Effect of Human Flavin-Containing Monooxygenase 3 Polymorphism on the Metabolism of Aurora Kinase Inhibitors




Abstract
:
1. Introduction
2. Results and Discussion
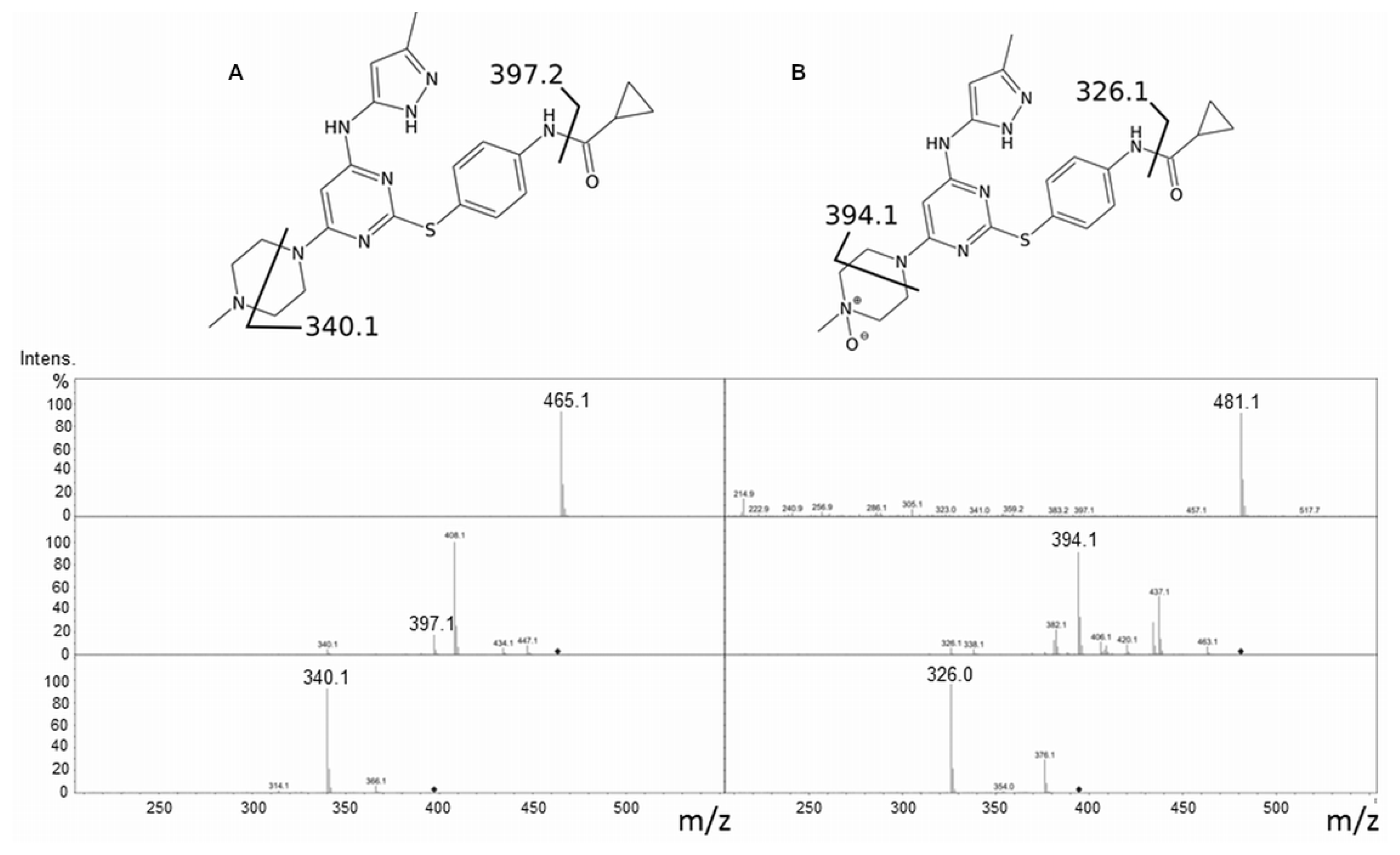
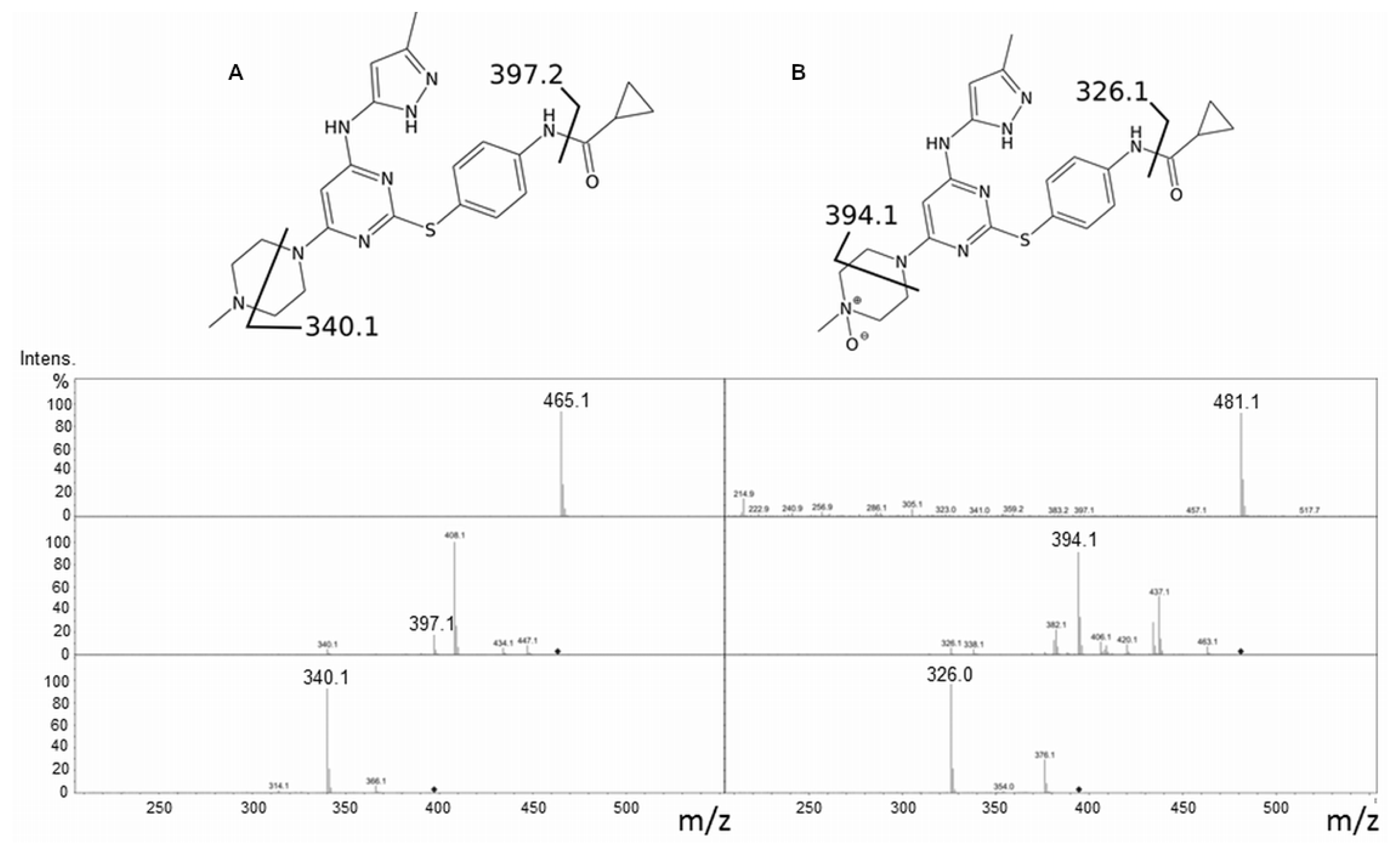
2.1. Tozasertib Metabolism by hFMO3
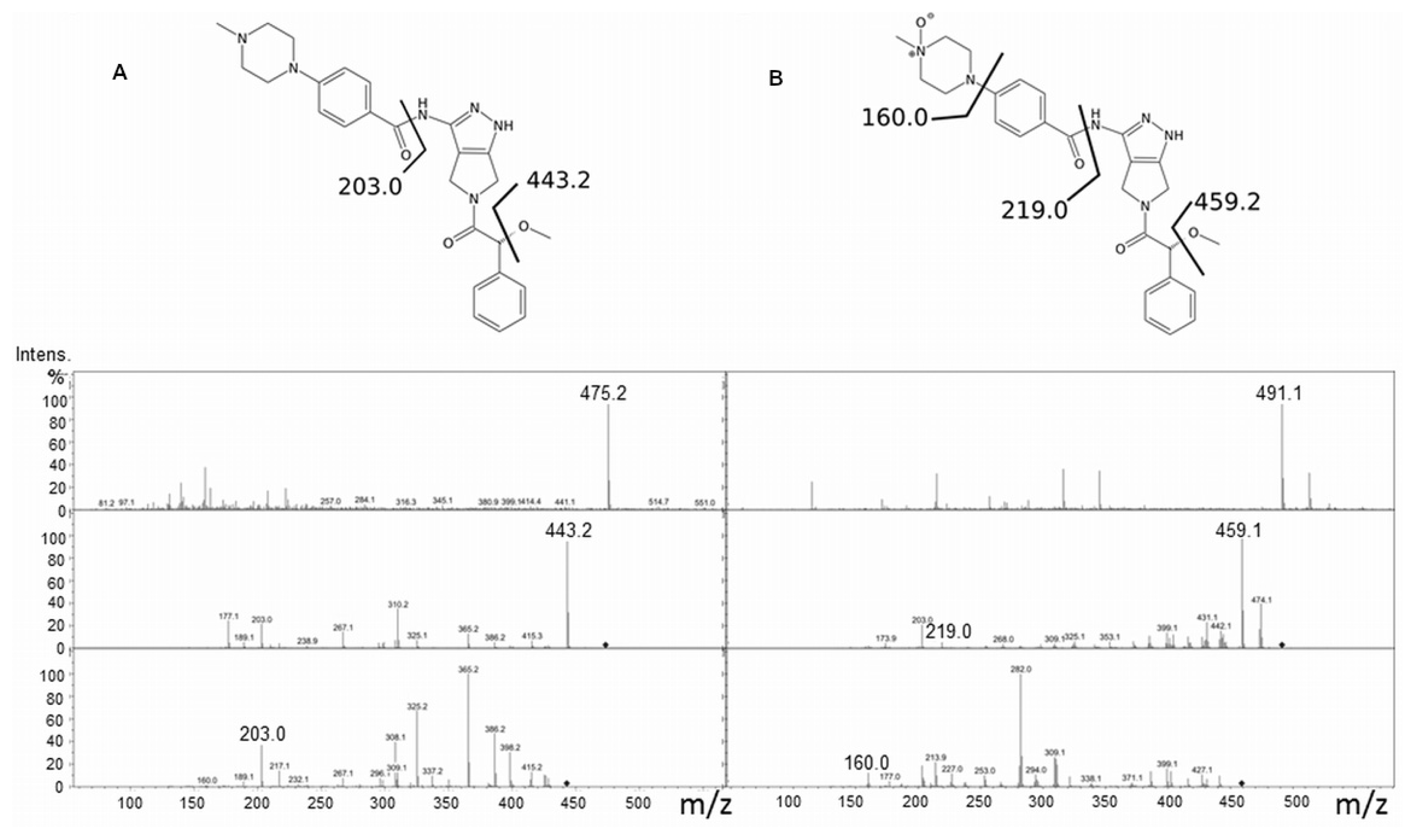
2.2. Danusertib Metabolism by hFMO3
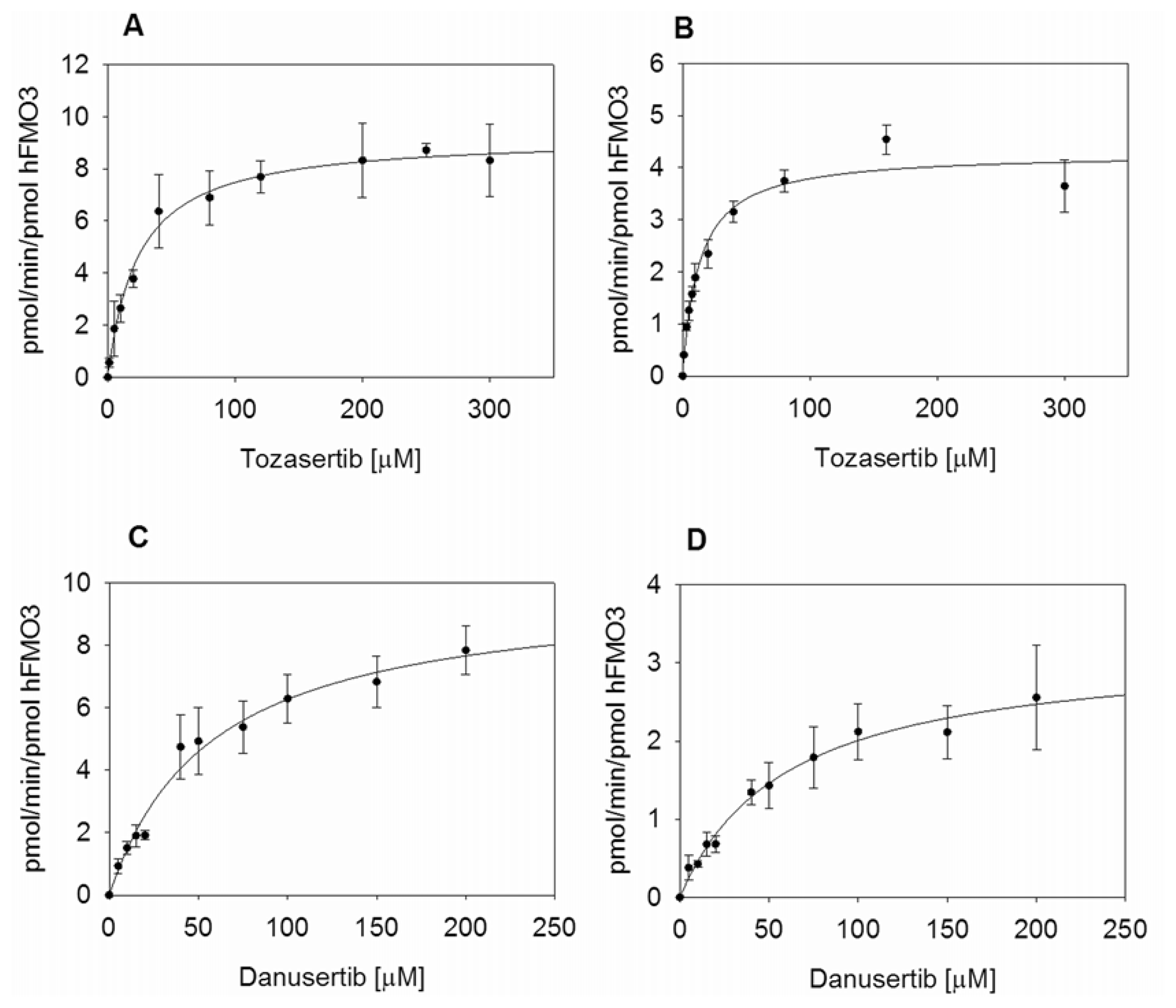
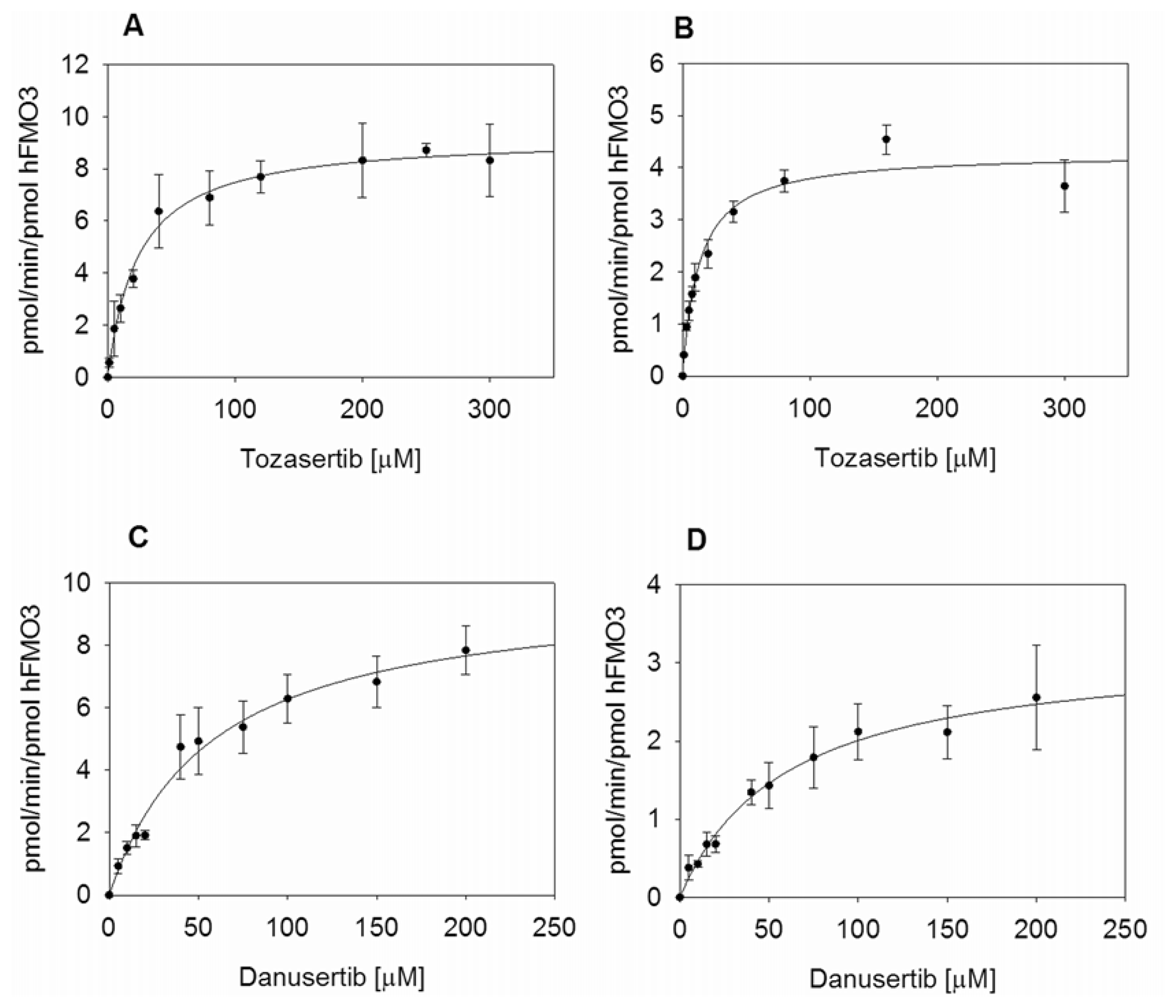
2.3. Kinetic Measurements of hFMO3 with Aurora Kinase Inhibitors
3. Experimental Section
3.1. Reagents
3.2. Recombinant FMO3 Protein Preparation
5′ CT GAC TGG TTG TAC ATG AAG CAG ATG AAT GC 3′5′ GC ATT CAT CTG CTT CAT GTA CAA CCA GTC AG 3′
3.3. Enzyme Assays
3.4. LC-MS Analyses of Danusertib, Tozasertib and Corresponding N-oxides
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Krueger, S.K.; Williams, D.E. Mammalian FMO: Structure/function, genetic polymorphism and role in drug metabolism. Pharmacol. Ther 2005, 106, 357–387. [Google Scholar]
- Overby, L.H.; Carver, G.C.; Philpot, R.M. Quantitation and kinetic properties of hepatic microsomal and recombinant flavin-containing monooxygenase 3 and 5 from humans. Chem. Biol. Interact 1997, 106, 29–45. [Google Scholar]
- Yeung, C.K.; Lang, D.H.; Thummel, K.E.; Rettie, A.E. Immunoquantitation of FMO1 in human liver, kidney and intestine. Drug Metab. Dispos 2000, 28, 1107–1111. [Google Scholar]
- Koukouritaki, S.B.; Poch, M.T.; Henderson, M.C.; Siddens, L.K.; Krueger, S.K.; VanDyke, J.E.; Williams, D.E.; Pajewski, N.M.; Wang, T.; Hines, R.N. Identification and functional analysis of common human flavin-containing monooxygenase 3 genetic variants. J. Pharm. Exp. Therap 2007, 320, 266–273. [Google Scholar]
- Koukouritaki, S.B.; Hines, R.N. Flavin-containing monooxygenase genetic polymorphism: Impact on chemical metabolism and drug development. Pharmacogenomics 2005, 6, 807–822. [Google Scholar]
- Cashman, J.R.; Zhang, J.; Leushner, J.; Braun, A. Population distribution of human flavin-containing monooxygenase form 3: Gene polymorphisms. Drug Metab. Dispos 2001, 29, 1629–1637. [Google Scholar]
- Cashman, J.R.; Akerman, B.R.; Forrest, S.M.; Treacy, E.P. Population-specific polymorphisms of the human FMO3 gene: Significance for detoxication. Drug Metab. Dispos 2000, 2, 169–173. [Google Scholar]
- Li, Y.; Zhang, Z.F.; Chen, J.; Huang, D.; Ding, Y.; Tan, M.H.; Qian, C.N.; Resau, J.H.; Kim, H.; Teh, B.T. VX-680/MK-0457, a potent and selective Aurora kinase inhibitor, targets both tumor and endothelial cells in clear cell renal cell carcinoma. Am. J. Transl. Res 2000, 3, 296–308. [Google Scholar]
- Gautschi, O.; Heighway, J.; Mack, P.C.; Purnell, P.R.; Lara, P.N., Jr; Gandara, D.R. Aurora kinases as anticancer drug targets. Clin. Cancer Res. 2008, 14, 1639–1648. [Google Scholar]
- Vader, G.; Lens, S.M. The Aurora kinase family in cell division and cancer. Biochim. Biophys. Acta 2008, 1786, 60–72. [Google Scholar]
- Martinelli, G.; Iacobucci, I.; Papayannidis, C.; Soverini, S. New targets for Ph+ leukaemia therapy. Best Pract. Res. Clin. Haematol 2009, 3, 445–454. [Google Scholar]
- Harrington, E.A.; Bebbington, D.; Moore, J.; Rasmussen, R.K.; Ajose-Adeogun, A.O.; Nakayama, T.; Graham, J.A.; Demur, C.; Hercend, T.; Diu-Hercend, A.; et al. VX-680 A potent and selective small-molecule inhibitor of aurora kinases suppresses tumor growth in vivo. Nat. Med 2004, 10, 262–267. [Google Scholar]
- Kitzen, J.J.; de Jonge, M.J.; Verweij, J. Aurora kinase inhibitors. Crit. Rev. Oncol. Hemat 2010, 2, 99–110. [Google Scholar]
- Fancelli, D.; Moll, J.; Varasi, M.; Bravo, R.; Artico, R.; Berta, D.; Bindi, S.; Cameron, A.; Candiani, I.; Cappella, P.; et al. 1,4,5,6-Tetrahydropyrrolo[3,4-c]pyrazoles: Identification of a potent Aurora kinase inhibitor with a favorable antitumor kinase inhibition profile. J. Med. Chem 2006, 49, 7247–7251. [Google Scholar]
- Carpinelli, P.; Ceruti, R.; Giorgini, M.L.; Cappella, P.; Gianellini, L.; Croci, V.; Degrassi, A.; Texido, G.; Rocchetti, M.; Vianello, P.; et al. PHA-739358, a potent inhibitor of Aurora kinases with a selective target inhibition profile relevant to cancer. Mol. Cancer Ther 2007, 12, 3158–3168. [Google Scholar]
- Ballard, J.E.; Prueksaritanont, T.; Tang, C. Hepatic metabolism of MK-0457, a potent aurora kinase inhibitor: Interspecies comparison and role of human cytochrome P450 and flavin-containing monooxygenase. Drug Metab. Dispos 2007, 9, 1447–1451. [Google Scholar]
- Roses, A.D. Pharmacogenetics and drug development: The path to safer and more effective drugs. Nat. Rev. Genet 2004, 5, 645–656. [Google Scholar]
- Panicco, P.; Dodhia, V.R.; Fantuzzi, A.; Gilardi, G. First enzyme-based amperometric platform to determine the polymorphic response in drug metabolism by cytochromes P450. Anal. Chem 2011, 83, 2179–2186. [Google Scholar]
- Sadeghi, S.J.; Meirinhos, R.; Catucci, G.; Dodhia, V.R.; di Nardo, G.; Gilardi, G. Direct electrochemistry of drug metabolizing hFMO3: Electrochemical turnover of benzydamine and tamoxifen. J. Am. Chem. Soc 2010, 132, 458–459. [Google Scholar]
- Castrignanò, S.; Sadeghi, S.J.; Gilardi, G. Electro-catalysis by immobilised human flavin-containing monooxygenase isoform 3 (hFMO3). Anal. Bioanal. Chem 2010, 398, 1403–1409. [Google Scholar]
- Catucci, G.; Gilardi, G.; Jeuken, L.; Sadeghi, S.J. In vitro drug metabolism by C-terminally truncated human flavin-containing monooxygenase 3. Biochem. Pharmacol 2012, 83, 551–558. [Google Scholar]
- Castrignanò, S.; Sadeghi, S.J.; Gilardi, G. Entrapment of human flavin-containing monooxygenase 3 in the presence of gold nanoparticles: TEM, FTIR and electrocatalysis. Biochim. Biophys. Acta 2012, 1820, 2072–2078. [Google Scholar]
- Yeung, C.K.; Adman, E.T.; Rettie, A.E. Functional characterization of genetic variants of human FMO3 associated with TMAU. Arch. Biochem. Biophys 2007, 464, 251–259. [Google Scholar]
- Steeghs, N.; Mathijssen, R.H.; Wessels, J.A.; de Graan, A.J.; van der Straaten, T.; Mariani, M.; Laffranchi, B.; Comis, S.; de Jonge, M.J.A.; Gelderblom, H.; et al. Influence of pharmacogenetic variability on the pharmacokinetics and toxicity of the aurora kinase inhibitor danusertib. Invest. New Drugs 2010, 5, 953–962. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WT hFMO3 | V257M hFMO3 | |||||||
|---|---|---|---|---|---|---|---|---|
| Km | Vmax | kcat | kcat/Km | Km | Vmax | kcat | kcat/Km | |
| μM | nmol/min/mg FMO3 | min−1 | min−1 μM−1 | μM | nmol/min/mg FMO3 | nmol/min/mg FMO3 | min−1 μM−1 | |
| Tozasertib | 23.8 ± 3.4 | 0.54 ± 0.02 | 9.3 ± 0.3 | 0.39 ± 0.06 | 12.9 ± 1.3 | 0.25 ± 0.01 | 4.3 ± 0.1 | 0.33 ± 0.04 |
| Danusertib | 57.3 ± 8.3 | 0.57 ± 0.03 | 9.9 ± 0.6 | 0.17 ± 0.03 | 60.1 ± 11.4 | 0.18 ± 0.01 | 3.2 ± 0.3 | 0.05 ± 0.01 |
© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Catucci, G.; Occhipinti, A.; Maffei, M.; Gilardi, G.; Sadeghi, S.J. Effect of Human Flavin-Containing Monooxygenase 3 Polymorphism on the Metabolism of Aurora Kinase Inhibitors. Int. J. Mol. Sci. 2013, 14, 2707-2716. https://doi.org/10.3390/ijms14022707
Catucci G, Occhipinti A, Maffei M, Gilardi G, Sadeghi SJ. Effect of Human Flavin-Containing Monooxygenase 3 Polymorphism on the Metabolism of Aurora Kinase Inhibitors. International Journal of Molecular Sciences. 2013; 14(2):2707-2716. https://doi.org/10.3390/ijms14022707
Chicago/Turabian StyleCatucci, Gianluca, Andrea Occhipinti, Massimo Maffei, Gianfranco Gilardi, and Sheila J. Sadeghi. 2013. "Effect of Human Flavin-Containing Monooxygenase 3 Polymorphism on the Metabolism of Aurora Kinase Inhibitors" International Journal of Molecular Sciences 14, no. 2: 2707-2716. https://doi.org/10.3390/ijms14022707