Oxidative Folding in the Mitochondrial Intermembrane Space in Human Health and Disease

{kind=link}
{kind=link}
Abstract
:1. Introduction
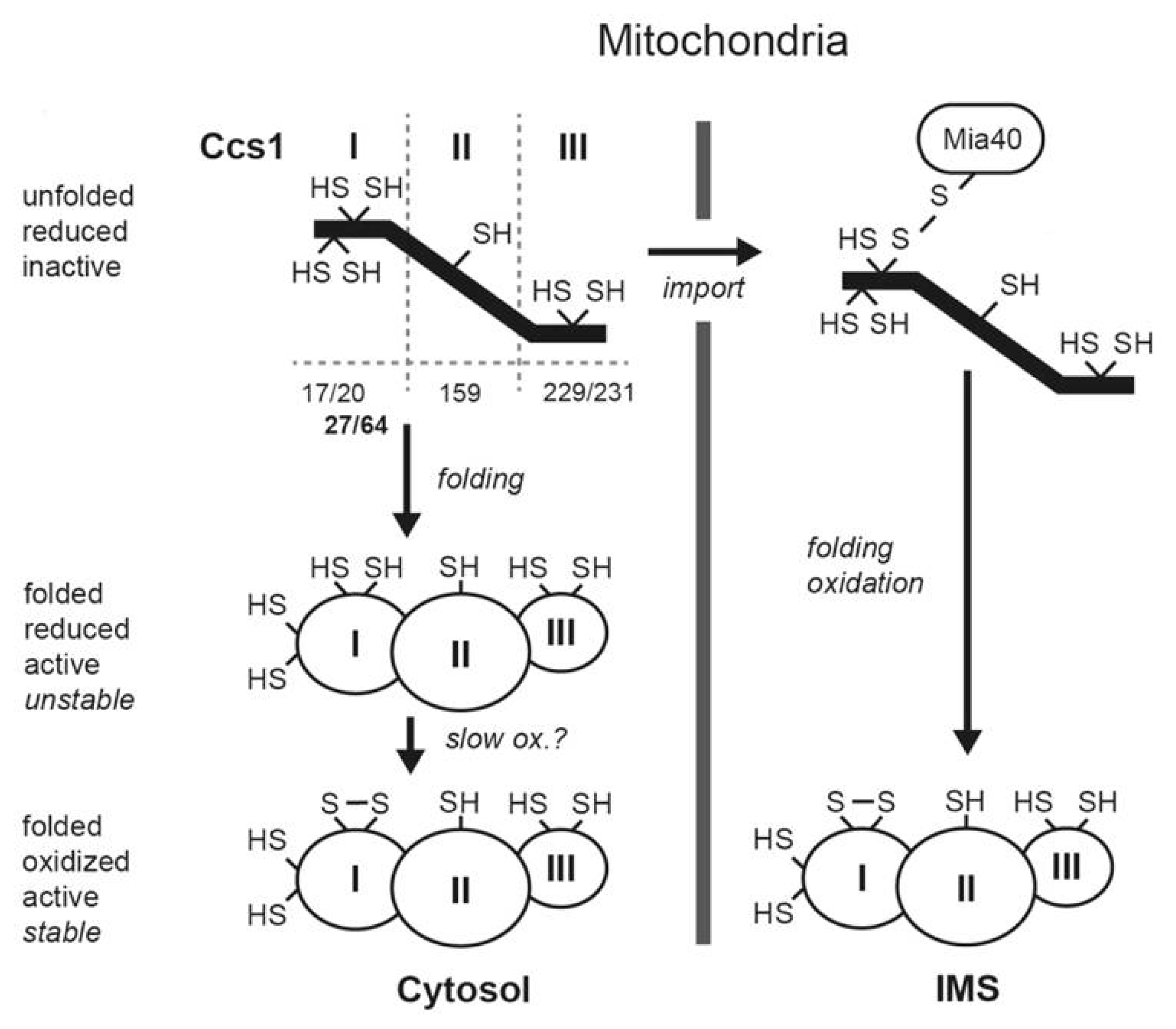
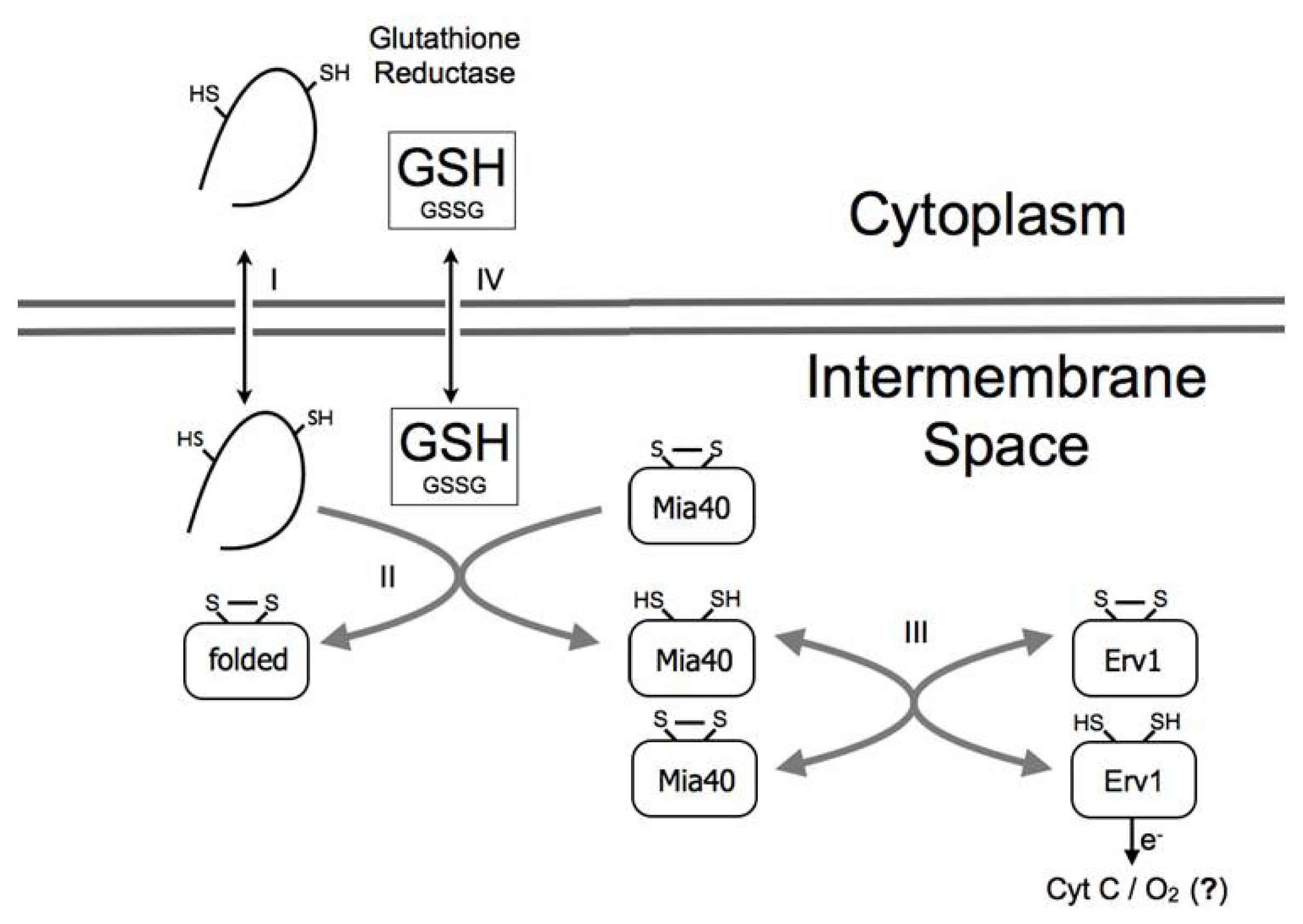
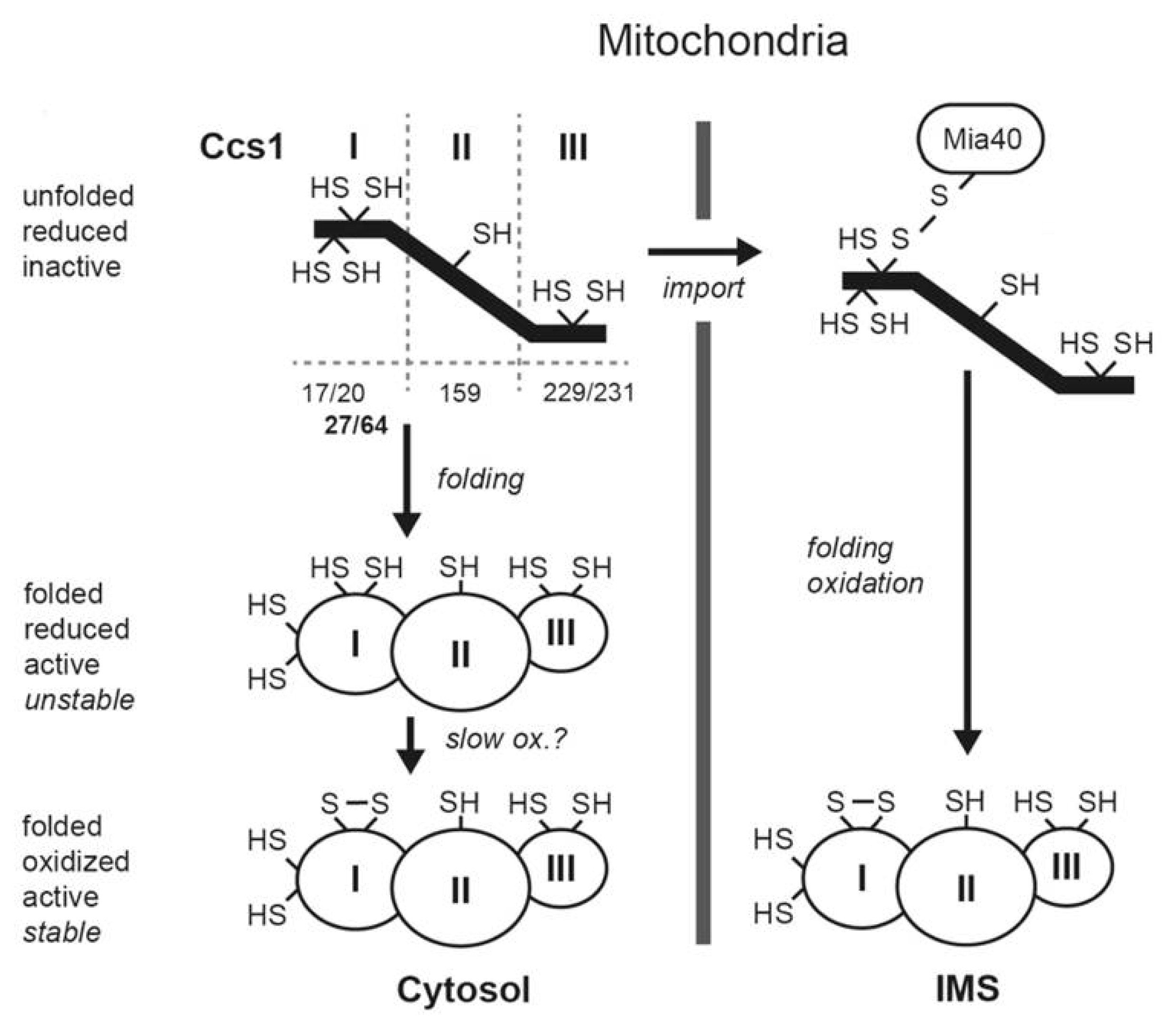
2. The Mia40 Pathway
3. A Link between the Mia40 Pathway and the Redox State of the Cell
4. Genetic Diseases Associated with Mia40 Pathway
5. Conclusions
Acknowledgments
Conflict of Interest
References
- Schmidt, O.; Pfanner, N.; Meisinger, C. Mitochondrial protein import: From proteomics to functional mechanisms. Nat. Rev. Mol. Cell. Biol 2010, 11, 655–667. [Google Scholar]
- Baker, K.P.; Schatz, G. Mitochondrial proteins essential for viability mediate protein import into yeast mitochondria. Nature 1991, 349, 205–208. [Google Scholar]
- MacKenzie, J.A.; Payne, R.M. Mitochondrial protein import and human health and disease. Biochim. Biophys. Acta 2007, 1772, 509–523. [Google Scholar]
- Dessi, P.; Whelan, J. Temporal regulation of in vitro import of precursor proteins into tobacco mitochondria. FEBS. Lett 1997, 415, 173–178. [Google Scholar]
- Dudley, P.; Wood, C.K.; Pratt, J.R.; Moore, A.L. Developmental regulation of the plant mitochondrial matrix located Hsp70 chaperone and its role in protein import. FEBS Lett 1997, 417, 321–324. [Google Scholar]
- Craig, E.E.; Chesley, A.; Hood, D.A. Thyroid hormone modifies mitochondrial phenotype by increasing protein import without altering degradation. Am. J. Physiol 1998, 275, C1508–C1515. [Google Scholar]
- Takahashi, M.; Chesley, A.; Freyssenet, D.; Hood, D.A. Contractile activity-induced adaptations in the mitochondrial protein import system. Am. J. Physiol 1998, 274, C1380–C1387. [Google Scholar]
- Neupert, W.; Herrmann, J.M. Translocation of proteins into mitochondria. Annu. Rev. Biochem 2007, 76, 723–749. [Google Scholar]
- Riemer, J.; Bulleid, N.; Herrmann, J.M. Disulfide formation in the ER and mitochondria: Two solutions to a common process. Science 2009, 234, 1284–1287. [Google Scholar]
- Endo, T.; Yamano, K. Multiple pathways for mitochondrial protein traffic. Biol. Chem 2009, 390, 723–730. [Google Scholar]
- Deponte, M.; Hell, K. Disulphide bond formation in the intermembrane space of mitochondria. J. Biochem 2009, 146, 599–608. [Google Scholar]
- Riemer, J.; Fischer, M.; Herrmann, J.M. Oxidation-driven protein import into mitochondria: Insights and blind spots. Biochim. Biophys. Acta 2011, 1808, 981–989. [Google Scholar]
- Koehler, C.M.; Beverly, K.N.; Leverich, E.P. Redox pathways of the mitochondrion. Antioxid. Redox Signal 2006, 8, 813–822. [Google Scholar]
- Chacinska, A.; Pfannschmidt, S.; Wiedemann, N.; Kozjak, V.; Sanjuán Szklarz, L.K.; Schulze-Specking, A.; Truscott, K.N.; Guiard, B.; Meisinger, C.; Pfanner, N. Essential role of Mia40 in import and assembly of mitochondrial intermembrane space proteins. EMBO J 2004, 23, 3735–3746. [Google Scholar]
- Naoé, M.; Ohwa, Y.; Ishikawa, D.; Ohshima, C.; Nishikawa, S.; Yamamoto, H.; Endo, T. Identification of Tim40 that mediates protein sorting to the mitochondrial intermembrane space. J. Biol. Chem 2004, 279, 47815–47821. [Google Scholar]
- Mesecke, N.; Terziyska, N.; Kozany, C.; Baumann, F.; Neupert, W.; Hell, K.; Herrmann, J.M. A disulfide relay system in the intermembrane space of mitochondria that mediates protein import. Cell 2005, 121, 1059–1069. [Google Scholar]
- Hell, K. The Erv1-Mia40 disulfide relay system in the intermembrane space of mitochondria. Biochim. Biophys. Acta 2008, 1783, 601–609. [Google Scholar]
- Weckbecker, D.; Longen, S.; Riemer, J.; Herrmann, J.M. Atp23 biogenesis reveals a chaperone-like folding activity of Mia40 in the IMS of mitochondria. EMBO J 2012, 31, 4348–4358. [Google Scholar]
- Chatzi, A.; Tokatlidis, K. The mitochondrial intermembrane space: A hub for oxidative folding linked to protein biogenesis. Antioxid. Redox Signal 2012, in press. [Google Scholar]
- Herrmann, J.M.; Hell, K. Chopped, trapped or tacked—Protein translocation into the IMS of mitochondria. Trends Biochem. Sci 2005, 30, 205–212. [Google Scholar]
- Herrmann, J.M.; Riemer, J. The intermembrane space of mitochondria. Antioxid. Redox Signal 2010, 13, 1341–1358. [Google Scholar]
- Herrmann, J.M.; Kohl, R. Catch me if you can! Oxidative protein trapping in the intermembrane space of mitochondria. J. Cell Biol 2007, 176, 559–563. [Google Scholar]
- Sideris, D.P.; Tokatlidis, K. Oxidative folding of small Tims is mediated by site-specific docking onto Mia40 in the mitochondrial intermembrane space. Mol. Microbiol 2007, 65, 1360–1373. [Google Scholar]
- Bihlmaier, K.; Mesecke, N.; Terziyska, N.; Bien, M.; Hell, K.; Herrmann, J.M. The disulfide relay system of mitochondria is connected to the respiratory chain. J. Cell Biol 2007, 179, 389–395. [Google Scholar]
- Tienson, H.L.; Dabir, D.V.; Neal, S.E.; Loo, R.; Hasson, S.A.; Boontheung, P.; Kim, S.K.; Loo, J.A.; Koehler, C.M. Reconstitution of the Mia40-Erv1 oxidative folding pathway for the small tim proteins. Mol. Biol. Cell 2009, 20, 3841–3490. [Google Scholar]
- Kojer, K.; Bien, M.; Gangel, H.; Morgan, B.; Dick, T.P.; Riemer, J. Glutathione redox potential in the mitochondrial intermembrane space is linked to the cytosol and impacts the Mia40 redox state. EMBO J 2012, 31, 3169–3182. [Google Scholar]
- Bottinger, L.; Gornicka, A.; Czerwik, T.; Bragoszewski, P.; Loniewska-Lwowska, A.; Schulze-Specking, A.; Truscott, K.N.; Guiard, B.; Milenkovic, D.; Chacinska, A. In vivo evidence for cooperation of Mia40 and Erv1 in the oxidation of mitochondrial proteins. Mol. Biol. Cell 2012, 23, 3957–3969. [Google Scholar]
- Bourens, M.; Dabir, D.V.; Tienson, H.L.; Sorokina, I.; Koehler, C.M.; Barrientos, A. Role of twin cys-xaa9-cys motif cysteines in mitochondrial import of the cytochrome c oxidase biogenesis factor cmc1. J. Biol. Chem 2012, 287, 31258–31269. [Google Scholar]
- Di Fonzo, A.; Ronchi, D.; Lodi, T.; Fassone, E.; Tigano, M.; Lamperti, C.; Corti, S.; Bordoni, A.; Fortunato, F.; Nizzardo, M.; et al. The mitochondrial disulfide relay system protein GFER is mutated in autosomal-recessive myopathy with cataract and combined respiratory-chain deficiency. Am. J. Hum. Genet 2009, 84, 594–604. [Google Scholar]
- Koehler, C.M. Import of mitochondrial carriers mediated by essential proteins of the intermembrane space. Science 1998, 279, 369–373. [Google Scholar]
- Wrobel, L.; Trojanowska, A.; Sztolsztener, M.E.; Chacinska, A. Mitochondrial protein import: Mia40 facilitates Tim22 translocation into the inner membrane of mitochondria. Mol. Biol. Cell 2013. [Google Scholar] [CrossRef]
- Chacinska, A.; Koehler, C.M.; Milenkovic, D.; Lithgow, T.; Pfanner, N. Importing mitochondrial proteins: Machineries and mechanisms. Cell 2009, 138, 628–644. [Google Scholar]
- Hu, J.; Dong, L.; Outten, C.E. The redox environment in the mitochondrial intermembrane space is maintained separately from the cytosol and matrix. J. Biol. Chem 2008, 283, 29126–29134. [Google Scholar]
- Bien, M.; Longen, S.; Wagener, N.; Chwalla, I.; Herrmann, J.M.; Riemer, J. Mitochondrial disulfide bond formation is driven by intersubunit electron transfer in Erv1 and proofread by glutathione. Mol. Cell 2010, 37, 516–528. [Google Scholar]
- Wright, G.; Terada, K.; Yano, M.; Sergeev, I.; Mori, M. Oxidative stress inhibits the mitochondrial import of preproteins and leads to their degradation. Exp. Cell Res 2001, 263, 107–117. [Google Scholar]
- Von Stedingk, E.; Pavlov, P.; Grinkevich, V.; Glaser, E. Mitochondrial protein import: Modification of sulfhydryl groups of the inner mitochondrial membrane import machinery in Solanum tuberosum inhibits protein import. Plant Mol. Biol 1997, 35, 809–820. [Google Scholar]
- Sturtz, L.A.; Diekert, K.; Jensen, L.T.; Lill, R.; Culotta, V.C. A fraction of yeast Cu,Zn-superoxide dismutase and its metallochaperone, Ccs, localize to the intermembrane space of mitochondria. J. Biol. Chem 2001, 276, 38084–38089. [Google Scholar]
- Reddehase, S.; Grumbt, B.; Neupert, W.; Hell, K. The disulfide relay system of mitochondria is required for the biogenesis of mitochondrial Ccs1 and Sod1. J. Mol. Biol 2009, 385, 331–338. [Google Scholar]
- Kloppel, C.; Suzuki, Y.; Kojer, K.; Petrungaro, C.; Longen, S.; Fiedler, S.; Keller, S.; Riemer, J. Mia40-dependent oxidation of cysteines in domain i of Ccs1 controls its distribution between mitochondria and the cytosol. Mol. Biol. Cell 2011, 22, 3749–3757. [Google Scholar]
- Kawamata, H.; Manfredi, G. Import, maturation and function of Sod1 and its cooper chaperone Ccs in the mitochondrial intermembrane space. Antioxid. Redox Signal 2010, 13, 1375–1384. [Google Scholar]
- Culotta, V.C.; Klomp, L.W.J.; Strain, J.; Casareno, R.L.B.; Krems, B.; Gitlin, J.D. The copper chaperone for superoxide dismutase. J. Biol. Chem 1997, 272, 23469–23472. [Google Scholar]
- Lamb, A.L.; Wernimont, A.K.; Pufahl, R.A.; Culotta, V.C.; O’Halloran, T.V.; Rosenzweig, A.C. Crystal structure of the copper chaperone for superoxide dismutase. Nat. Struct. Mol. Biol 1999, 6, 724–729. [Google Scholar]
- Rae, T.D.; Torres, A.S.; Pufahl, R.A.; O’Halloran, T.V. Mechanism of Cu,Zn-superoxide dismutase activation by the human metallochaperone hCcs. J. Biol. Chem 2001, 276, 5166–5176. [Google Scholar]
- Schmidt, P.J.; Rae, T.D.; Pufahl, R.A.; Hamma, T.; Strain, J.; O’Halloran, T.V.; Culotta, V.C. Multiple protein domains contribute to the action of the copper chaperone for superoxide dismutase. J. Biol. Chem 1999, 274, 23719–23725. [Google Scholar]
- Lamb, A.L.; Torres, A.S.; O’Halloran, T.V.; Rosenzweig, A.C. Heterodimeric structure of superoxide dismutase in complex with its metallochaperone. Nat. Struct. Mol. Biol 2001, 8, 751–755. [Google Scholar]
- Lamb, A.L.; Wernimont, A.K.; Pufahl, R.A.; O’Halloran, T.V.; Rosenzweig, A.C. Crystal structure of the second domain of the human copper chaperone for superoxide dismutase. Biochemistry 2000, 39, 1589–1595. [Google Scholar]
- GroB, D.P.; Burgard, C.A.; Reddehase, S.; Leitch, J.M.; Culotta, V.C.; Hell, K. Mitochondrial Ccs1 contains a structural disulfide bond crucial for the import of this unconventional substrate by the disulfide relay system. Mol. Biol. Cell 2011, 22, 3758–3767. [Google Scholar]
- Field, L.S. Factors controlling the uptake of yeast copper/zinc superoxide dismutase into mitochondria. J. Biol. Chem 2003, 278, 28052–28059. [Google Scholar]
- Klöppel, C.; Michels, C.; Zimmer, J.; Herrmann, J.M.; Riemer, J. In yeast redistribution of Sod1 to the mitochondrial intermembrane space provides protection against respiration derived oxidative stress. Biochem. Biophys. Res. Commun 2010, 403, 114–119. [Google Scholar]
- Fischer, L.R.; Igoudjil, A.; Magrane, J.; Li, Y.; Hansen, J.M.; Manfredi, G.; Glass, J.D. SOD1 targeted to the mitochondrial intermembrane space prevents motor neuropathy in the Sod1 knockout mouse. Brain 2010, 134, 196–209. [Google Scholar]
- Kawamata, H.; Manfredi, G. Different regulation of wild-type and mutant Cu,Zn superoxide dismutase localization in mammalian mitochondria. Hum. Mol. Gen 2008, 17, 3303–3317. [Google Scholar]
- Yang, J.; Staples, O.; Thomas, L.W.; Briston, T.; Robson, M.; Poon, E.; Simões, M.L.; El-Emir, E.; Buffa, F.M.; Ahmed, A.; et al. Human CHCHD4 mitochondrial proteins regulate cellular oxygen consumption rate and metabolism and provide a critical role in hypoxia signaling and tumor progression. J. Clin. Invest 2012, 122, 600–611. [Google Scholar]
- Semenza, G.L. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 2009, 29, 625–634. [Google Scholar]
- Hofmann, S. The C66W mutation in the Deafness Dystonia Peptide 1 (DDP1) affects the formation of functional DDP1.TIM13 complexes in the mitochondrial intermembrane space. J. Biol. Chem 2002, 277, 23287–23293. [Google Scholar]
- Roesch, K.; Curran, S.P.; Tranebjaerg, L.; Koehler, C.M. Human Deafness Dystonia syndrome is caused by a defect in assembly of the DDP1/TIMM8a-TIMM13 complex. Hum. Mol. Gen 2002, 11, 477–486. [Google Scholar]
- Jin, H.; Kendall, E.; Freeman, T.C.; Roberts, R.G.; Vetrie, D.L.P. The Human Family of Deafness/Dystonia peptide (DDP) related mitochondrial import proteins. Genomics 1999, 61, 259–267. [Google Scholar]
- Davis, A.J.; Sepuri, N.B.; Holder, J.; Johnson, A.E.; Jensen, R.E. Two intermembrane space TIM complexes interact with different domains of Tim23p during its import into mitochondria. J. Cell Biol 2000, 150, 1271–1282. [Google Scholar]
- Donzeau, M.; Káldi, K.; Adam, A.; Paschen, S.; Wanner, G.; Guiard, B.; Bauer, M.F.; Neupert, W.; Brunner, M. Tim23 links the inner and outer mitochondrial membranes. Cell 2000, 101, 401–412. [Google Scholar]
- Roesch, K. The calcium-binding aspartate/glutamate carriers, citrin and aralar1, are new substrates for the DDP1/TIMM8a-TIMM13 complex. Hum. Mol. Gen 2004, 13, 2101–2111. [Google Scholar]
- Sirrenberg, C.; Endres, M.; Folsch, H.; Stuart, R.A.; Neupert, W.; Brunner, M. Carrier protein import into mitochondria mediated by the intermembrane proteins Tim10/Mrs11 and Tim12/Mrs5. Nature 1998, 391, 912–915. [Google Scholar]
- Daithankar, V.N.; Schaefer, S.A.; Dong, M.; Bahnson, B.J.; Thorpe, C. Structure of the human sulfhydryl oxidase augmenter of liver regeneration and characterization of a human mutation causing an autosomal recessive myopathy. Biochemistry 2010, 49, 6737–6745. [Google Scholar]
- Lisowsky, T. Dual function of a new nuclear gene for oxidative phosphorylation and vegetative growth in yeast. Mol. Gen. Genet 1992, 232, 58–64. [Google Scholar]
- Becher, D.; Kricke, J.; Stein, G.; Lisowsky, T. A mutant for the yeast scErv1 gene displays a new defect in mitochondrial morphology and distribution. Yeast 1999, 15, 1171–1181. [Google Scholar]
- Sztolsztener, M.E.; Brewinska, A.; Guiard, B.; Chacinska, A. Disulfide bond formation: Sulfhydryl oxidase ALR controls mitochondrial biogenesis of human mia40. Traffic 2012. [Google Scholar] [CrossRef]
- Napoli, E.; Wong, S.; Hung, C.; Ross-Inta, C.; Bomdica, P.; Giulivi, C. Defective mitochondrial disulfide relay system, altered mitochondrial morphology and function in huntington’s disease. Hum. Mol. Gen. 2012. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fraga, H.; Ventura, S. Oxidative Folding in the Mitochondrial Intermembrane Space in Human Health and Disease. Int. J. Mol. Sci. 2013, 14, 2916-2927. https://doi.org/10.3390/ijms14022916
Fraga H, Ventura S. Oxidative Folding in the Mitochondrial Intermembrane Space in Human Health and Disease. International Journal of Molecular Sciences. 2013; 14(2):2916-2927. https://doi.org/10.3390/ijms14022916
Chicago/Turabian StyleFraga, Hugo, and Salvador Ventura. 2013. "Oxidative Folding in the Mitochondrial Intermembrane Space in Human Health and Disease" International Journal of Molecular Sciences 14, no. 2: 2916-2927. https://doi.org/10.3390/ijms14022916