Green Approach—Multicomponent Production of Boron—Containing Hantzsch and Biginelli Esters

Abstract
:
1. Introduction
2. Results and Discussion
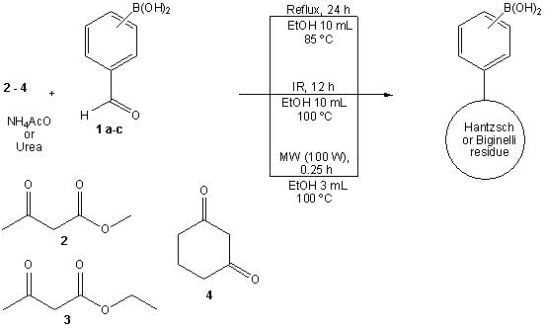
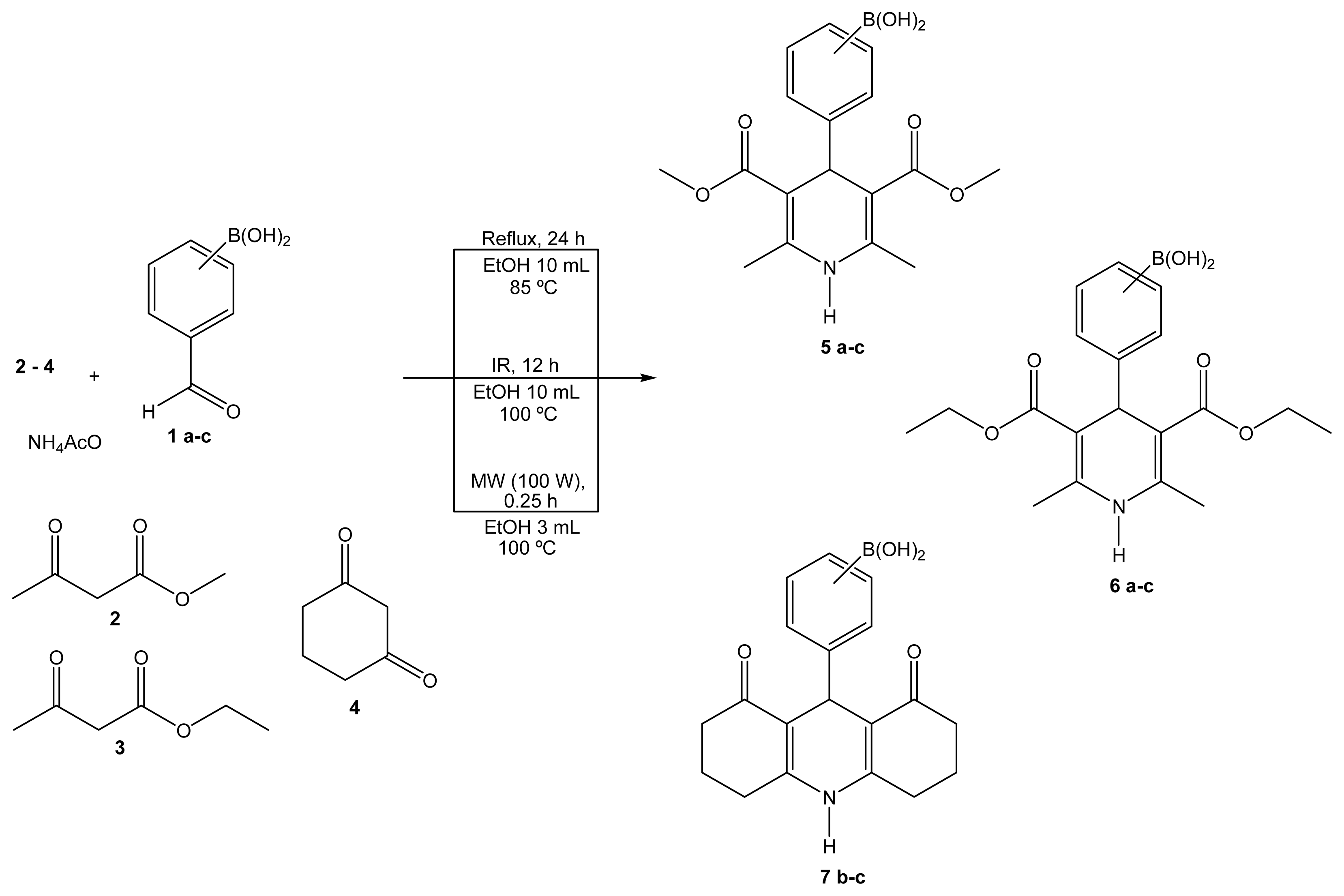
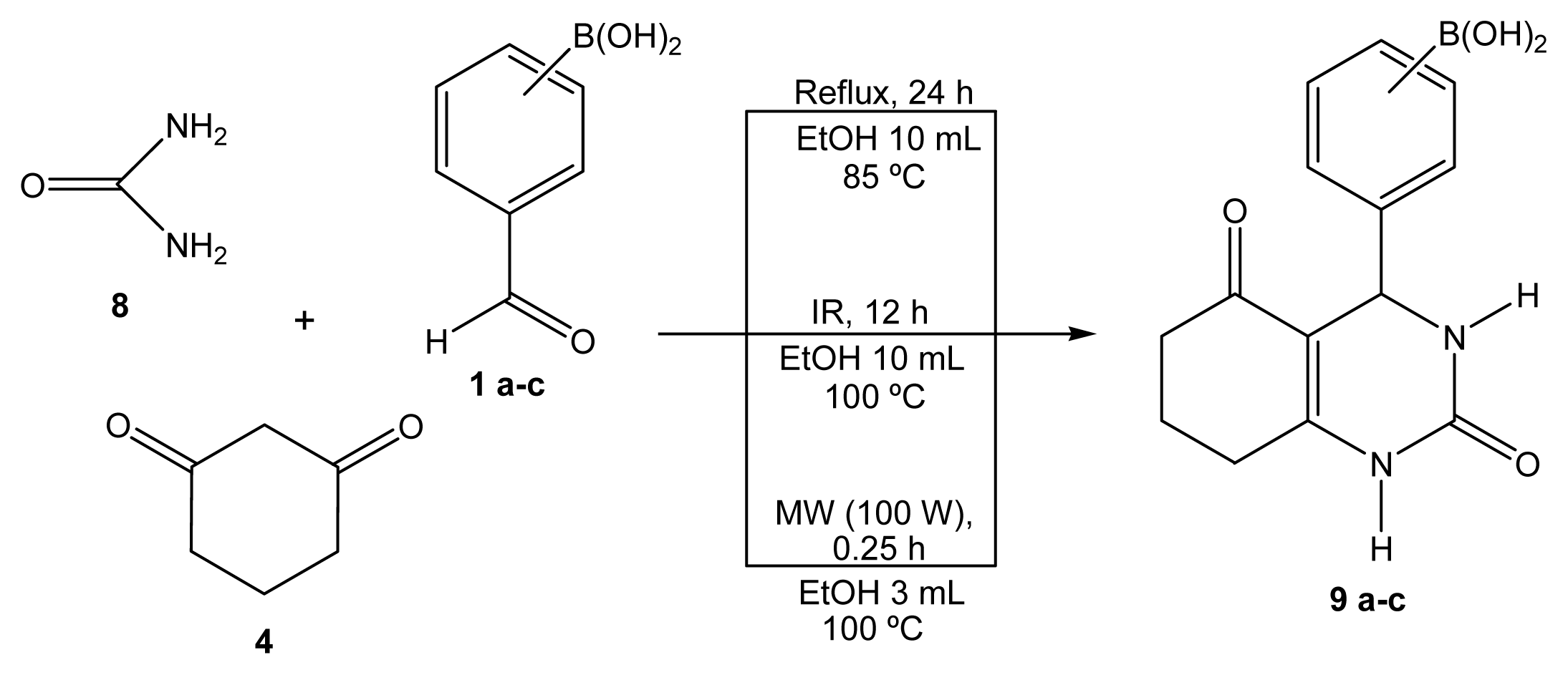
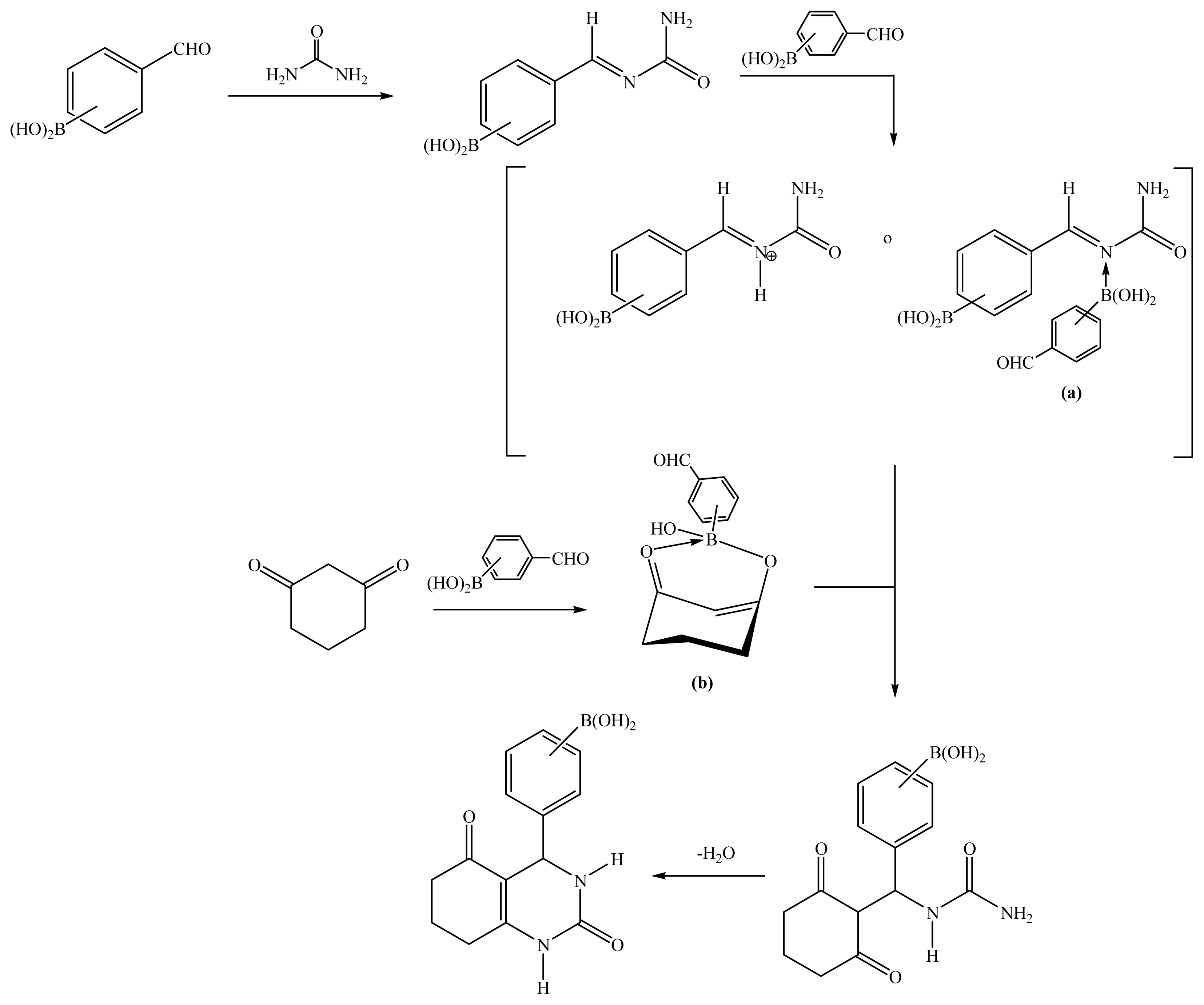
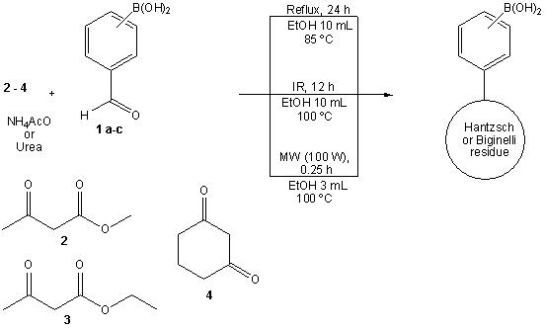
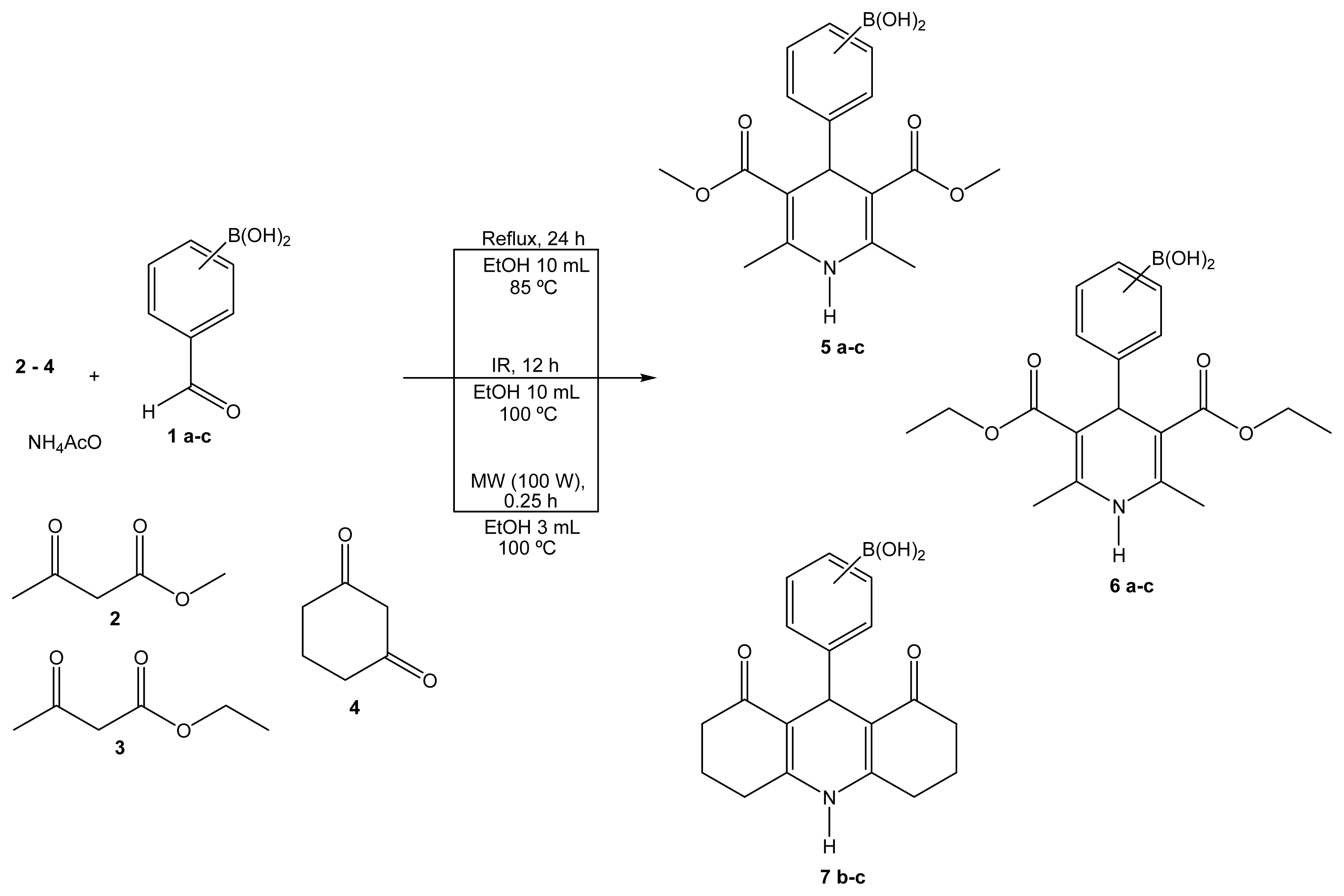
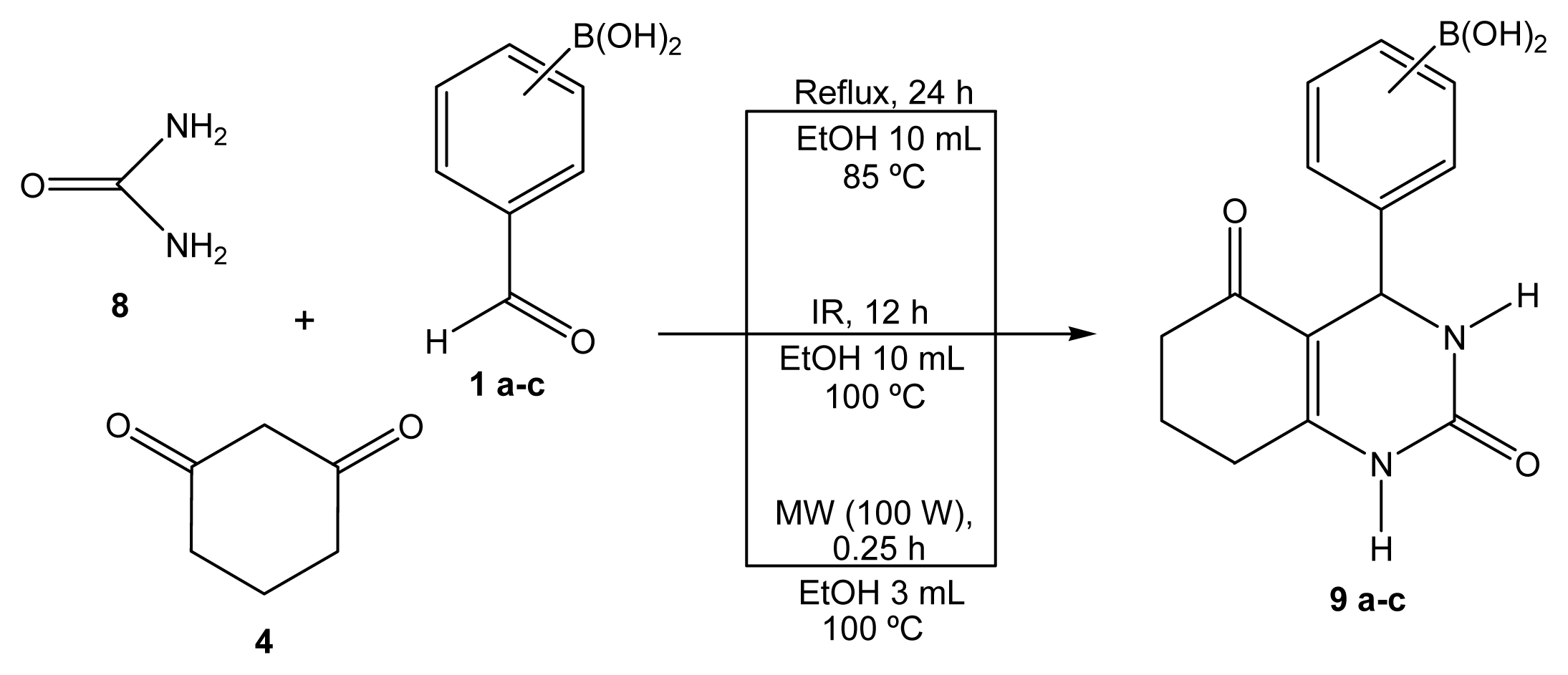
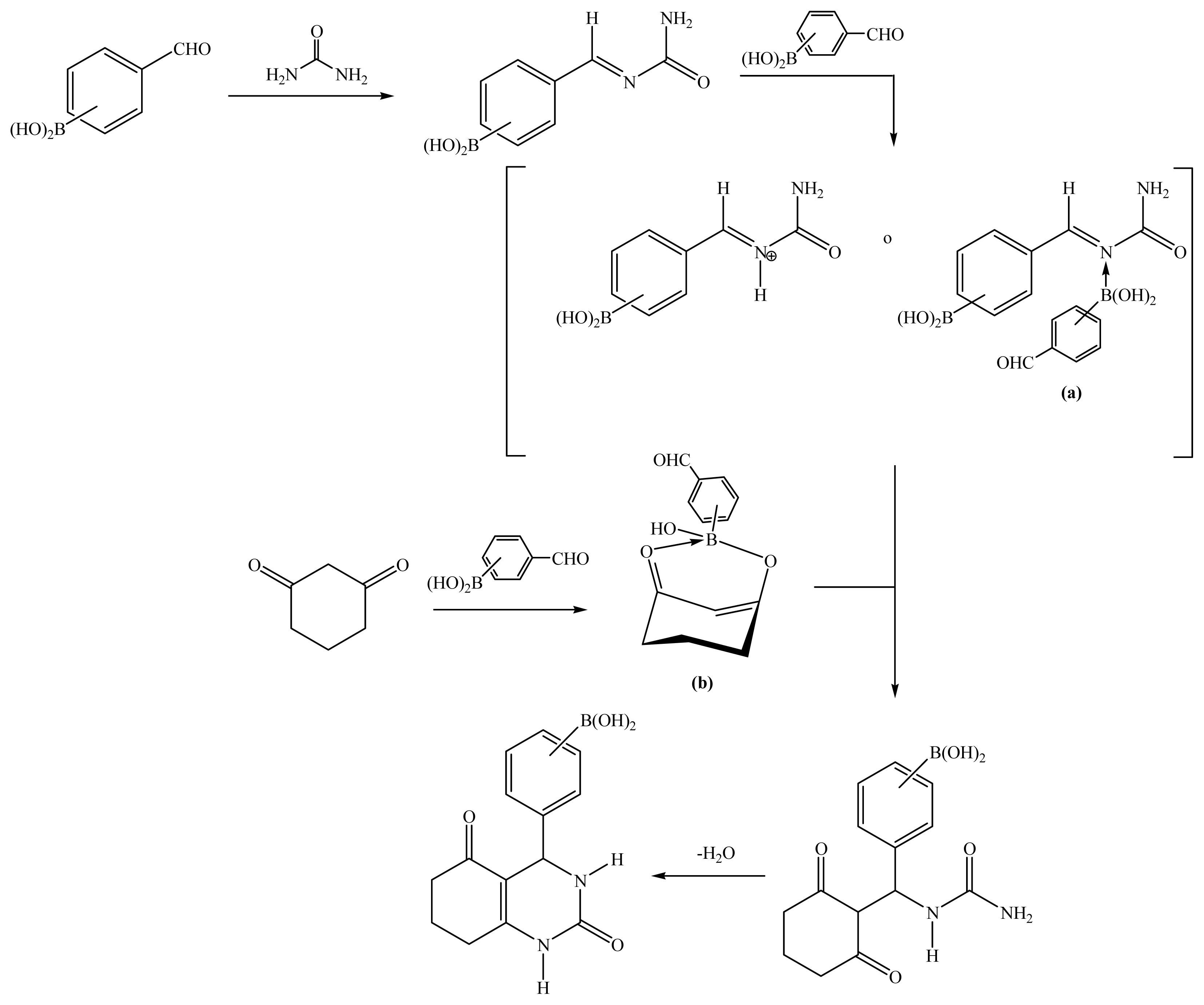
2.1. Synthesis
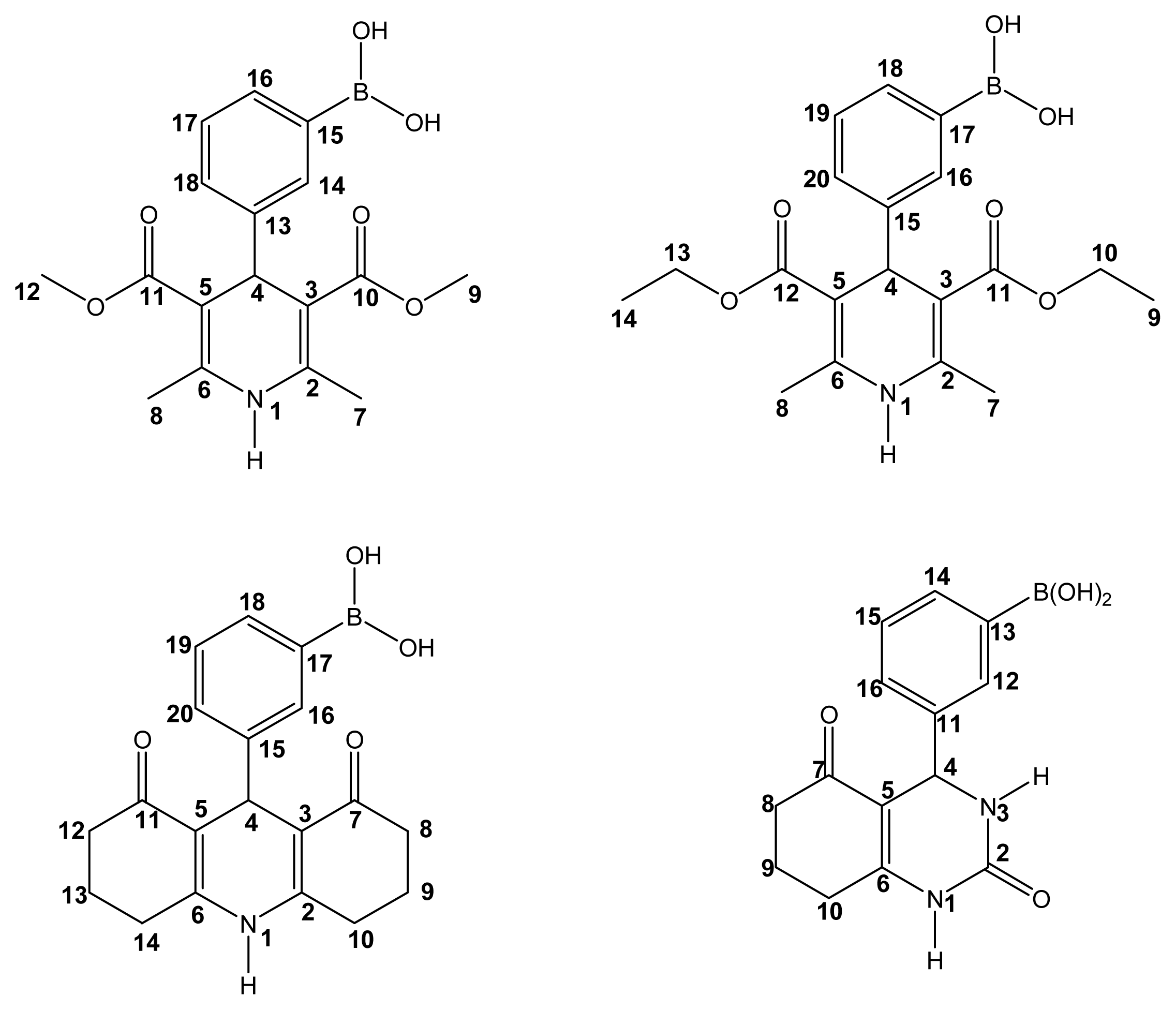
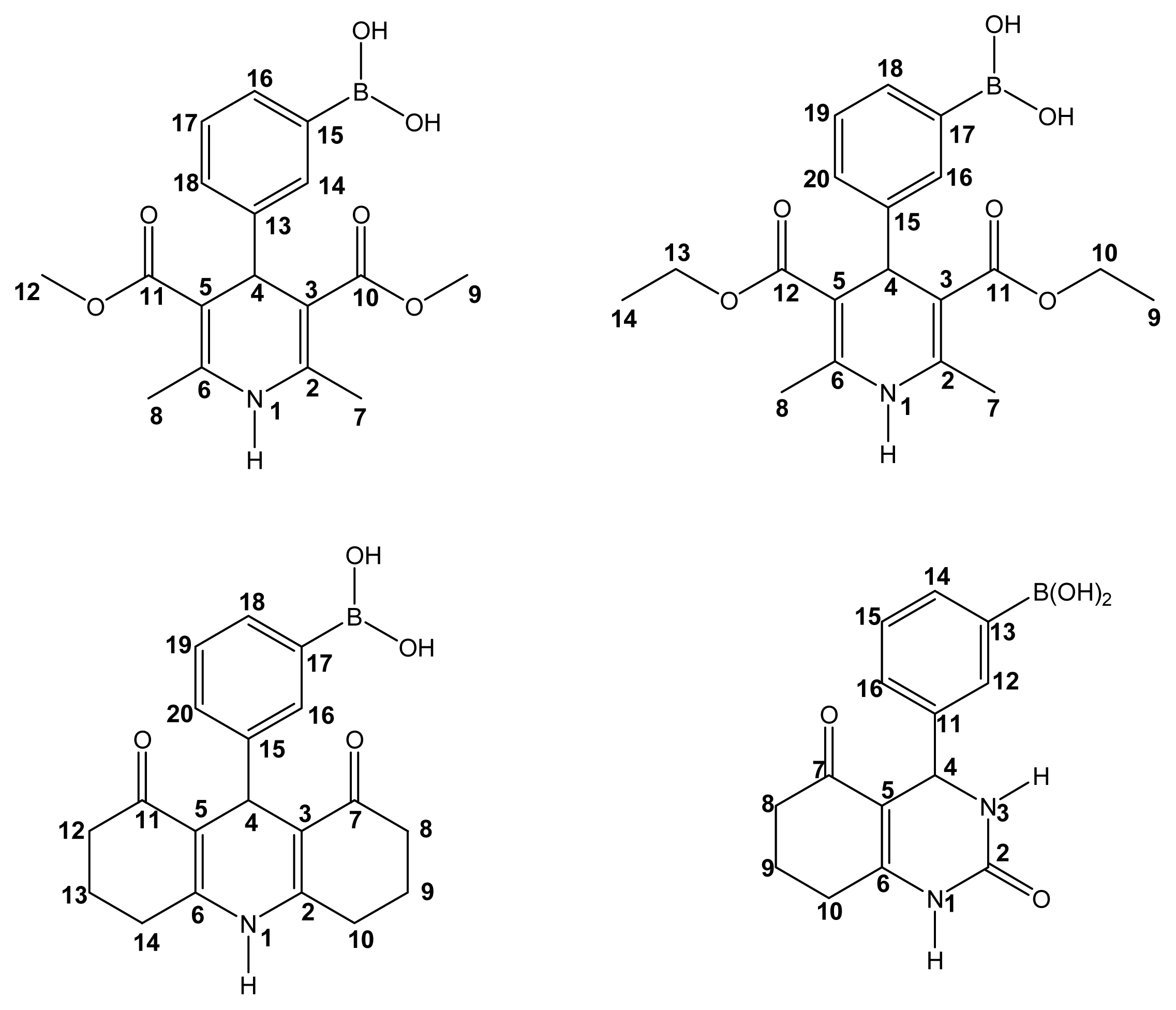
2.2. Spectroscopic Characterization
Boron-containing-Hantzsch esters
Boron-containing-Biginelli esters
Mass spectral data for BCDHPy and BCDHPm
3. Experimental Section
3.1. General
3.2. General Synthesis
Boron-containing-Hantzsch esters
2-(3,5-Bis(methoxycarbonyl)-2,6-dimethyl-1,4-dihydropyridin-4-yl)phenylboronic acid (5a)
3-(3,5-Bis(methoxycarbonyl)-2,6-dimethyl-1,4-dihydropyridin-4-yl)phenylboronic acid (5b)
4-(3,5-Bis(methoxycarbonyl)-2,6-dimethyl-1,4-dihydropyridin-4-yl)phenylboronic acid (5c)
2-(3,5-Bis(ethoxycarbonyl)-2,6-dimethyl-1,4-dihydropyridin-4-yl)phenylboronic acid (6a)
3-(3,5-Bis(ethoxycarbonyl)-2,6-dimethyl-1,4-dihydropyridin-4-yl)phenylboronic acid (6b)
4-(3,5-Bis(ethoxycarbonyl)-2,6-dimethyl-1,4-dihydropyridin-4-yl)phenylboronic acid (6c)
3-(1,1′-Dioxociclohex-2-en[3,2-b,2′,3′-e]-1,4-dihydropyridin-4-yl)phenylboronic acid (7b)
4-(1,1′-Dioxociclohex-2-en[3,2-b,2′,3′-e]-1,4-dihydropyridin-4-yl)phenylboronic acid (7c)
Boron-containing-Biginelli esters
2-(1-Oxociclohex-2-en[2,3-e]-2(1H)-oxo-3,4-dihydropiridimidin-4-yl)phenylboronic acid (9a)
3-(1-Oxociclohex-2-en[2,3-e]-2(1H)-oxo-3,4-dihydropiridimidin-4-yl)phenylboronic acid (9b)
4-(1-Oxociclohex-2-en[2,3-e]-2(1H)-oxo-3,4-dihydropiridimidin-4-yl)phenylboronic acid (9c)
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Zhu, J.; Bienaymé, H. Multicomponent Reactions; Wiley-VCH: Weinheim, Germany, 2005. [Google Scholar]
- Pandey, G.; Singh, R.; Gary, A.; Singh, V. Synthesis of Mannich type products via a three-component coupling reaction. Tetrahedron Lett 2005, 46, 2137–2140. [Google Scholar]
- Ugi, I.; Werner, B.; Domling, A. The chemistry of isocyanides, their multicomponent reactions and their libraries. Molecules 2003, 8, 53–66. [Google Scholar]
- Kappe, C. Recent advances in the Biginelli dihydropyrimidine synthesis. New tricks from an old dog. Acc. Chem. Res 2000, 33, 879–888. [Google Scholar]
- Anastas, P.T.; Warner, J.C. Green Chemistry: Theory and Practice; Oxford University Press: New York, NY, USA, 1998. [Google Scholar]
- Janis, R.A.; Triggle, D.J. New developments in calcium ion channel antagonists. J. Med. Chem 1983, 26, 775–785. [Google Scholar]
- Miri, R.; Javidnia, K.; Sarkarzadeh, H.; Hemmateenejad, B. Synthesis, study of 3D structures, and pharmacological activities of lipophilic nitroimidazolyl-1,4-dihydropyridines as calcium cannel antagonist. Bioorg. Med. Chem 2006, 14, 4842–4849. [Google Scholar]
- Gordeev, M.F.; Patel, D.V.; Gordon, E.M. Approaches to combinatorial synthesis of heterocycles: A solid-phase synthesis of 1,4-dihydropyridines. J. Org. Chem 1996, 61, 924–928. [Google Scholar]
- Coburn, R.A.; Wierzba, M.; Suto, M.J.; Solo, A.J.; Triggle, A.M.; Triggle, D.J. 1,4-Dihydropyridines antagonist activities at the calcium channel: A quantitative structure-activity relationship approach. J. Med. Chem 1988, 31, 2103–2107. [Google Scholar]
- Godfrain, T.; Miller, R.; Wibo, M. Calcium antagonism and calcium entry blockade. Pharmacol. Rev 1986, 38, 321–416. [Google Scholar]
- Bokaeva, S.S. Effects of some pyrimidine derivates on the growth of transplanted tumors in animals. Tr. Kaz. Nauch.-Issled. Inst.-Onkol. Radiol 1967, 3, 305–309. [Google Scholar]
- Maruyama, S.; Kawanishi, Y. Synthesis and emission properties of novel violet-blue emissive aromatic bis(diazaboroles). J. Mater. Chem 2002, 12, 2245–2249. [Google Scholar]
- Velasco, B.; Trujillo, J.; Miranda, R. Preparation of apoptotic inducers, 2,2-diphenyl-1,3,2-oxazaborolidin-5-ones, under alkaline conditions. Synlett 2007, 6, 921–924. [Google Scholar]
- Soriano-Úrsua, M.A.; Mancilla-Percino, T.; Correa-Basurto, J.; Querejeta, E.; Trujillo-Ferrara, J.G. Give boron a chance: Boron containing compounds reach ionotropic and metabotropic transmembrane receptors. Mini Rev. Med. Chem 2011, 11, 1031–1038. [Google Scholar]
- Gómez, R.; Ramírez-San Juan, E.; Miranda, R.; Villalobos-Molina, R.; Delgado, F.; Osnaya, R.; Trujillo-Ferrara, J. Vasodilator effects of bis-dihydropyridines structurally related to nifedipine. Med. Chem 2006, 2, 527–534. [Google Scholar]
- Velasco-Bejarano, B.; Trujillo-Ferrara, J.; Fabila, L.H.; Miranda, R.; Sánchez-Torres, L.E. In vitro apoptotic activity of 2,2-diphenyl-1,3,2-oxazaborolidin-5-ones in L5178Y cells. Life Sci 2007, 80, 1007–1013. [Google Scholar]
- Noguez, M.O.; García, A.; Ibarra, C.; Cabrera, A.; Aceves, J.M.; Miranda, R. Green synthesis of bis-Biginelli esters, with vasodilatory effects, their mass spectrometric and physical studies. Trends Org. Chem 2009, 13, 75–82. [Google Scholar]
- Reyes, L.; Corona, S.; Arroyo, G.; Delgado, F.; Miranda, R. Eco-Contribution for the production of N-arylnitrones: Solvent-Free and assisted by microwaves. Int. J. Mol. Sci 2010, 11, 2576–2583. [Google Scholar]
- Noguez, M.O.; Marcelino, V.; Rodríguez, H.; Martín, O.; Martínez, J.O.; Arroyo, G.A.; Pérez, J.F.; Suárez, M.; Miranda, R. Infrared assisted production of 3,4-dihydro-2(1H)-pyridones in solvent-free conditions. Int. J. Mol. Sci 2011, 12, 2641–2649. [Google Scholar]
- Toxics Release Inventory (TRI) Program. Available online: http://www.epa.gov/tri/trichemicals/index.htm accessed on 18 October 2012.
- Kappe, C.O. Controlled microwave heating in modern organic synthesis. Angew. Chem. Int. Ed 2004, 43, 6250–6284. [Google Scholar]
- Hayes, B. Microwave Synthesis: Chemistry at the Speed of Light; CEM Publishing: Matthews, NC, USA, 2002. [Google Scholar]
- Doxsee, M.; Hutchison, E. Green Organic Chemistry Strategies, Tools, and Laboratory Experiments; Thomson Brooks/Cole: Minster, OH, USA, 2004. [Google Scholar]
- Hall, D. Boronic Acids: Preparation and Applications in Organic Synthesis and Medicine; Wiley-VCH: Weinheim, Germany, 2005. [Google Scholar]
- Altamore, T.; Duggan, P.; Krippner, G. Improving the membrane permeability of sialic acid derivatives. Bioorg. Med. Chem 2006, 14, 1126–1133. [Google Scholar]
- Debache, A.; Boumoud, B.; Amimour, M.; B-Elfaitah, A.; Rhouati, S.; Carboni, B. Phenylboronic acid as a mild and efficient catalyst for Biginelli reaction. Tetrahedron Lett 2006, 47, 5697–5699. [Google Scholar]
- Martínez, J.; Abarca, V.; Pérez, F.J.; Carranza, V.; Miranda, R. Thioglycerol matrix interactions in the FAB+MS of several Hantzsch’s and Biginelli’s esters derivates of boronic acids. Rapid Commun. Mass Spectrom. 2012. submitted. [Google Scholar]
- Miranda, R.; Noguez, O.; Velasco, B.; Arroyo, G.; Penieres, G.; Martínez, J.; Delgado, F. Irradiación infrarrojo: Nueva manera para activar reacciones, un acercamiento al protocolo de la química verde. Educ. Quim 2009, 20, 421–425. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Solid color | Time (h)/Temp (°C) | Yield (%) * | mp (°C) | ||||
|---|---|---|---|---|---|---|---|---|
| Reflux | IR | MW | Reflux | IR | MW | |||
| 5a | yellow | 24/85 | 12/100 | 0.25/100 | 3.3 | 11.8 | 56.3 | 190–195 |
| 5b | yellow | 24/85 | 12/100 | 0.25/100 | 3.2 | 11.7 | 72.5 | 155–158 |
| 5c | yellow | 24/85 | 12/100 | 0.25/100 | 5.9 | 17.2 | 80.2 | 144–146 |
| 6a | yellow | 24/85 | 12/100 | 0.25/100 | 4.3 | 13.4 | 31.5 | 156–160 |
| 6b | yellow | 24/85 | 12/100 | 0.25/100 | 6.3 | 16.8 | 72.1 | 198–201 |
| 6c | yellow | 24/85 | 12/100 | 0.25/100 | 4.9 | 11.9 | 68.9 | 150–154 |
| 7b | brown | 24/85 | 12/100 | 0.25/100 | 6.9 | 14.6 | 60.0 | >300 |
| 7c | brown | 24/85 | 12/100 | 0.25/100 | 5.4 | 13.4 | 43.5 | >300 |
| 9a | brown | 24/85 | 12/100 | 0.33/140 | 3.3 | 11.7 | 45.4 | >300 |
| 9b | brown | 24/85 | 12/100 | 0.33/140 | 5.0 | 12.2 | 48.9 | >300 |
| 9c | brown | 24/85 | 12/100 | 0.33/140 | 3.4 | 11.8 | 17.8 | >300 |
© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Martínez, J.; Romero-Vega, S.; Abeja-Cruz, R.; Álvarez-Toledano, C.; Miranda, R. Green Approach—Multicomponent Production of Boron—Containing Hantzsch and Biginelli Esters. Int. J. Mol. Sci. 2013, 14, 2903-2915. https://doi.org/10.3390/ijms14022903
Martínez J, Romero-Vega S, Abeja-Cruz R, Álvarez-Toledano C, Miranda R. Green Approach—Multicomponent Production of Boron—Containing Hantzsch and Biginelli Esters. International Journal of Molecular Sciences. 2013; 14(2):2903-2915. https://doi.org/10.3390/ijms14022903
Chicago/Turabian StyleMartínez, Joel, Stephany Romero-Vega, Rita Abeja-Cruz, Cecilio Álvarez-Toledano, and René Miranda. 2013. "Green Approach—Multicomponent Production of Boron—Containing Hantzsch and Biginelli Esters" International Journal of Molecular Sciences 14, no. 2: 2903-2915. https://doi.org/10.3390/ijms14022903