/ijms14022431
Chicago/Turabian StyleLi, Xiaoman, Hongde Xu, Chongan Xu, Meina Lin, Xiaoyu Song, Fei Yi, Yanling Feng, Kathleen A. Coughlan, William Chi-shing Cho, Sang Soo Kim, and et al. 2013. "The Yin-Yang of DNA Damage Response: Roles in Tumorigenesis and Cellular Senescence" International Journal of Molecular Sciences 14, no. 2: 2431-2448. https://doi.org/10.3390/ijms14022431