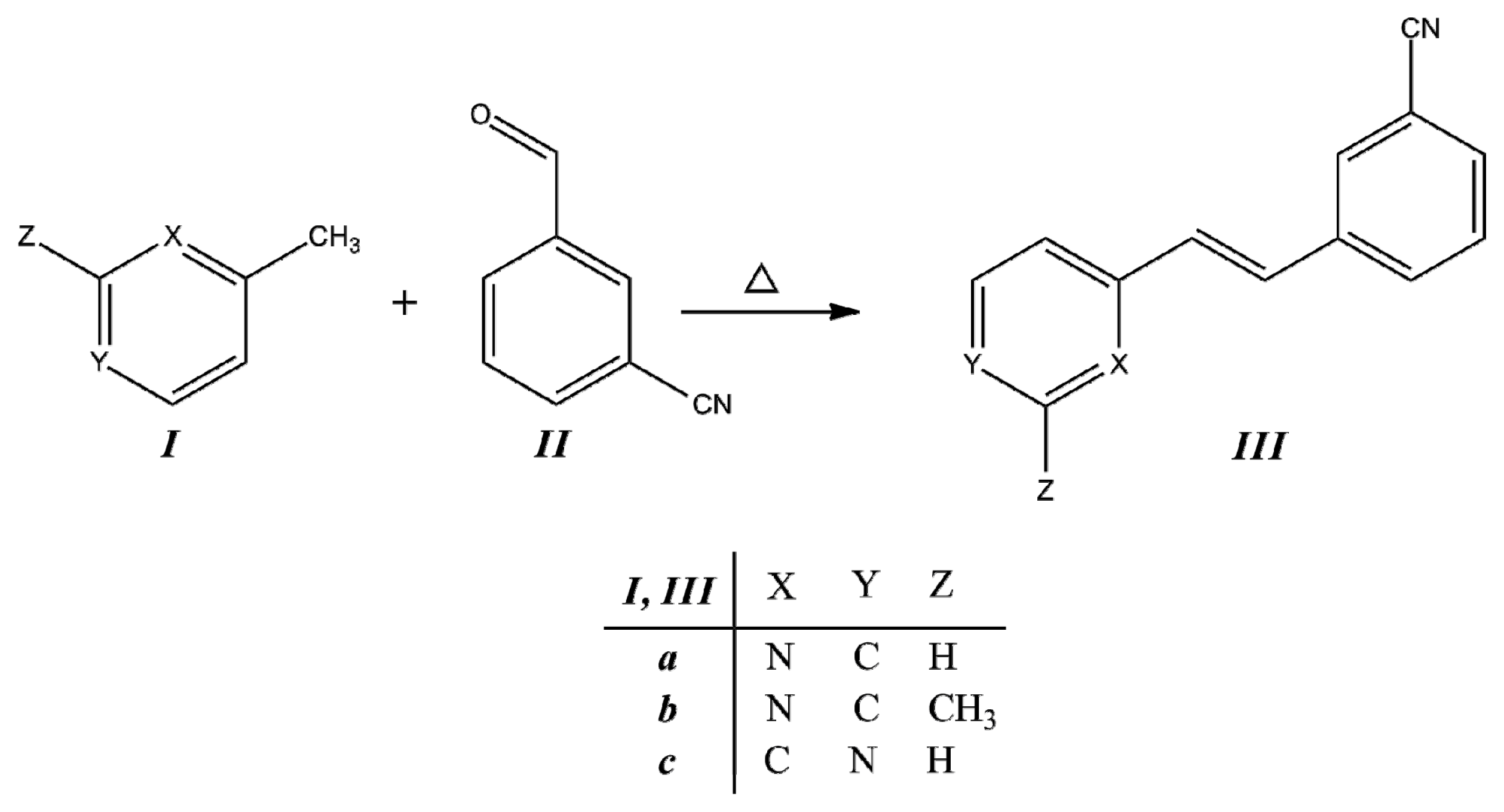
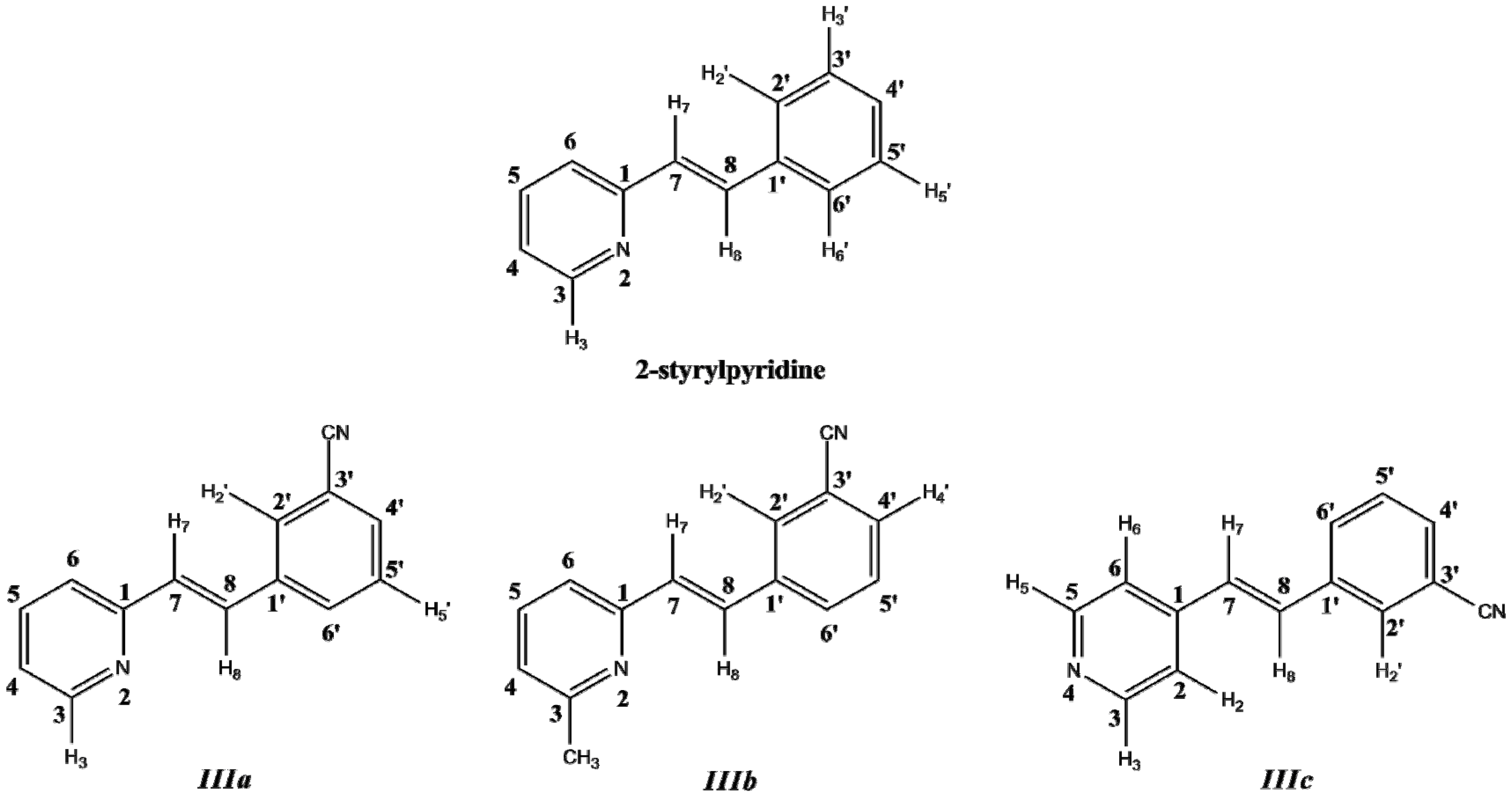
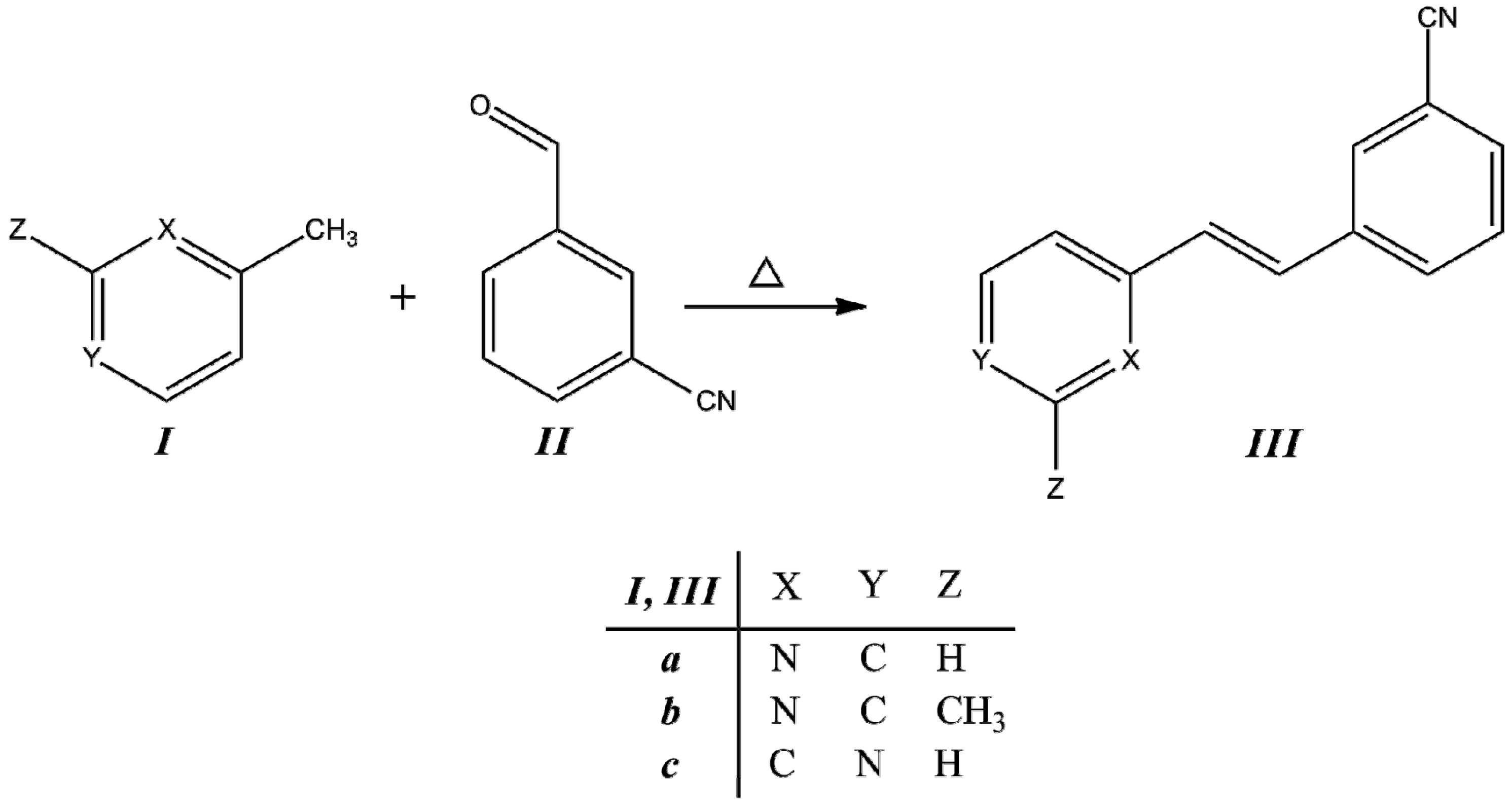
3.1. Synthesis
Compounds
IIIa–
IIIc were obtained from the corresponding methylpyridine
I with the corresponding aldehyde
II, as is described in the Experimental section. The condensation reaction scheme is presented in
Figure 2.
Figure 2.
Synthesis of trans-2-(m-cyanostyryl)pyridine (IIIa), trans-2-[3-methyl-(m-cyanostyryl)]pyridine (IIIb) and trans-4-(m-cyanostyryl)pyridine (IIIc).
Figure 2.
Synthesis of trans-2-(m-cyanostyryl)pyridine (IIIa), trans-2-[3-methyl-(m-cyanostyryl)]pyridine (IIIb) and trans-4-(m-cyanostyryl)pyridine (IIIc).
2-Styrylpyridine was synthesized according to the previously reported method [
24,
27]. The temperature, reaction time and the presence of the –C≡N and –CH
3 substituents play important roles in the product formation, as well as the presence of either the
ortho- or
para-position pyridine.
Table 1 shows the yields and measured properties of each compound.
Table 1.
Conditions, yields and properties of the styrylpyridine-like model compounds (IIIa–IIIc).
Table 1.
Conditions, yields and properties of the styrylpyridine-like model compounds (IIIa–IIIc).
| Compound | Temp | Time | Yield | Appearance | Melting point | Solubility |
|---|
| | °C | h | % | | °C | |
| IIIa | 120 | 30 | 72 | white powder | 77–78 | toluene, CHCl3, THF, acetone, EtOH, MeOH, DMSO |
| IIIb | 140 | 22 | 59 | beige powder | 68–78 | CHCl3, THF, acetone, MeOH, DMSO |
| IIIc | 120 | 30 | 63 | beige powder | 63–65 | hexane, cyclohexane, toluene, CHCl3, THF, acetone, EtOH, MeOH, DMSO |
Under these reaction conditions, we obtained
IIIa–
IIIc in almost the same yield as reported for 2-styrylpyridine [
24,
27] under the same conditions. Recent studies have described the use of Knoevenagel condensation in the synthesis of several substituted stilbenes in the absence of catalyst and solvent [
19,
22,
24,
25,
26,
27,
28,
29,
30]. However, compared with the conditions used for reactions of pyridylacetonitrile with benzaldehyde, we observed that the effect of the –C≡N group on the reactivity was dependent on its site of attachment in the structure. The reactivity was more pronounced for pyridylacetonitrile than for picoline in obtaining the
IIIa–
IIIc compounds. The reactions conduced without catalyst and solvent gave better yields, even at room temperature [
19,
30], when the –C≡N group was located on the –CH
2CN than when located in the aromatic aldehyde. According to the results from the three reactions of
Table 1, the reactivity behavior is –CH
3 moiety <
p-position <
o-position on pyridine ring due to the inductive effect. Systems with π–conjugated units provide an effective pathway for the efficient push-pull charge transfer between donor and acceptor groups. Styrylpyridine-like materials are expected to have different applications in photochemistry and fluorescence processes when the donor-acceptor or acceptor-acceptor is introduced in the phenyl rings in order to extend the conjugation over the whole molecule.
3.2. Molecular Structures
Optimized parameters were obtained by the B3LYP/6-311 + G(d,p) level for the three model compounds. Previous tests were carried out for the isomers of these molecules with the –C≡N group attached to the equivalent phenyl ring in the
meta-position [
34]. The rotamers showed energy values similar to each other (~1 kJ mol
−1); however, the energy barriers were ~20 to 25 kJ mol
−1 [
34] for the torsion motion of the cyano-substituted phenyl ring. The energetically most stable isomers of molecules
IIIa–
IIIc were those with the –C≡N group attached in position C(3'). Molecules
IIIa and
IIIb displayed an orientation
anti to the –C≡N group with respect to the
N-atom of the pyridine ring, but for
IIIc, the orientation was different (
Figure 1).
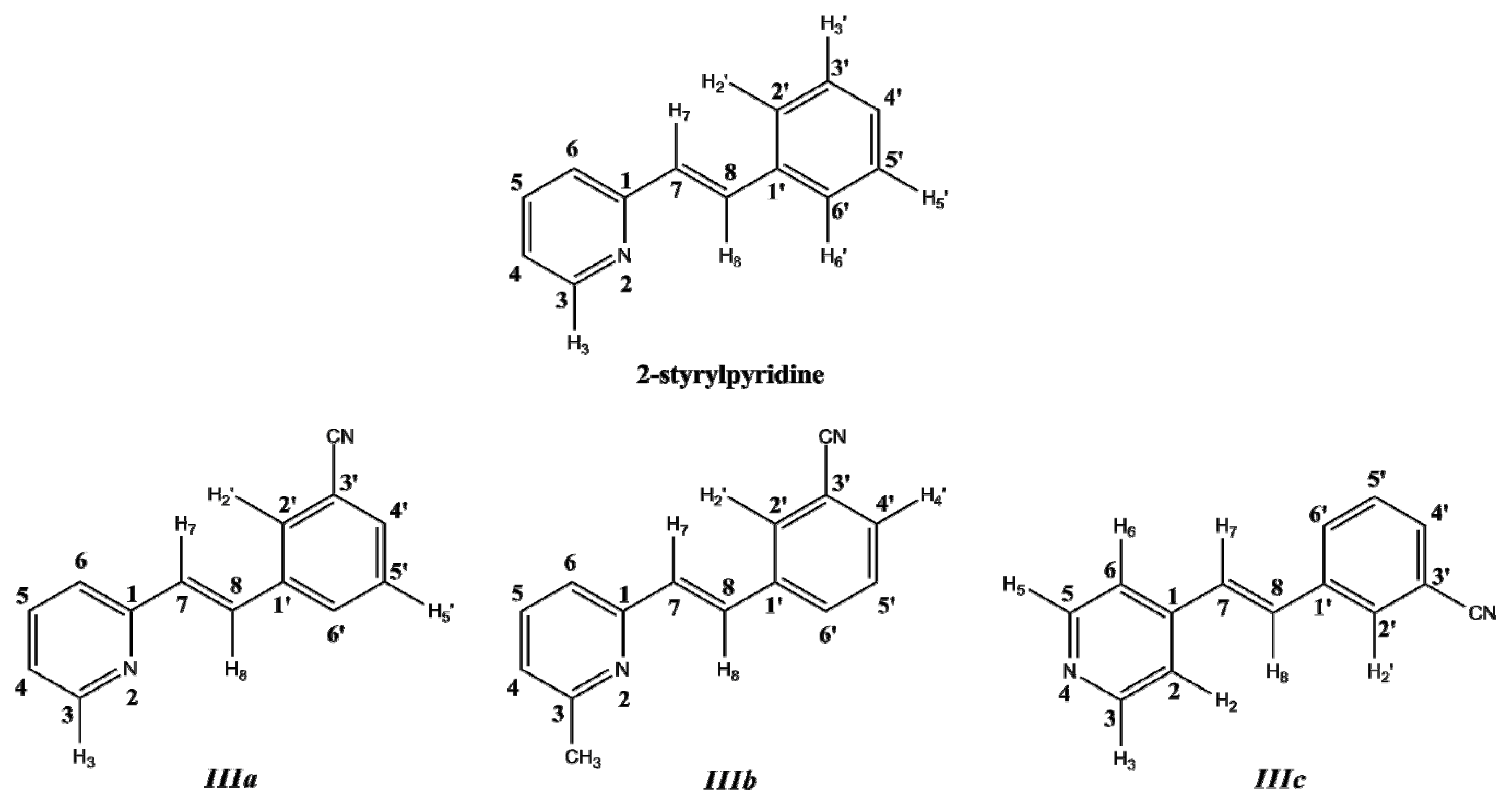
In this work, a two-dimensional conformational analysis was carried out for evaluating simultaneously the effect of the torsion of phenyl group, θ1, and the pyridine group, θ2. The two conformational coordinates were defined as dihedrals θ1, between atoms C(7)–C(8)–C(1')–C(2') for 2-styrylpyridine,
IIIa and
IIIb and C(7)–C(8)–C(1')–C(6') for
IIIc and θ2, between atoms N(2)–C(1)–C(7)–C(8) for 2-styrylpyridine and
IIIa and
IIIb and C(2)–C(1)–C(7)–C(8) for
IIIc (
Figure 1). For the four molecules, a grid of points was generated on each conformational coordinate using increments of 30° in a range of 0°-180°. At each grid point, the conformational coordinates were kept frozen, whereas the rest of the structure was fully relaxed.
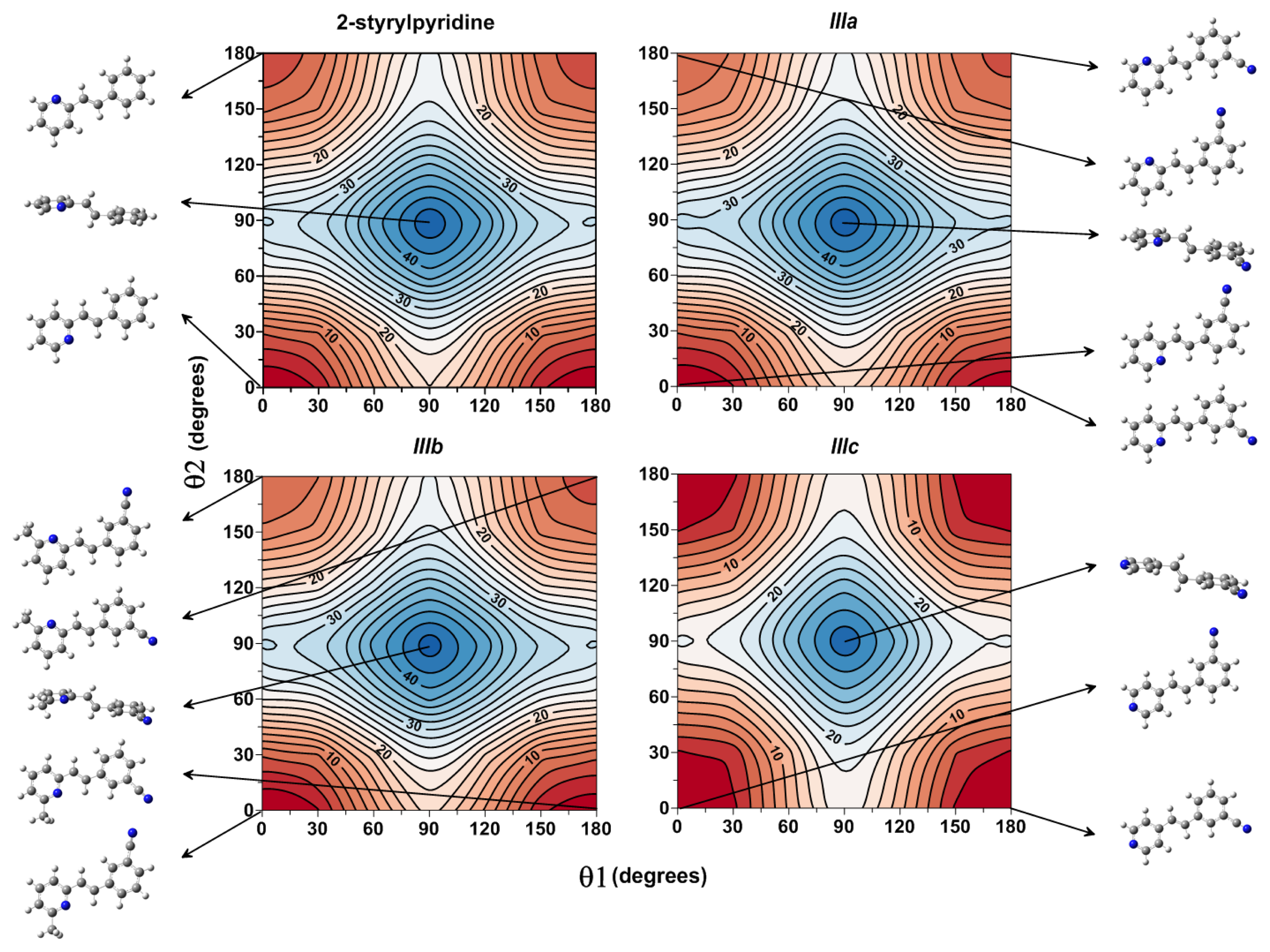
Figure 3 shows the isocontour potential energy maps as the function θ1 and θ2 torsional angles described. The interval between isocontour lines is 10 kJ mol
−1, and the data refer to the minimum value. Red zones correspond to lower energy zones, whereas blue zones correspond to higher energy zones. The structures of the minima and maximum are included in the figure.
Figure 3, for the case of 2-styrylpyridine, shows double minima arising from the symmetric torsion of the phenyl ring; therefore, equivalent minima for (θ1, θ2) (0°, 0° = 180°, 0° and 0°, 180° = 180°, 180°) are found. The global minimum (0°, 0°) and the local minimum (0°, 180°) were located on the potential energy map for the torsion of the pyridine group with a relative energy between rotamers of 4.65 kJ mol
−1; see
Table 2. For
IIIa and
IIIb, in addition to the minima due to the torsion of the pyridine ring, another two minima were located due to the torsion of the cyano-substituted phenyl. So, the rotamer (180°, 0°) is almost energetically equivalent to global minimum (0°, 0°) in less than 1 kJ mol
−1, while two local minima in (0°, 180°) and (180°, 180°) were found with energy differences of about 1 kJ mol
−1 between them (
Table 2). For
IIIc, the position of the
N-atom in the pyridine ring provided two symmetric minima with respect to the pyridine torsion, while the global minimum was found for the (0°, 180°) and the local minimum for (0°, 0°) rotamers. For the four molecules, the activation barriers were found at ~34 to 46 kJ mol
−1, corresponding to structures with (θ1, θ2) = (90°, 90°).
Figure 3.
Isocontour potential energy maps as a function of θ1 and θ2 torsional angles of 2-styrylpyridine, trans-2-(m-cyanostyryl)pyridine (IIIa), trans-2-[3-methyl-(m-cyanostyryl)]pyridine (IIIb) and trans-4-(m-cyanostyryl)pyridine (IIIc) obtained at the B3LYP/6-311 + G(d,p) theory level. The structures at the minima and maximum are included.
Figure 3.
Isocontour potential energy maps as a function of θ1 and θ2 torsional angles of 2-styrylpyridine, trans-2-(m-cyanostyryl)pyridine (IIIa), trans-2-[3-methyl-(m-cyanostyryl)]pyridine (IIIb) and trans-4-(m-cyanostyryl)pyridine (IIIc) obtained at the B3LYP/6-311 + G(d,p) theory level. The structures at the minima and maximum are included.
In order to complement the conformational study, which provides results based essentially on enthalpy values, we achieved a thermostatistical analysis to account for entropic effects. The population of the different rotamers was calculated at room temperature by using the technique developed by Niño
et al. [
52]. The populations (in percent) were calculated from the relative energies obtained at the B3LYP/6-311 + G(d,p) theory level in
Figure 3. The results collected in
Table 2 show that for 2-styrylpyridine, the population for the global minimum is considerably larger (11.85%) than that for the local minimum (1.80%), confirming that the rotamer that is energetically more stable (0°, 0°) is the most populated. For
IIIa and
IIIb, it was found that the minima (0°, 0°) and (180°, 0°) energetic equivalents had populations with values of less than 1.5% of the difference between them, while the minima corresponding to the rotamers (0°, 180°) and (180°, 180°) gave small population values (
Table 2). Finally, for
IIIc, the difference in population between the global and local minima is less than 1%. In all cases, the rotamers with maximum energies gave a population of 0%, as was expected.
Table 2.
Relative energies in (kJ mol−1) calculated at the B3LYP/6-311 + G(d,p) theory level and populations in (%) of the styrylpyridine-like model compounds (IIIa–IIIc) for different rotamers, with θ1 and θ2 dihedral angles in (°).
Table 2.
Relative energies in (kJ mol−1) calculated at the B3LYP/6-311 + G(d,p) theory level and populations in (%) of the styrylpyridine-like model compounds (IIIa–IIIc) for different rotamers, with θ1 and θ2 dihedral angles in (°).
| 2-Styrylpyridine | IIIa |
|---|
| θ1, θ2 | Relative energy | Population | θ1, θ2 | Relative energy | Population |
|---|
| 0.0, 0.0 | 0.000 | 11.847 | 0.0, 0.0 | 0.000 | 6.854 |
| 0.0, 180.0 | 4.647 | 1.797 | 0.0, 180.0 | 6.459 | 0.076 |
| 30.0, 30.0 | 8.113 | 0.087 | 30.0, 30.0 | 7.903 | 0.064 |
| 60.0, 60.0 | 31.821 | 0.002 | 60.0, 60.0 | 31.532 | 0.001 |
| 90.0, 0.0 | 20.033 | 0.008 | 90.0, 0.0 | 19.088 | 0.003 |
| 90.0, 90.0 | 45.815 | 0.000 | 90.0, 90.0 | 45.658 | 0.000 |
| 90.0, 180.0 | 25.074 | 0.009 | 90.0, 180.0 | 24.916 | 0.003 |
| 180.0, 0.0 | 0.000 | 11.847 | 180.0, 0.0 | 0.630 | 5.254 |
| 180.0, 180.0 | 4.647 | 1.797 | 180.0, 180.0 | 5.277 | 1.231 |
| IIIb | IIIc |
| θ1, θ2 | Relative energy | Population | θ1, θ2 | Relative energy | Population |
| 0.0, 0.0 | 0.000 | 6.581 | 0.0, 0.0 | 0.000 | 5.246 |
| 0.0, 180.0 | 6.091 | 0.011 | 0.0, 180.0 | 0.053 | 4.348 |
| 30.0, 30.0 | 7.850 | 0.025 | 30.0, 30.0 | 2.757 | 0.077 |
| 60.0, 60.0 | 31.296 | 0.007 | 60.0, 60.0 | 19.770 | 0.004 |
| 90.0, 0.0 | 19.114 | 0.005 | 90.0, 0.0 | 18.168 | 0.002 |
| 90.0, 90.0 | 45.290 | 0.001 | 90.0, 90.0 | 33.712 | 0.001 |
| 90.0, 180.0 | 24.627 | 0.013 | 90.0, 180.0 | 25.074 | 0.005 |
| 180.0, 0.0 | 0.525 | 5.541 | 180.0, 0.0 | 0.000 | 5.056 |
| 180.0, 180.0 | 5.094 | 0.897 | 180.0, 180.0 | 0.053 | 4.348 |
The results mentioned showed the minima (0°, 0°) and (180°, 0°) of IIIa and IIIb can exist in equivalent proportions, demonstrating that different isomer states can have similar populations.
On the other hand, the compounds involving symmetric groups in their structures, 2-styrylpyridine and IIIc, showed opposing behaviors. For example, in 2-styrylpyridine, the global and local minima, arising from the torsion of pyridine, had conformational energies and populations larger than in IIIc, in which both minima arise from the torsion of the cyano-substituted phenyl with similar values between them.
Selected optimized parameters in the gas phase and those, including the solvent CHCl
3 effect, for model styrylpyridine-like compounds
IIIa–
IIIc are summarized in
Table 3.
B3LYP/6-311 + G(d,p) has been shown to be an adequate level of theory for the geometry calculation for molecules of this kind [
34]. The internuclear distances, valence angles and dihedral angles did not show significant changes among these three molecules. No important differences were found between the gas phase values and those with the solvent effect for the three molecules (
Table 3). Three compounds showed planar structures according to the X-ray data of the title compounds 2-styrylpyridine [
24] and 4-styrylpyridine [
53].
Table 3.
Theoretical structural parameters of the equilibrium structures of the styrylpyridine-like model compounds (
IIIa–
IIIc) calculated at the B3LYP/6-311 + G(d,p) theory level. The numbering convention is shown in
Figure 1. Internuclear distances in (Å), valence and dihedral angles in (°).
Table 3.
Theoretical structural parameters of the equilibrium structures of the styrylpyridine-like model compounds (IIIa–IIIc) calculated at the B3LYP/6-311 + G(d,p) theory level. The numbering convention is shown in Figure 1. Internuclear distances in (Å), valence and dihedral angles in (°).
| | IIIa | IIIb | IIIc |
|---|
| Parameter | Gas | PCM | Gas | PCM | Gas | PCM |
| 7–8 | 1.344 | 1.344 | 1.344 | 1.344 | 1.344 | 1.345 |
| 7–1 | 1.467 | 1.467 | 1.468 | 1.468 | 1.465 | 1.465 |
| 8–1' | 1.465 | 1.465 | 1.464 | 1.465 | 1.466 | 1.466 |
| 4–3 | 1.397 | 1.397 | 1.339 | 1.402 | 1.335 | 1.338 |
| 3–2 | 1.330 | 1.332 | 1.335 | 1.337 | 1.392 | 1.391 |
| 2–1 | 1.347 | 1.349 | 1.346 | 1.346 | 1.402 | 1.403 |
| 3'–C | 1.433 | 1.432 | 1.432 | 1.432 | 1.432 | 1.431 |
| C≡N | 1.156 | 1.156 | 1.155 | 1.156 | 1.155 | 1.156 |
| 1–7–8 | 124.1 | 124.5 | 124.1 | 124.5 | 126.5 | 126.1 |
| 2–1–7 | 118.5 | 118.7 | 118.4 | 118.7 | 119.3 | 119.3 |
| 7–8–1' | 127.1 | 127.0 | 127.2 | 127.0 | 126.9 | 126.7 |
| 2'–3'–C | 119.6 | 119.5 | 119.6 | 119.5 | 119.7 | 119.6 |
| 3'–C≡N | 180.0 | 179.9 | 179.9 | 179.9 | 179.9 | 179.9 |
| 2–1–7–8 | 0.1 | 0.0 | 0.4 | 0.1 | 179.3 | 176.9 |
| 1–7–8–1' | 180.0 | 180.0 | 180.0 | 180.0 | 180.0 | 180.0 |
| 7–8–1'–2' a | 0.4 | 0.3 | 0.7 | 0.2 | 179.2 | 176.6 |
| C–3–2–1 | --- | --- | 179.9 | 180.0 | --- | --- |
| 1'–2'–3'–C | 180.0 | 180.0 | 180.0 | 180.0 | 180.0 | 179.9 |
3.3. IR Spectroscopy
Theoretical IR spectra of 2-styrylpyridine and
IIIa–
IIIc compounds showed similar characteristic infrared band frequencies in the gas phase and when including the solvent CDCl
3 effect. Theoretical scaled frequencies were obtained by using the scale factor of 0.9648 [
44]. The results of the calculated and experimental harmonic frequencies are collected in
Table 4.
Table 4.
Calculated B3LYP/6-311 + G(d,p) and experimental harmonic frequencies in (cm−1) of the 2-styrylpyridine and IIIa–IIIc compounds.
Table 4.
Calculated B3LYP/6-311 + G(d,p) and experimental harmonic frequencies in (cm−1) of the 2-styrylpyridine and IIIa–IIIc compounds.
| | 2-Styrylpyridine | IIIa | IIIb | IIIc |
|---|
| Assignment | Gas | PCM | Exp a | Gas | PCM | Exp b | Gas | PCM | Exp b | Gas | PCM | Exp b |
| δ(C–H) out of plane in
trans config. | 981.5 | 982.5 | 985s | 971.4 | 972.5 | 983 s | 968.3 | 972.3 | 973s | 970.3 | 973.0 | 975 s |
| ν(C=C)Py | 1407.5 | 1405.7 | 1426 s | 1410.0 | 1408.8 | 1431 s | 1550.6 | 1546.7 | 1481 w | 1390.2 | 1389.7 | 1418 s |
| ν(C=N)Py | 1446.8 | 1446.5 | 1468 s | 1457.6 | 1457.1 | 1476 s | 1576.6 | 1574.2 | 1580 s | 1471.6 | 1472.2 | 1481 s |
| [ν(C=N) + ν(C=C)]Py | 1471.9 | 1470.6 | 1494 s | 1544.0 | 1541.6 | 1491 w | ------ | ------ | ------ | 1530.6 | 1526.4 | 1549 s |
| ν(C=N)Py + ν(C=C)Ph | 1556.1 | 1553.7 | 1580 s | 1576.9 | 1574.6 | 1584s | ------ | ------ | ------ | 1578.0 | 1575.9 | 1594 s |
| ν(C=C) | 1625.9 | 1624.1 | 1635 m | 1628.3 | 1626.7 | 1638 m | 1628.9 | 1626.8 | 1641 m | 1628.2 | 1625.7 | 1640 m |
| ν(C≡N) | --- | --- | --- | 2250.5 | 2242.6 | 2232 s | 2250.3 | 2242.5 | 2231 s | 2252.3 | 2244.0 | 2230 s |
| ν(C–H)Py | 3055.7 | 3061.4 | 3074 m | 3034.2 | 3036.2 | 3066 m | 3079.4 | 3081.3 | 3032 w | 3043.1 | 3046.6 | 3065 m |
| ν(C–H)Ph | 3078.8 | 3080.1 | 3141 w | 3090.5 | 3092.0 | 3146 w | 3089.7 | 3091.8 | 3079 m | 3090.5 | 3093.3 | 3154 w |
| δ−as(CH3) | --- | --- | --- | --- | --- | --- | 1020.9 | 1019.8 | 1036 m | --- | --- | --- |
| δs(CH3) | --- | --- | --- | --- | --- | --- | 1360.8 | 1356.7 | 1374 m | --- | --- | --- |
| δ+as(CH3) | --- | --- | --- | --- | --- | --- | 1446.0 | 1439.6 | 1451 s | --- | --- | --- |
| νs(CH3) | --- | --- | --- | --- | --- | --- | 2928.2 | 2926.9 | 2925 m | --- | --- | --- |
| νas(CH3) | --- | --- | --- | --- | --- | --- | 2982.6 | 2980.0 | 2959 s | --- | --- | --- |
Of the ring vibrations, the CH bond stretching of the aromatic ring appeared at the 3100.0–3000.0 cm
−1 region. The ν(C–H) mode for pyridine was found in the range of 3079.4–3034.2 cm
−1 in the gas phase and at the 3081.3–3036.2 cm
−1 range in the solution phase. The ν(C–H) mode for the phenyl ring was calculated at 3090.5-3078.8cm
−1 and 3093.3–3080.1 cm
−1 ranges in gas and solution phases, respectively, (
Table 4). The experimental values at 3074–3032 cm
−1 for ν(C–H)Py and at 3154–3079 cm
−1 for ν(C–H)Ph were in accord with an earlier report for aromatic compounds [
54].
Spectral IR characterization of halogen-substituted compounds showed bands between 3004 and 3076 cm
−1 for ν(C–H) ring vibrations [
30]. These ranges were in good agreement with the values obtained for our cyano-substituted 2-styrylpyridine (
IIIa and
IIIb) and 4-styrylpyridine (
IIIc).
Stretching modes ν(C=C) and ν(C=N) of the pyridine ring were calculated at 1550.6–1390.2 cm
−1 and 1576.6–1446.8 cm
−1, respectively, in the gas phase, whilst values of 1546.7–1389.7 cm
−1 and 1574.2–1446.5 cm
−1 were calculated in the solution phase (
Table 4). In the experimental IR spectrum, the bands at the 1580–1418 cm
−1 range were also assigned to these stretching modes due to ν(C=C)Py and ν(C=N)Py of the pyridine ring (1600–1430 cm
−1 range) [
55].
For 2-styrylpyridine and
IIIa–
IIIc compounds, a combination of bands resulting from the interaction between two vibrations were found coupled in the 1544.0–1471.9 cm
−1 and 1541.6–1470.6 cm
−1 regions corresponding to the stretching modes [ν(C=N) + ν(C=C)] for the pyridine, in the gas, as well as the solution phase. On the other hand, coupled stretching modes ν(C=N) of pyridine + ν(C=C) of phenyl were found in the 1578.0–1556.1 cm
−1 and 1575.9–1553.7 cm
−1 regions in both phases (
Table 4). In the 1600–1500 cm
−1 region [
54] the most of the six-membered aromatic ring systems is reported.
In contrast, the characteristic ν(C=C) mode of the alkene double bond when it is conjugated with aromatic rings was obtained at 1628.9–1625.9 cm
−1 and 1626.8–1624.1 cm
−1, in gas and solution phases, respectively, and the experimental measurement appeared at 1641–1635 cm
−1. These values were in accord with 1625 cm
−1 reported [
55] for this mode.
The band in the regions of 981.5–968.3 cm
−1 and 982.5–972.3 cm
−1 was assigned to the vibration associated with the out of plane deformation d(C-H) due to the protons of a double bond –CH=CH– in the
trans configuration, as calculated in both gas and solution conditions. The experimental values appeared at the 985–973 cm
−1 region, in good agreement with the calculated values and with the values of 980–960 cm
−1 reported [
55], as well as with the values reported previously for 2- and 4-styrylpyridines with halogen substituents [
30].
The mode ν(C≡N) for the cyano-substituted styrylpyridine compounds
IIIa–
IIIc was found at 2252.3–2250.3 cm
−1 and 2242.5 cm
−1 in the gas and solution phases, respectively. The experimental spectra for
IIIa–
IIIc compounds showed the characteristic band shape in the 2232–2230 cm
−1 range, which is the characteristic vibration frequency value for this mode assigned to compounds containing a cyano moiety [
54].
For compound
IIIb, the band corresponding to the CH stretching modes of the –CH
3 appeared at 2982.6–2928.2 cm
−1 region in gas phase and the 2980.0–2926.9 cm
−1 region in solution phase, and the experimental values were two bands at 2959 and 2925 cm
−1. These values agreed with the experimental value reported of 2960 cm
−1 for the asymmetric stretching and 2870 cm
−1 for the symmetric one [
56].
Alternatively, out-of-plane CH deformations for the methyl group for the compound
IIIb were found in the ranges of 1020.9–1019.8 cm
−1 and 1446.0–1439.6 cm
−1 for the asymmetric flexions δ
−as(CH
3) and δ
+as(CH
3), respectively, whilst the symmetric bending δ
s(CH
3) was found at 1360.8–1356.7 cm
−1. These values matched with the values of 1375 and 1450 cm
−1, respectively, for symmetric and asymmetric flexions reported in the literature [
55] and in agreement with the observed values for the
IIIb spectrum of 1374 and 1451 cm
−1, respectively.
3.4. UV Spectroscopy
The electronic transition energies have been calculated within the framework of the Time-Dependent Density Functional Theory method (TD-DFT) [
45,
46,
47,
48]. These calculations have been performed on the lowest energy structures of each model compound as obtained from B3LYP/6-311 + G(d,p) calculations in gas phase and including the solvent CHCl
3 effect. The TD-DFT approach accounts for the dynamic electron correlation caused by the coupling with the correlated ground state function. A previous comparative study [
34] using ZINDO/S, TD-B3LYP and TD-PBE0 methods with different basis sets showed that the theoretical λ
max bands related to the electronic transition between S
0→S
1 states were more exact, as compared with the available experimental data, using the TD-B3LYP/6-31G(d,p)//B3LYP/6-311 + G(d,p) theory level in gas phase.
The results for the electronic transitions, their assignments, the maxima absorption and the oscillator strengths of the styrylpyridine-like model compounds in the gas phase and including the solvent CHCl
3 effect are shown in
Table 5. The major MO→MO excitations involved in the three lowest-lying transitions are also indicated.
The experimental maximum absorptions, λ
max, were observed at 311.0, 309.6, 312.1 and 293.6 nm for 2-styrylpyridine and molecules
IIIa–
IIIc, respectively (
Table 5). The TD-B3LYP/6-31G(d,p)//B3LYP/6-311 + G(d,p) calculations carried out correctly predicted the maximum absorption wavelengths, λ
max (nm), with errors of 0.19%, 0.10%, 0.22% and 4.63% in the gas phase. While the values for 2-styrylpyridine,
IIIa and
IIIb were reproduced correctly, the λ
max for
IIIc was shifted ~14 nm toward the shorter wavelength region of the spectrum in the experimental data. In the solution phase, similar errors of 3.95%, 3.64% and 3.87% were found for 2-styrylpyridine and
IIIa and
IIIb, respectively, while a considerable increment of 8.41% was obtained for
IIIc.
Table 5.
Electronic transition, their assignments, the absorption maxima and oscillator strengths of the 2-styrylpyridine and IIIa–IIIc compounds calculated at the B3LYP/6-31G(d)//B3LYP/6-311 + G(d,p) theory level.
Table 5.
Electronic transition, their assignments, the absorption maxima and oscillator strengths of the 2-styrylpyridine and IIIa–IIIc compounds calculated at the B3LYP/6-31G(d)//B3LYP/6-311 + G(d,p) theory level.
| | Electronic transition | Absorption maxima (nm (eV)) | Oscillator strengths | MO/Character (% Coefficient) | Experimental (nm (eV)) |
|---|
| Gas | PCM | Gas | PCM |
|---|
| 2- styrylpyridine | S0–S1 | 311.6 (3.98) | 323.3 (3.83) | 0.8922 | 1.0597 | HOMO→LUMO (98%) | 311.0 (3.99) |
| S0–S2 | 291.2 (4.26) | 288.1 (4.30) | 0.0014 | 0.0014 | HOMO-2→LUMO (97%) |
| S0–S3 | 270.9 (4.58) | 271.6 (4.57) | 0.0030 | 0.0049 | HOMO-1→LUMO (74%) + HOMO→LUMO+2 (25%) |
| IIIa | S0–S1 | 309.3 (4.01) | 320.9 (3.86) | 0.9656 | 1.1193 | HOMO→LUMO (98%) | 309.6 (4.01) |
| S0–S2 | 298.4 (4.16) | 300.5 (4.13) | 0.0010 | 0.0133 | HOMO-1→LUMO (96%) |
| S0–S3 | 295.7 (4.19) | 295.2 (4.20) | 0.0116 | 0.0012 | HOMO→LUMO + 1 (86%) + HOMO-2→LUMO (12%) |
| IIIb | S0–S1 | 312.7 (3.96) | 324.1 (3.83) | 0.9432 | 1.0916 | HOMO→LUMO (97%) | 312.0 (3.98) |
| S0–S2 | 302.7 (4.10) | 302.8 (4.10) | 0.0010 | 0.0141 | HOMO-1→LUMO (96%) |
| S0–S3 | 297.7 (4.16) | 300.3 (4.13) | 0.0088 | 0.0012 | HOMO→LUMO + 1 (82%) + HOMO-2→LUMO (14%) |
| IIIc | S0–S1 | 307.2 (4.04) | 318.3 (3.89) | 0.7615 | 0.9422 | HOMO→LUMO (98%) | 293.6 (4.23) |
| S0–S2 | 285.4 (4.34) | 296.9 (4.18) | 0.1545 | 0.1452 | HOMO→LUMO + 1 (71%) + HOMO-3→LUMO (24%) |
| S0–S3 | 309.0 (4.01) | 288.1 (4.30) | 0.0021 | 0.0018 | HOMO-1→LUMO (93%) |
The results in
Table 5 indicated that the maximum absorption wavelength for the 2-styrylpyridine and
IIIa–
IIIc compounds are mainly associated with the S
0–S
1 transition, with the largest oscillator strength values. This transition involves the promotion of one electron from the bonding highest-occupied MO (HOMO) into the antibonding lowest-unoccupied MO (LUMO),
i.e., it is due to the π→π* transition in the
trans configuration. In the case of 2-styrylpyridine, the contribution of the HOMO→LUMO excitation was 98% for the S
0–S
1 transition, while for the S
0–S
2 transition, the main contribution (97%) was derived from the excitation of HOMO-2→LUMO. The S
0–S
3 transition in 2-styrylpyridine was characterized by two excitations contributed by HOMO-1→LUMO (74%) + HOMO→LUMO + 2 (25%). For the cyano-substituted compounds, the excitation of HOMO→LUMO was the main contribution to the first S
0–S
1 transition (98%, 97% and 98%, for
IIIa,
IIIb and
IIIc, respectively). The excitation of HOMO-1→LUMO contributed 96% to the second lowest-lying transition S
0–S
2 in the molecules
IIIa and
IIIb. The third lowest-lying transition S
0–S
3, corresponding to the HOMO→LUMO + 1 + HOMO-2→LUMO excitations, contributed 86% and 12%, respectively, in
IIIa and 82% and 14%, respectively, in
IIIb. For compound
IIIc, the HOMO→LUMO+1 (71%) + HOMO-3→LUMO (24%) and HOMO-1→LUMO (93%) excitations made the greatest contributions, the second and third lowest-lying transitions, respectively. This behavior explains the transition bands observed in the experimental spectra for all compounds.
On the other hand, Percino
et al. recently reported absorption spectra of Cl- and F-substituted styrylpyridines that showed one strong absorption signal in the range of 308–318 nm [
30], assigned to the π→π* transition for the double bond with substituents in the
trans position.
ortho-pyridine vinylene compounds containing –F and –Cl attached in the
meta-position on the phenyl ring that were analogous to
IIIa showed absorption maxima at 309 and 308 nm, respectively [
30]. These authors did not observe a shift of the absorption wavelength owing to the presence of –F or –Cl with respect to the maximum absorption wavelength of 2-styrylpyridine. These results were similar to the value of 309 nm obtained for compound
IIIa containing –C≡N (
Table 5). For molecule
IIIc, with the
N-atom position in the
para-position, we observed hypsochromic shifts of ~16 nm in the experimentally determined spectrum and ~3 nm in the theoretically derived data with respect to
IIIa. These results are at odds with the data of Percino
et al. [
30], who reported a red shift in λ
max for Cl-substituted 4-styrylpyridine to 318 nm, which indicated a bathochromic effect of the electron-withdrawing halogen substituent.
Daku
et al. [
33] obtained a value of 314.5 nm for
trans 4-styrylpyridine by using LR-TDDFT calculations for the first excited state S
0–S
1 with an oscillator strength value of 0.90418. This value is approximate to that obtained in the present work for the
trans 4-styrylpyridine cyano-substituted (
IIIc) compound by ~7 nm. In general, the results are in agreement with previous experimental and theoretical results for compounds with a double bond in the
trans position.
On the other hand, the analysis of the energies of the frontier molecular orbitals and, therefore, the band gap energies, give useful information about the optical and electronic properties of the conjugated compounds. The energies of the main molecular orbitals (HOMO, HOMO-1, LUMO and LUMO + 1 in au) calculated by using the TD-B3LYP/6-311 + G(d,p) theory level in gas and solution phases are collected in
Table 6. We also present the calculated HOMO-LUMO gap energies (∆
E = Є
LUMO − Є
HOMO, in eV) of the compounds.
Table 6.
Orbital energies (au) and Gap energies (eV) of the 2-styrylpyridine and IIIa–IIIc compounds calculated at the B3LYP/6-31G(d)//B3LYP/6-311 + G(d,p) theory level.
Table 6.
Orbital energies (au) and Gap energies (eV) of the 2-styrylpyridine and IIIa–IIIc compounds calculated at the B3LYP/6-31G(d)//B3LYP/6-311 + G(d,p) theory level.
| | ЄHOMO-1 | ЄHOMO | ЄLUMO | ЄLUMO+1 | Gap energy |
|---|
| | Gas | PCM | Gas | PCM | Gas | PCM | Gas | PCM | Gas | PCM |
| 2-stypy | −0.2625 | −0.2654 | −0.2201 | −0.2234 | −0.0714 | −0.0747 | −0.0340 | −0.0361 | 4.04 | 4.04 |
| IIIa | −0.2742 | −0.2737 | −0.2366 | −0.2340 | −0.0867 | −0.0841 | −0.0638 | −0.0647 | 4.08 | 4.08 |
| IIIb | −0.2702 | −0.2698 | −0.2338 | −0.2317 | −0.0848 | –0.0828 | −0.0629 | −0.0643 | 4.05 | 4.05 |
| IIIc | −0.2727 | −0.2749 | −0.2491 | −0.2429 | −0.0979 | −0.0920 | −0.0684 | −0.0646 | 4.11 | 4.10 |
The results indicated similar values for molecular orbital and gap energies for both phases. The electron-withdrawing effects of the cyano group stabilized to the HOMO and LUMO orbitals of
IIIa–
IIIc with respect to 2-styrylpyridine. Є
HOMO and Є
LUMO were slightly lower in the gas than in the solution phase, except for 2-styrylpyridine. The smallest gap values were 4.04 and 4.05 eV for 2-styrylpyridine and molecule
IIIb, respectively (
Table 6). The observation may be due to the inductive effect caused by the electron-donating properties of the –CH
3 group that counteract the electron-withdrawing effects of the –C≡N.
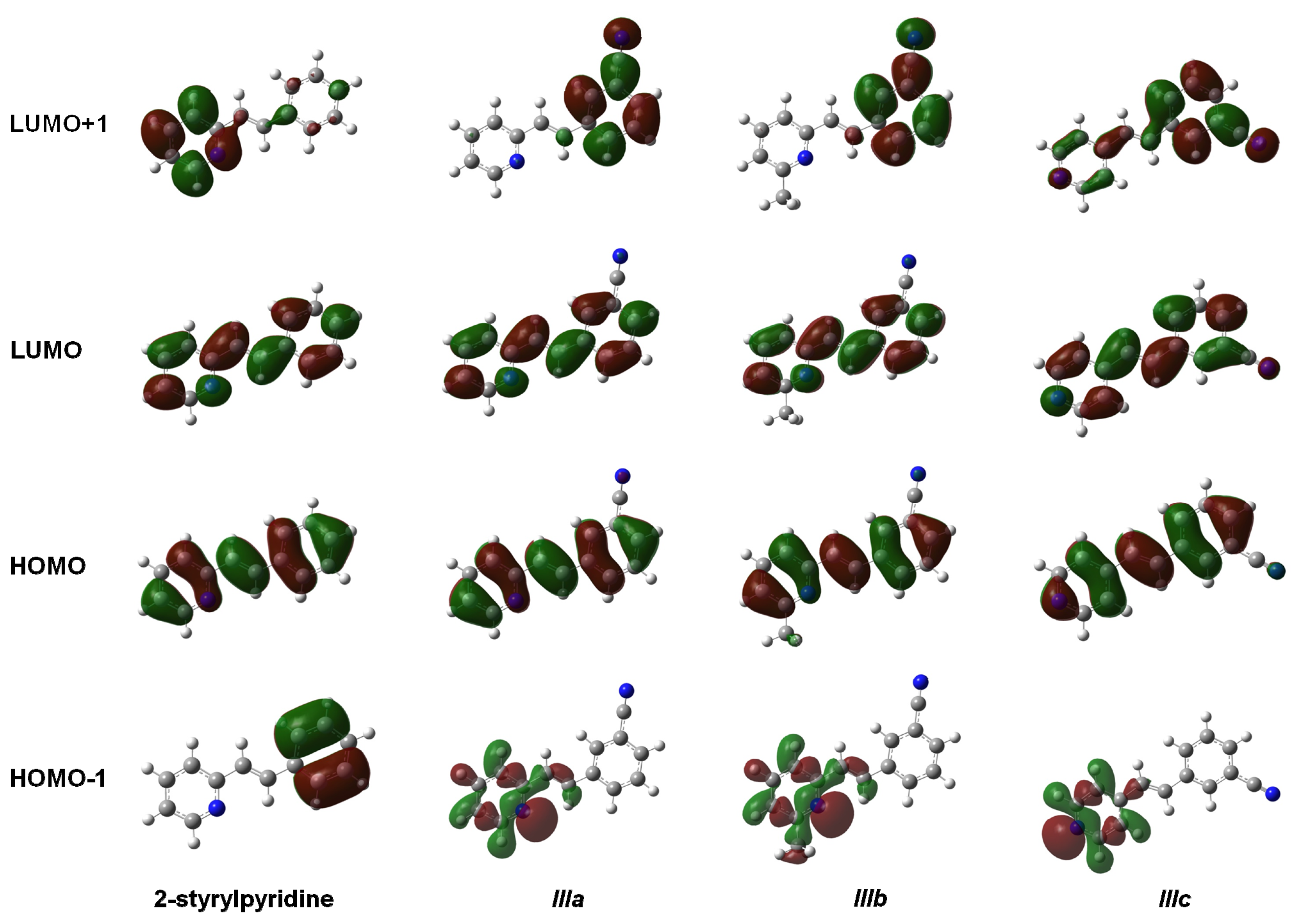
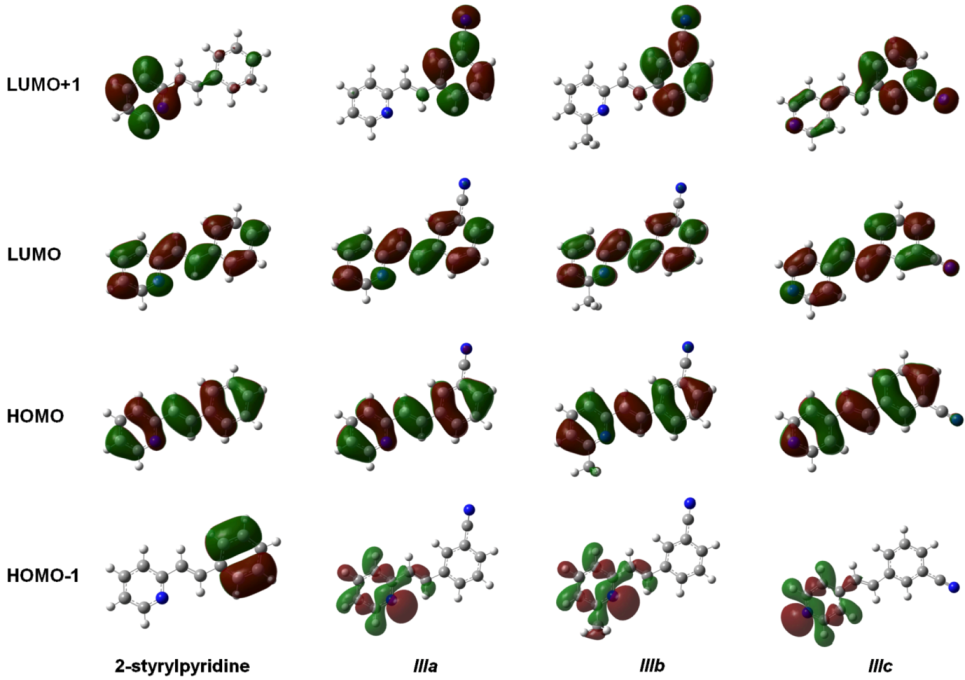
The isosurfaces of the frontier molecular orbitals related to the electronic transition between S
0→S
1 states (transition between molecular orbitals HOMO→LUMO) are depicted in
Figure 4 for the four molecules at the TD-B3LYP/6-311 + G(d,p) level in the gas phase. Similar orbital plots were obtained when the solvent CHCl
3 effect was taken into account. In all cases, HOMO and LUMO π MOs were delocalized over the entire molecule. The isosurfaces of the orbital HOMO were localized in four molecules through the central double bond and the double bonds on the rings, whereas the cyano group made a slight contribution to the electronic distribution of the HOMO in molecules
IIIa–
IIIc. In contrast, LUMO distribution was also similar in the four compounds and was localized on the nitrogen and carbon atoms of the rings and on the central carbons in the molecules. This study showed that the methyl group attached to the pyridine ring in molecule
IIIb had no effect on either the HOMO or the LUMO distributions. The electron distributions of the HOMO-1 were not uniform over the whole molecule. In 2-stryrylpyridine, the electron distribution was concentrated in the phenyl ring, but in molecules
IIIa–
IIIc, it was concentrated in the atoms of the pyridine ring, particularly at the lone pair electrons of the
N-atom. In the case of LUMO + 1, the electron distribution for 2-styrylpyridine was more concentrated on the atoms of the pyridine ring, with a slight concentration on the phenyl ring. The electron distributions for molecules
IIIa and
IIIb were somewhat similar to 2 styrylpyridine, while for
IIIc, the density was moved onto the phenyl ring and to a lesser extent onto the double bonds of the pyridine ring (
Figure 4).
Thus, through the analysis of UV-Vis spectra, molecular orbital energies and isosurfaces, we are able to make predictions about whether a substituent will cause a blue or a red shift in the maximum absorption peaks, quantitatively and qualitatively, particularly if a compound has the same substituent in the structure.
Figure 4.
Main molecular orbitals of 2-styrylpyridine, trans-2-(m-cyanostyryl)pyridine (IIIa), trans-2-[3-methyl-(m-cyanostyryl)]pyridine (IIIb) and trans-4-(m-cyanostyryl)pyridine (IIIc) obtained at the TD-B3LYP/6-311 + G(d,p) theory level.
Figure 4.
Main molecular orbitals of 2-styrylpyridine, trans-2-(m-cyanostyryl)pyridine (IIIa), trans-2-[3-methyl-(m-cyanostyryl)]pyridine (IIIb) and trans-4-(m-cyanostyryl)pyridine (IIIc) obtained at the TD-B3LYP/6-311 + G(d,p) theory level.
3.5. 1H NMR Spectroscopy
The theoretical
1H NMR results of the compounds were calculated by using the Gauge-Independent Atomic Orbital (GIAO) method [
49,
50] at the B3LYP/6-311 + G(2d,p) level from the optimized structures calculated at the B3LYP/6-311 + G(d,p) level in gas phase and the solvent CDCl
3 effect for the 2-styrylpyridine and the
IIIa–
IIIc compounds. In order to express the chemical shifts, δ (ppm), the geometry was optimized, and the
1H NMR spectrum of the tetramethylsilane (TMS) molecule was calculated using the same method and basis set for use as a reference. The calculated isotropic shielding constants σ
i were then transformed to chemical shifts relative to TMS using δ
i = σ
TMS − σ
I, where σ
TMS = 31.88. The
1H NMR data obtained at 400 MHz in CDCl
3 and theoretical chemical shifts of
1H, in ppm, are presented and compared in
Table 7.
Table 7.
Theoretical and experimental 1H NMR chemical shifts (ppm) of the 2-styrylpyridine and IIIa–IIIc compounds calculated at the B3LYP/6-311 + G(2d,p) //B3LYP/6-311 + G(d,p) theory level. All values are referenced to the chemical shift of TMS computed at the same theory level.
Table 7.
Theoretical and experimental 1H NMR chemical shifts (ppm) of the 2-styrylpyridine and IIIa–IIIc compounds calculated at the B3LYP/6-311 + G(2d,p) //B3LYP/6-311 + G(d,p) theory level. All values are referenced to the chemical shift of TMS computed at the same theory level.
| Chemical shifts, d (ppm) |
|---|
| | 2–styrylpyridine | | IIIa |
| Gas | PCM | Expa | Gas | PCM | Expb |
| H3 (d, 1H) | 8.964 | 8.993 | 8.653–8.639 | H3 (d, 1H) | 8.916 | 8.942 | 8.620–8.609 |
| H6', H2' (m, 2H) | 8.148–7.645 | 8.285–7.752 | 7.630–7.613 | H2' (s, 1H) | 8.425 | 8.582 | 7.818 |
| H7 (d, 1H) | 7.418 | 7.594 | 7.240–7.186 | H7 (m, 2H) | 7.462 | 7.629 | 7.204–7.164 |
| H8 (d, 1H) | 8.461 | 8.362 | 7.708–7.654 | H8 (d, 2H) | 8.406 | 8.335 | 7.632–7.592 |
| H5', H3' (m, 3H) | 7.593–7.544 | 7.716–7.684 | 7.416–7.310 | H5' (m, 1H) | 7.818 | 8.021 | 7.488–7.442 |
| | IIIb | | | IIIc | |
| Gas | PCM | Expb | Gas | PCM | Expb |
| H (s, 3H, CH3) | 2.714 | 2.704 | 2.599 | H3, H5 (d, 2H) | 8.952 | 8.936 | 8.605–8.585 |
| H2' (s, 2H) | 8.419 | 8.577 | 7.827 | H2' (s, 1H) | 7.783 | 7.971 | 7.791 |
| H7 (d, 1H) | 7.408 | 7.569 | 7.220–7.180 | H7 (d, 1H) | 7.574 | 7.746 | 7.074–7.020 |
| H8 (d, 1H) | 8.379 | 8.304 | 7.605–7.564 | H8 (d, 1H) | 7.365 | 7.525 | 7.261–7.207 |
| H4' (d, 1H) | 7.677 | 7.837 | 7.796–7.777 | H2, H6 (m, 2H) | 8.220 | 8.444 | 7.365–7.350 |
As it is shown in
Table 7, the theoretical δ values were in good agreement with experimental data for the 2-styrylpyridine and the
IIIa–
IIIc compounds. For 2-styrylpyrydine,
IIIa and
IIIc, the δ for H in position 3 (H
3) was calculated at 8.9 ppm in both gas phase and CDCl
3 solvent, and the experimental signal was found in 8.6 ppm. Both values agreed very well in the range of chemical shift values for the typical protons for
ortho position-substituted pyridine compounds at 8.5 ppm [
55]. Also, the values were consistent with those reported in the literature for the ring current in Hückel aromatic systems containing heteroatoms, which appeared at 8.59 ppm [
57]. On the other hand, the protons of the –CH
3 at the 3 position for
IIIb (
Figure 1) had a calculated δ of 2.7 ppm in both phases and experimentally was a single signal at 2.6 ppm, as typically reported for methyl moieties [
57].
The proton attached in position 2' for 2-styrylpyridine and compounds
IIIa–
IIIc showed theoretical chemical shifts in agreement with the experimental measurements. Error values calculated with respect to experimental data were 2.9%, 7.8%, 7.6% and 0.1%, respectively, in the gas phase. These error percentages slightly increased by using the PCM model in the calculation. In the case of a proton next to a –C≡N group in mono-substituted benzene, the δ value reported is 7.87 ppm [
57]. This value can be roughly compared with δ of H
2' proton on the phenyl ring adjoining the –C≡N group in compounds
IIIa–
IIIc; see
Figure 1. The results summarized in
Table 7, for the theoretical and experimental values obtained in this work, showed that the best approach was for the compound
IIIc.
Interesting features were found in the assignment of the protons H
7 and H
8. From the
1H NMR spectra of all compounds, two doublets corresponding to two protons in the
trans position with J
H–H = 16 Hz were found. It was difficult to determine which signal represented the H
7 or the H
8 proton, but the
1H NMR calculation results allowed us to assign them adequately. H
7 and H
8 protons were experimentally assigned in the 7.2–7.0 ppm and 7.7–7.2 ppm ranges, respectively. The theoretical values were larger than those obtained in the experimental measurements. H
7 was assigned in a range of approximately 7.6–7.4 ppm, while H
8 appeared in a range of 8.5–7.4 ppm for the four molecules (
Table 7). Using the PCM model, similar ranges were calculated. For H
7 in gas phase, the theoretical errors with respect to the experimental values were 2.9%, 3.9%, 2.9% and 7.5%, for 2-styrylpyridine and the molecules
IIIa–
IIIc, respectively. Error percentages increased by ~2% for all four molecules by using the PCM model. Errors of 10.1%, 10.4%, 10.5% and 1.8% were obtained for H
8 in 2-styrylpyridine and
IIIa–
IIIc, respectively, in gas phase, while by using the PCM model, the errors decreased in ~1% for 2-styrylpyridine,
IIIa and
IIIb; however, for
IIIc, it increased by ~2% with respect to that obtained in the gas phase.
The characteristic chemical shift values for the H
7 and H
8 protons attached to a double bond in the
trans position were influenced toward the lower field due to the extended conjugation caused by the presence of substituents on the aromatic rings [
57].
Theoretically, the ppm found for the H
7 and H
8 protons showed that H
8 appeared at chemical shifts larger than H
7 in 2-styrylpyridine and molecules
IIIa and
IIIb, which could be explained by the different steric effects due to the presence of the lone pair of electrons at the
N-atom of the pyridine ring on the H
7 and H
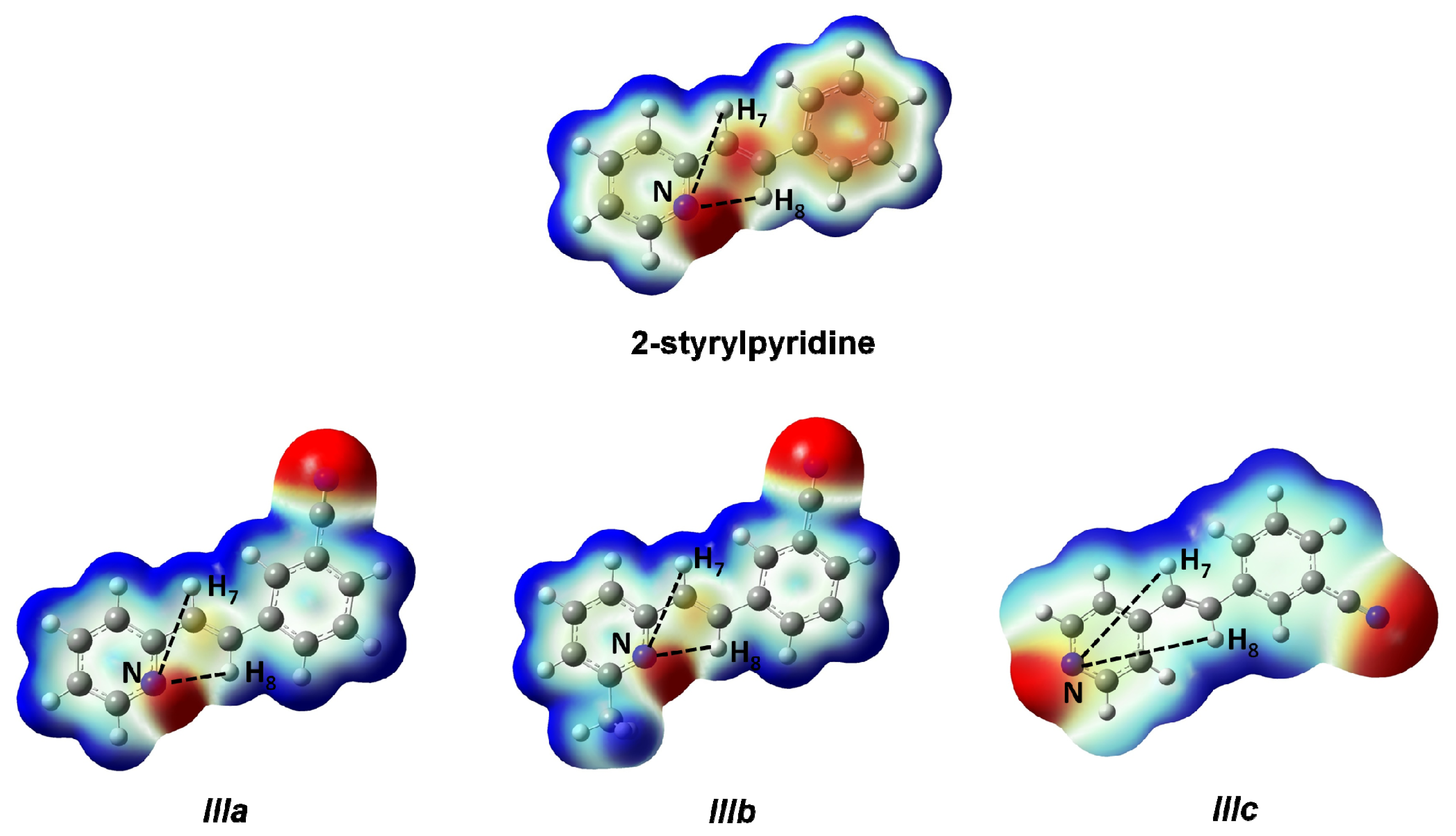
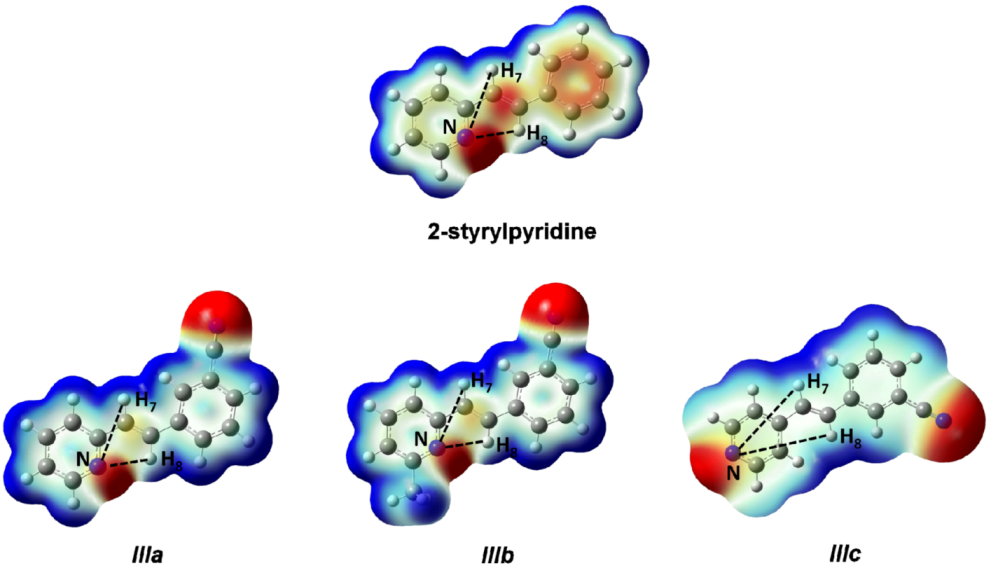
8 protons. On the other hand, the electronic density distribution throughout the whole molecule was useful for the correct assignment of d of these protons. The total electron density mapped with the electrostatic potential surface is presented (isoval = 0.003) in
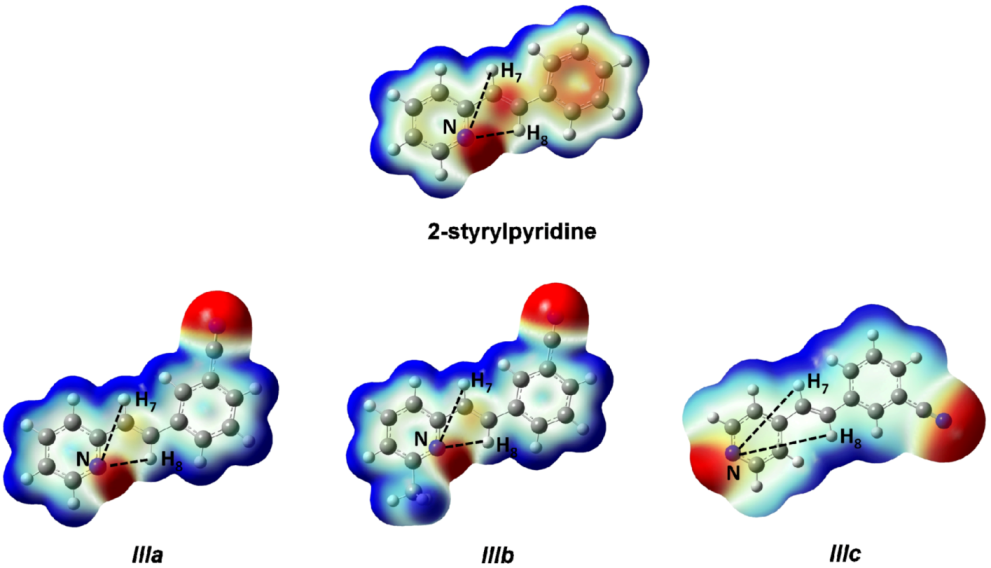
Figure 5. Red regions indicate negative charge, and blue regions indicate positive charge. Yellow regions correspond to an intermediate value between the extremes red and blue.
The distances H
7–N and H
8–N were calculated for all four molecules. For 2-styrylpyridine,
IIIa and
IIIb, the values were 3.36 and 2.50 Å, respectively. The smaller distance indicates the strong electron-withdrawing effect of the pyridine
N-atom, which renders the proton more acidic, and therefore, the chemical shift for H
8 was moved toward the downfield zone in the spectrum. PCM calculation did not take into account this effect, because the distances H
7–N (3.36 Å) and H
8–N (2.52 Å) remained almost constant with respect to gas phase calculation. The distribution of the electron density shown in
Figure 4 indicated that the presence of the electro-withdrawing –C≡N group delocalized the electron distribution, thus decreasing the negative charge (in red color) on the double bond (–CH=CH–) and on the bonds of the phenyl ring with respect to 2-styrylpyridine. In the
IIIa and
IIIb molecules, proton H
8 was more profoundly affected by the proximity of the lone electron pair of the
N-atom, which was manifested as a shorter distance (H
8–N = 2.52 Å), leaving the proton more labile or unshielded. This fact might explain the larger chemical shift value found with respect to the proton H
7, located at a farther distance from the
N-atom.
For molecule IIIc, the behavior of protons H7 and H8 could be due to the A–π–A structure rather than by the presence of the lone electron pair of the N-atom, because the distances were longer: H8–N = 5.13 Å and H7–N = 4.82 Å. So, the influence of the N-atom in the para-position and the symmetry in the molecule played a more important role than the N-proton distances in explaining the calculated and measured chemical shifts. The electron density and chemical environment about both protons was very similar in the molecule, and therefore, the δ values were very similar.
Figure 5.
Total electron density mapped with the electrostatic potential of 2-styrylpyridine, trans-2-(m-cyanostyryl)pyridine (IIIa), trans-2-[3-methyl-(m-cyanostyryl)]pyridine (IIIb) and trans-4-(m-cyanostyryl) pyridine (IIIc) obtained at the TD-B3LYP/6-311 + G(d,p) theory level.
Figure 5.
Total electron density mapped with the electrostatic potential of 2-styrylpyridine, trans-2-(m-cyanostyryl)pyridine (IIIa), trans-2-[3-methyl-(m-cyanostyryl)]pyridine (IIIb) and trans-4-(m-cyanostyryl) pyridine (IIIc) obtained at the TD-B3LYP/6-311 + G(d,p) theory level.
From
1H NMR of the analogous halogen-substituted compounds, the principal signals for protons in the
trans position of a double bond were reported at 7.604–7.550 ppm and 7.079–7.025 ppm for the
p-fluoro-2-styrylpyridine, while for
p-chloro-2-styrylpyridine, signals appeared at 7.294–7.240 and 7.033–6.979 [
30]. These results are in agreement with our results for CN-substituted compounds
IIIa and
IIIb (
Table 7). On the other hand, our results for the molecule
IIIc are in good agreement with respect to previously reported data for 4-styrylpyridine, as well as for the Cl-substituted derivative. The chemical shifts for protons in the double bond were at 7.591–7.537 and 7.126–7.072 ppm [
30].
In
Table 7, the values of chemical shifts for the other protons theoretically characterized and reported, for 2-styrylpyridine (H5' and H3', error = 2.8%),
IIIa (H3', error = 4.8%),
IIIb (H4', error = 1.4%) and
IIIc (H2 and H6, error = 11.7%) were in agreement with the experimental values obtained in the laboratory. These errors were increased by ~2% when the solvent effect was included, except for the molecule
IIIb, whose error value decreased in 0.4% by using the PCM calculation.
The theoretical 1H NMR data for the compounds were extremely useful for the assignment of the double signals in experimental spectra and above all for the assignments for the H7 and H8 positions, which in the experimental spectrum were not easily distinguished.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}