Development and Validation of Single Nucleotide Polymorphisms (SNPs) Markers from Two Transcriptome 454-Runs of Turbot (Scophthalmus maximus) Using High-Throughput Genotyping

Abstract
:1. Introduction
2. Results and Discussion
2.1. Database Exploitation and SNP Detection
2.2. SNP Performance
2.3. SNP Diversity
2.4. SNP Position within Genes: Synonymous vs. Non-Synonymous Substitutions
3. Experimental Section
3.1. EST Database, SNP Detection and Annotation
3.2. SNP Genotyping and Validation
3.3. Gene Diversity and Population Analysis
3.4. Detection of Synonymous/Non-Synonymous SNPs
4. Conclusions
Acknowledgments
References
- APROMAR. La Acuicultura Marina en Espana; Asociación Empresarial de Productores de Cultivos Marinos: Chiclana, Spain, 2012; p. 84. Available online: http://www.apromar.es/Informes/informe%202012/Informe-APROMAR-2012.pdf accessed on 10 July 2012.
- Bouza, C.; Presa, P.; Castro, J.; Sanchez, L.; Martinez, P. Allozyme and microsatellite diversity in natural and domestic populations of turbot (Scophthalmus maximus) in comparison with other Pleuronectiformes. Can. J. Fish. Aquat. Sci 2002, 59, 1460–1473. [Google Scholar]
- Castro, J.; Bouza, C.; Sanchez, L.; Cal, R.M.; Piferrer, F.; Martinez, P. Gynogenesis assessment using microsatellite genetic markers in turbot (Scophthalmus maximus). Mar. Biotechnol 2003, 5, 584–592. [Google Scholar]
- Castro, J.; Bouza, C.; Presa, P.; Pino-Querido, A.; Riaza, A.; Ferreiro, I.; Sanchez, L.; Martinez, P. Potential sources of error in parentage assessment of turbot (Scophthalmus maximus) using microsatellite loci. Aquaculture 2004, 242, 119–135. [Google Scholar]
- Sanchez-Molano, E.; Cerna, A.; Toro, M.A.; Bouza, C.; Hermida, M.; Pardo, B.G.; Cabaleiro, S.; Fernandez, J.; Martinez, P. Detection of growth-related QTL in turbot (Scophthalmus maximus). BMC Genomics 2011, 12, 473. [Google Scholar]
- Rodriguez-Ramilo, S.T.; Toro, M.A.; Bouza, C.; Hermida, M.; Pardo, B.G.; Cabaleiro, S.; Martinez, P.; Fernandez, J. QTL detection for Aeromonas salmonicida resistance related traits in turbot (Scophthalmus maximus). BMC Genomics 2011, 12, 541. [Google Scholar]
- Bouza, C.; Hermida, M.; Pardo, B.G.; Vera, M.; Fernandez, C.; de la Herran, R.; Navajas-Perez, R.; Alvarez-Dios, J.A.; Gomez-Tato, A.; Martinez, P. An Expressed Sequence Tag (EST)-enriched genetic map of turbot (Scophthalmus maximus): A useful framework for comparative genomics across model and farmed teleosts. BMC Genetics 2012, 13, 54. [Google Scholar]
- Vilas, R.; Bouza, C.; Vera, M.; Millan, A.; Martinez, P. Variation in anonymous and EST-microsatellites suggests adaptive population divergence in turbot. Mar. Ecology-Prog. Ser 2010, 420, 231–239. [Google Scholar]
- Pardo, B.G.; Fernandez, C.; Millan, A.; Bouza, C.; Vazquez-Lopez, A.; Vera, M.; Alvarez-Dios, J.A.; Calaza, M.; Gomez-Tato, A.; Vazquez, M.; et al. Expressed sequence tags (ESTs) from immune tissues of turbot (Scophthalmus maximus) challenged with pathogens. BMC Vet. Res 2008, 4, 37. [Google Scholar]
- Pereiro, P.; Balseiro, P.; Romero, A.; Dios, S.; Forn-Cuni, G.; Fuste, B.; Planas, J.V.; Beltran, S.; Novoa, B.; Figueras, A. High-Throughput sequence analysis of turbot (Scophthalmus maximus) transcriptome using 454-pyrosequencing for the discovery of antiviral immune genes. PLoS One 2012, 7, e35369. [Google Scholar]
- Ribas, L.; Pardo, B.G.; Fernandez, C.; Alvarez-Dios, J.A.; Gomez-Tato, A.; Quiroga, M.I.; Planas, J.; Sitja-Bobadilla, A.; Martinez, P.; Piferrer, F. A combined strategy involving Sanger and 454 pyrosequencing increases genomic resources to aid in the management of reproduction; disease control and genetic selection in the turbot (Scophthalmus maximus). BMC Genomics 2013. submitted. [Google Scholar]
- Metzker, M.L. Applications of next-generation sequencing: Sequencing technologies—The next generation. Nat. Rev. Genetics 2010, 11, 31–46. [Google Scholar]
- Voelkerding, K.V.; Dames, S.A.; Durtschi, J.D. Next-Generation sequencing: From basic research to diagnostics. Clin. Chem 2009, 55, 641–658. [Google Scholar]
- Adams, M.D.; Kelley, J.M.; Gocayne, J.D.; Dubnick, M.; Polymeropoulos, M.H.; Xiao, H.; Merril, C.R.; Wu, A.; Olde, B.; Moreno, R.F.; et al. Complementary DNA sequencing— Expressed sequence tags and human genome project. Science 1991, 252, 1651–1656. [Google Scholar]
- Marra, M.A.; Hillier, L.; Waterston, R.H. Expressed sequence tags-ESTablishing bridges between genomes. Trends Genetics 1998, 14, 4–7. [Google Scholar]
- Liu, Z.J.; Li, P.; Kocabas, A.; Karsi, A.; Ju, Z.L. Microsatellite-containing genes from the channel catfish brain: Evidence of trinucleotide repeat expansion in the coding region of nucleotide excision repair gene RAD23B. Biochem. Biophys. Res. Commu 2001, 289, 317–324. [Google Scholar]
- Serapion, J.; Kucuktas, H.; Feng, J.A.; Liu, Z.J. Bioinformatic mining of type I microsatellites from expressed sequence tags of channel catfish (Ictalurus punctatus). Mar. Biotechnol 2004, 6, 364–377. [Google Scholar]
- He, C.; Chen, L.; Simmons, M.; Li, P.; Kim, S.; Liu, Z.J. Putative SNP discovery in interspecific hybrids of catfish by comparative EST analysis. Anim. Genetics 2003, 34, 445–448. [Google Scholar]
- Bouza, C.; Hermida, M.; Pardo, B.G.; Fernandez, C.; Fortes, G.G.; Castro, J.; Sanchez, L.; Presa, P.; Perez, M.; Sanjuan, A.; et al. A microsatellite genetic map of the turbot (Scophthalmus maximus). Genetics 2007, 177, 2457–2467. [Google Scholar]
- Moen, T.; Hayes, B.; Nilsen, F.; Delghandi, M.; Fjalestad, K.T.; Fevolden, S.E.; Berg, P.R.; Lien, S. Identification and characterisation of novel SNP markers in Atlantic cod: Evidence for directional selection. BMC Genetics 2008, 9, 18. [Google Scholar]
- Morin, P.A.; Luikart, G.; Wayne, R.K.; Grp, S.N.P.W. SNPs in ecology; evolution and conservation. Trends Ecol. Evol 2004, 19, 208–216. [Google Scholar]
- Ferber, S.; Reusch, T.B.H.; Stam, W.T.; Olsen, J.L. Characterization of single nucleotide polymorphism markers for eelgrass (Zostera marina). Mol. Ecol. Resour 2008, 8, 1429–1435. [Google Scholar]
- Stapley, J.; Reger, J.; Feulner, P.G.D.; Smadja, C.; Galindo, J.; Ekblom, R.; Bennison, C.; Ball, A.D.; Beckerman, A.P.; Slate, J. Adaptation genomics: The next generation. Trends Ecol. Evol 2010, 25, 705–712. [Google Scholar]
- Kim, S.; Misra, A. SNP genotyping: Technologies and biomedical applications. Ann. Rev. Biomed. Eng 2007, 9, 289–320. [Google Scholar]
- Bester, A.E.; Roodt-Wilding, R.; Whitaker, H.A. Discovery and evaluation of single nucleotide polymorphisms (SNPs) for Haliotis midae: A targeted EST approach. Anim. Genetics 2008, 39, 321–324. [Google Scholar]
- Sachidanandam, R.; Weissman, D.; Schmidt, S.C.; Kakol, J.M.; Stein, L.D.; Marth, G.; Sherry, S.; Mullikin, J.C.; Mortimore, B.J.; Willey, D.L.; et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 2001, 409, 928–933. [Google Scholar]
- Reich, D.E.; Gabriel, S.B.; Altshuler, D. Quality and completeness of SNP databases. Nat. Genetics 2003, 33, 457–458. [Google Scholar]
- Brumfield, R.T.; Beerli, P.; Nickerson, D.A.; Edwards, S.V. The utility of single nucleotide polymorphisms in inferences of population history. Trends Ecol. Evol 2003, 18, 249–256. [Google Scholar]
- Vera, M.; Alvarez-Dios, J.A.; Millan, A.; Pardo, B.G.; Bouza, C.; Hermida, M.; Fernandez, C.; de la Herran, R.; Molina-Luzon, M.J.; Martinez, P. Validation of single nucleotide polymorphism (SNP) markers from an immune Expressed Sequence Tag (EST) turbot; Scophthalmus maximus; database. Aquaculture 2011, 313, 31–41. [Google Scholar]
- Stickney, H.L.; Schmutz, J.; Woods, I.G.; Holtzer, C.C.; Dickson, M.C.; Kelly, P.D.; Myers, R.M.; Talbot, W.S. Rapid mapping of zebrafish mutations with SNPs and oligonucleotide microarrays. Genome Res 2002, 12, 1929–1934. [Google Scholar]
- Cenadelli, S.; Maran, V.; Bongioni, G.; Fusetti, L.; Parma, P.; Aleandri, R. Identification of nuclear SNPs in gilthead seabream. J. Fish. Biol 2007, 70, 399–405. [Google Scholar]
- Hayes, B.; Laerdahl, J.K.; Lien, S.; Moen, T.; Berg, P.; Hindar, K.; Davidson, W.S.; Koop, B.F.; Adzhubei, A.; Hoyheim, B. An extensive resource of single nucleotide polymorphism markers associated with Atlantic salmon (Salmo salar) expressed sequences. Aquaculture 2007, 265, 82–90. [Google Scholar]
- Wang, S.L.; Sha, Z.X.; Sonstegard, T.S.; Liu, H.; Xu, P.; Somridhivej, B.; Peatman, E.; Kucuktas, H.; Liu, Z.J. Quality assessment parameters for EST-derived SNPs from catfish. BMC Genomics 2008, 9, 450. [Google Scholar]
- Hubert, S.; Bussey, J.T.; Higgins, B.; Curtis, B.A.; Bowman, S. Development of single nucleotide polymorphism markers for Atlantic cod (Gadus morhua) using expressed sequences. Aquaculture 2009, 296, 7–14. [Google Scholar]
- Hubert, S.; Higgins, B.; Borza, T.; Bowman, S. Development of a SNP resource and a genetic linkage map for Atlantic cod (Gadus morhua). BMC Genomics 2010, 11, 191. [Google Scholar]
- Sauvage, C.; Bierne, N.; Lapegue, S.; Boudry, P. Single nucleotide polymorphisms and their relationship to codon usage bias in the Pacific oyster Crassostrea gigas. Gene 2007, 406, 13–22. [Google Scholar]
- Vera, M.; Pardo, B.G.; Pino-Querido, A.; Alvarez-Dios, J.A.; Fuentes, J.; Martinez, P. Characterization of single-nucleotide polymorphism markers in the Mediterranean mussel; Mytilus galloprovincialis. Aquac. Res 2010, 41, e568–e575. [Google Scholar]
- Zhang, L.S.; Guo, X.M. Development and validation of single nucleotide polymorphism markers in the eastern oyster Crassostrea virginica Gmelin by mining ESTs and resequencing. Aquaculture 2010, 302, 124–129. [Google Scholar]
- Du, Z.Q.; Ciobanu, D.C.; Onteru, S.K.; Gorbach, D.; Mileham, A.J.; Jaramillo, G.; Rothschild, M.F. A gene-based SNP linkage map for pacific white shrimp; Litopenaeus vannamei. Anim. Genetics 2010, 41, 286–294. [Google Scholar]
- Gorbach, D.M.; Hu, Z.L.; Du, Z.Q.; Rothschild, M.F. Mining ESTs to determine the usefulness of SNPs across shrimp species. Anim. Biotechnol 2010, 21, 100–103. [Google Scholar]
- Lepoittevin, C.; Frigerio, J.M.; Garnier-Gere, P.; Salin, F.; Cervera, M.T.; Vornam, B.; Harvengt, L.; Plomion, C. In vitro vs. in silico detected SNPs for the development of a genotyping array: What can we learn from a non-model species? PLoS One 2010, 5, e11034. [Google Scholar]
- Zhu, C.; Cheng, L.; Tong, J.; Yu, X. Development and characterization of new single nucleotide polymorphism markers from expressed sequence tags in common carp (Cyprinus carpio). Int. J. Mol. Sci 2012, 13, 7343–7353. [Google Scholar]
- Roche Diagnostics GmbH, cDNA Rapid Library Preparation Method Manual; Roche Applied Science: Manheim, Germany, 2009.
- Campbell, N.R.; Amish, S.J.; Pritchard, V.L.; McKelvey, K.S.; Young, M.K.; Schwartz, M.K.; Garza, J.C.; Luikart, G.; Narum, S.R. Development and evaluation of 200 novel SNP assays for population genetic studies of westslope cutthroat trout and genetic identification of related taxa. Mol. Ecol. Resour 2012, 12, 942–949. [Google Scholar]
- Koski, L.B.; Gray, M.W.; Lang, B.F.; Burger, G. AutoFACT: An (Auto)under-barmatic (F)under-barunctional (A)under-barnnotation and (C)under-barlassification (T)under-barool. BMC Bioinforma 2005, 6, 151. [Google Scholar]
- Kim, H.; Schmidt, C.J.; Decker, K.S.; Emara, M.G. A double-screening method to identify reliable candidate non-synonymous SNPs from chicken EST data. Anim. Genetics 2003, 34, 249–254. [Google Scholar]
- Wondji, C.S.; Hemingway, J.; Ranson, H. Identification and analysis of single nucleotide polymorphisms (SNPs) in the mosquito Anopheles funestus; malaria vector. BMC Genomics 2007, 8, 5. [Google Scholar]
- Chevreux, B.; Pfisterer, T.; Drescher, B.; Driesel, A.; Müller, W.E.G.; Wetter, T.; Suhai, S. Using the miraEST assembler for reliable and automated mRNA transcript assembly and dtection in sequenced ESTs. Genome Res 2004, 14, 1147–1159. [Google Scholar]
- Huang, X.; Madan, A. CAP3: A DNA sequence assembly program. Genome Res 1999, 9, 868–877. [Google Scholar]
- Ueno, S.; le Provost, G.; Leger, V.; Klopp, C.; Noirot, C.; Frigerio, J.-M.; Salin, F.; Salse, J.; Abrouk, M.; Murat, F.; et al. Bioinformatic analysis of ESTs collected by Sanger and pyrosequencing methods for a keystone forest tree species: Oak. BMC Genomics 2010, 11, 650. [Google Scholar]
- Tang, J.; Vosman, B.; Voorrips, R.E.; Linden, C.G.; van der Linden, C.G.; Leunissen, J.A.M. QualitySNP: A pipeline for detecting single nucleotide polymorphisms and insertions/deletions in EST data from diploid and polyploid species. BMC Bioinforma 2006, 7, 438. [Google Scholar]
- MySQL Home Page. Available online: http://www.mysql.com accessed on 1 July 2009.
- M-View Home Page. Available online: http://bio-mview.sourceforge.net accessed on 1 July 2009.
- BLAST Home Page. Available online: http://blast.ncbi.nlm.nih.gov/Blast.cgi? accessed on 2 November 2012.
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual, 1st ed; Press CSHL: New York, NY, USA, 1989. [Google Scholar]
- Buetow, K.H.; Edmonson, M.; MacDonald, R.; Clifford, R.; Yip, P.; Kelley, J.; Little, D.P.; Strausberg, R.; Koester, H.; Cantor, C.R.; et al. High-throughput development and characterization of a genomewide collection of gene-based single nucleotide polymorphism markers by chip-based matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Proc. Natl. Acad. Sci. USA 2001, 98, 581–584. [Google Scholar]
- Oeth, P.; del Mistro, G.; Marnellos, G.; Shi, T.; van den Boom, D. Qualitative and quantitative genotyping using single base primer extension coupled with matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MassARRAY). Methods Mol. Biol 2009, 578, 307–343. [Google Scholar]
- Goudet, J. FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.3). Available online: http://www.unil.ch/izea/softwares/fstat.html accessed on 1 March 2003.
- Raymond, M.; Rousset, F. GENEPOP (Version 1.2)—Population genetics software for exact tests and ecumenicism. J. Hered 1995, 86, 248–249. [Google Scholar]
- Rousset, F. GENEPOP’007: A complete re-implementation of the GENEPOP software for Windows and Linux. Mol. Ecol. Resour 2008, 8, 103–106. [Google Scholar]
- Louis, E.J.; Dempster, E.R. An exact test for Hardy-Weinberg and multiple alleles. Biometrics 1987, 43, 805–811. [Google Scholar]
- Rice, W.R. Analyzing tables of statistical tests. Evolution 1989, 43, 223–225. [Google Scholar]
- ORF Finder Home Page. Available online: http://www.ncbi.nlm.nih.gov/gorf/gorf.html accessed on 8 November 2012.
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W improving the sensitivity of progressive multiple sequence alignment through sequence weighting; position-specific gap penalties and weight matrix choice. Nucl. Acids Res 1994, 22, 4673–4680. [Google Scholar]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program fro Windows 95/98/NT. Nucl. Acids Symp. Ser 1999, 41, 95–98. [Google Scholar]
- QuickGO Home Page. Available online: http://www.ebi.ac.uk/QuickGO/ accessed on 15 October 2012.
- AmiGO Home Page. Available online: http://amigo.geneontology.org/cgi-bin/amigo/go.cgi accessed on 16 October 2012.
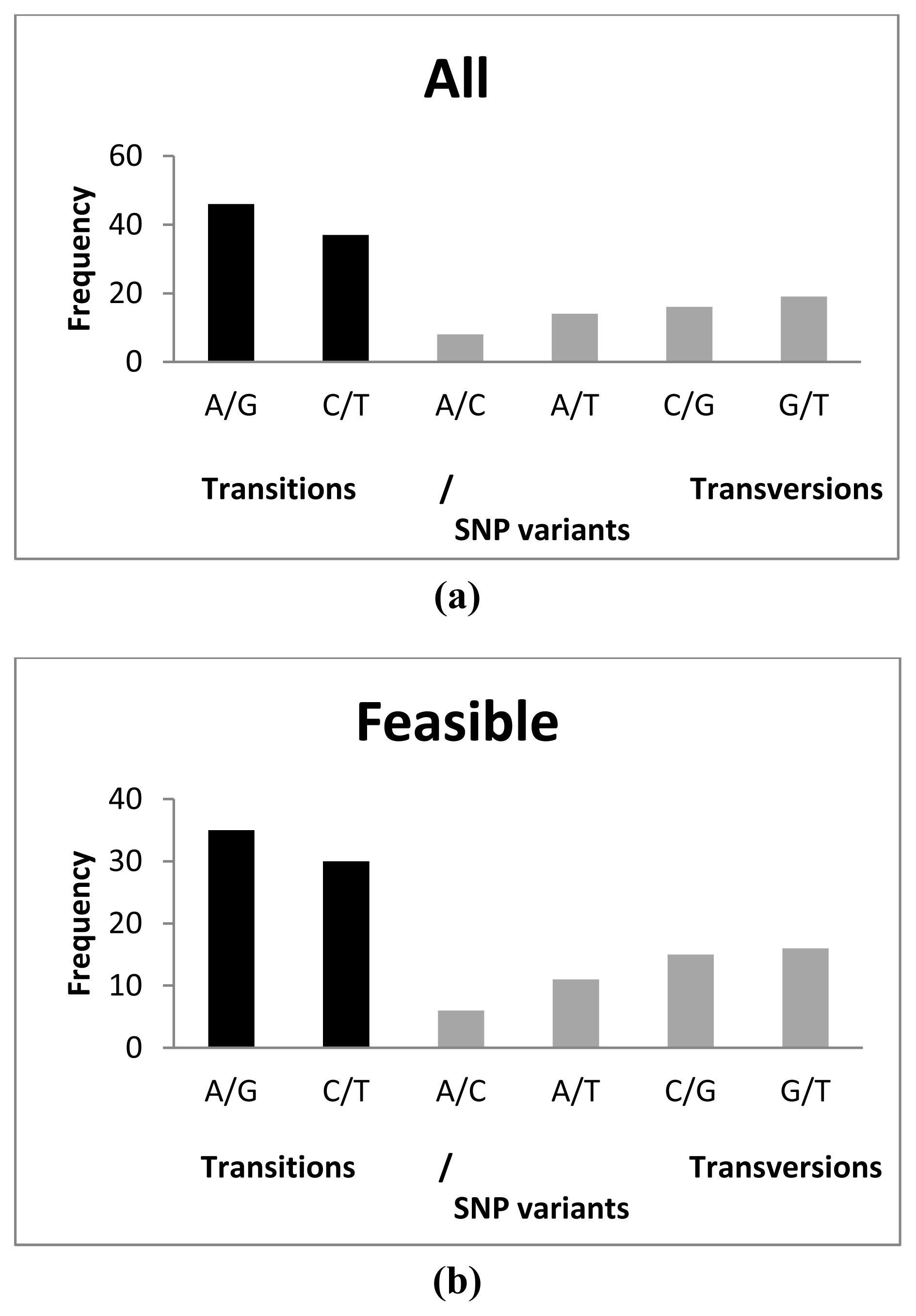

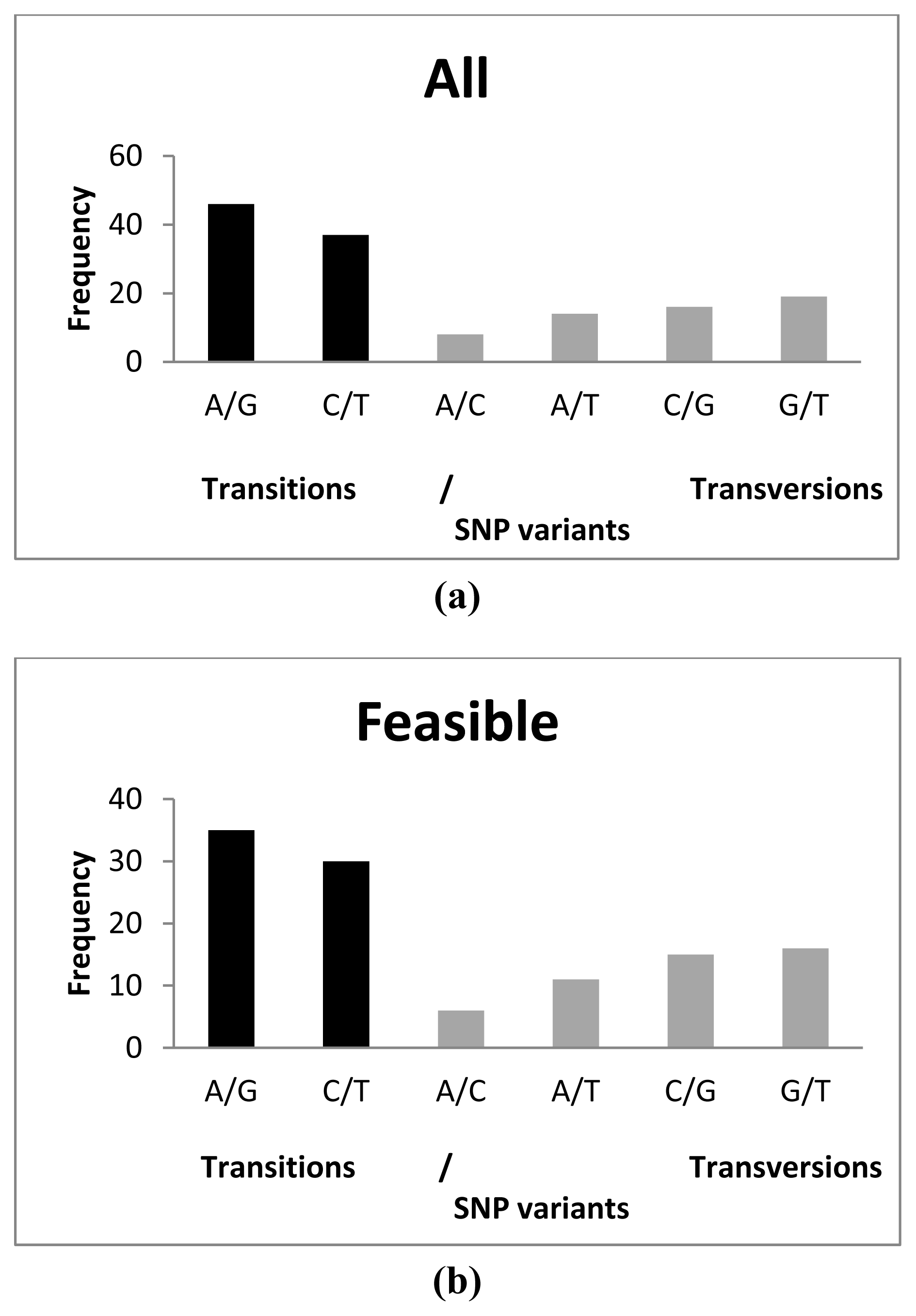{kind=link}
| SNP Name | Annotation | Variants | MAF | P (HW) | He | Fis |
|---|---|---|---|---|---|---|
| SmaSNP_211 | Cyclin-dependent kinase 2 interacting protein | A/T | A = 0.152 | 0.1307 | 0.262 | 0.307 |
| SmaSNP_212 | Zona pellucida sperm-binding protein 3 | A/G | A = 0.152 | 0.4198 | 0.265 | 0.179 |
| SmaSNP_215 | Mitotic specific cyclin-B1 | C/T | T = 0.212 | 0.2948 | 0.338 | −0.255 |
| SmaSNP_216 | Pre-mRNA branch site protein p14 | A/T | T = 0.348 | 0.7003 | 0.460 | −0.119 |
| SmaSNP_217 | Zona pellucida protein C1 | A/G | G = 0.258 | 0.1616 | 0.390 | 0.301 |
| SmaSNP_218 | Mitochondrial ribosomal protein S18A | G/T | T = 0.333 | 1.0000 | 0.452 | 0.061 |
| SmaSNP_219 | U3 small nucleolar ribonucleoprotein protein IMP3 | G/T | G = 0.409 | 1.0000 | 0.491 | −0.050 |
| SmaSNP_220 | Coatomer subunit epsilon isoform 1 | C/T | T = 0.197 | 0.5750 | 0.322 | 0.153 |
| SmaSNP_222 | Signal recognition particle 14 kDa protein | G/T | G = 0.203 | 1.0000 | 0.329 | −0.046 |
| SmaSNP_223 | Epithelial cell adhesion protein | A/T | T = 0.333 | 1.0000 | 0.452 | 0.061 |
| SmaSNP_224 | Transcription initiation factor TFIID subunit D11 | C/G | G = 0.182 | 1.0000 | 0.302 | −0.003 |
| SmaSNP_225 | Acidic ribosomal protein P1 | A/G | G = 0.480 | 1.0000 | 0.510 | 0.059 |
| SmaSNP_226 | Alcohol dehydrogenase Class-3 | C/T | T = 0.288 | 0.6913 | 0.416 | −0.093 |
| SmaSNP_227 | Thioredoxin protein 4A | A/G | A = 0.242 | 1.0000 | 0.373 | 0.025 |
| SmaSNP_228 | Novel protein similar to vertebrate THAP domain containing 4 (THAP4) | A/G | G = 0.212 | 0.6068 | 0.340 | 0.109 |
| SmaSNP_229 | Tumor suppressor candidate 2 | A/G | A = 0.031 | 1.0000 | 0.061 | −0.016 |
| SmaSNP_230 | Optic atrophy 3 protein | C/T | T = 0.266 | 0.6477 | 0.397 | 0.135 |
| SmaSNP_231 | RNA 3′-terminal phosphate cyclase | A/C | C = 0.266 | 0.6475 | 0.397 | 0.135 |
| SmaSNP_232 | RAD1 homolog | A/G | A = 0.438 | 0.4921 | 0.501 | 0.127 |
| SmaSNP_233 | Ubiquitin carrier protein | G/T | T = 0.409 | 1.0000 | 0.491 | −0.050 |
| SmaSNP_234 | chromatin accessibility complex protein 1 | A/G | G = 0.047 | 0.0504 | 0.092 | 0.659 |
| SmaSNP_235 | Nucleolar protein 16 | A/G | G = 0.258 | 0.4023 | 0.389 | 0.144 |
| SmaSNP_236 | Isopentenyl-diphosphate delta-isomerase 1 | C/G | G = 0.141 | 0.4763 | 0.246 | 0.111 |
| SmaSNP_237 | Ran-specific GTPase-activating protein | G/T | T = 0.030 | 1.0000 | 0.060 | −0.016 |
| SmaSNP_238 | Forkhead box H1 | A/G | A = 0.453 | 1.0000 | 0.503 | −0.056 |
| SmaSNP_239 | Stathmin | C/T | C = 0.258 | 0.6436 | 0.387 | −0.174 |
| SmaSNP_240 | Ubiquinol-cytochrome c reductase core I protein | C/T | C = 0.152 | 0.5521 | 0.261 | 0.072 |
| SmaSNP_241 | BolA-like protein 3 | C/G | G = 0.313 | 0.4371 | 0.438 | 0.143 |
| SmaSNP_243 | ce ceroid-lipofuscinosis neuronal protein 5 | G/T | G = 0.455 | 1.0000 | 0.504 | 0.038 |
| SmaSNP_244 | SSU rRNA; Psetta maxima (turbot) | C/T | C = 0.061 | 1.0000 | 0.116 | −0.049 |
| SmaSNP_245 | Chromobox protein homolog 3 | G/T | T = 0.030 | 1.0000 | 0.060 | −0.016 |
| SmaSNP_246 | Transmembrane protein 208 | A/C | A = 0.469 | 0.7198 | 0.505 | −0.114 |
| SmaSNP_247 | Ribosomal protein L18a | A/C | A = 0.234 | 1.0000 | 0.365 | 0.058 |
| SmaSNP_248 | Pre-mRNA-processing factor 19 | C/T | C = 0.318 | 1.0000 | 0.440 | −0.032 |
| SmaSNP_249 | Alpha-l-fucosidase | A/G | A = 0.500 | 0.7275 | 0.509 | 0.106 |
| SmaSNP_250 | Protein phosphatase 2 (Formerly 2A) | A/G | G = 0.406 | 0.0598 | 0.493 | 0.366 |
| SmaSNP_252 | LON peptidase N-terminal domain and RING finger protein 1 | G/T | T = 0.167 | 1.0000 | 0.282 | 0.034 |
| SmaSNP_253 | IK cytokine | A/G | A = 0.439 | 0.0348 | 0.497 | −0.402 |
| SmaSNP_256 | Ribonuclease UK114 | C/T | C = 0.232 | 0.6038 | 0.364 | 0.116 |
| SmaSNP_257 | Inner centromere protein | A/G | G = 0.303 | 0.4239 | 0.430 | 0.154 |
| SmaSNP_259 | Beta-galactoside-binding lectin | C/T | C = 0.379 | 0.7242 | 0.477 | −0.079 |
| SmaSNP_260 | Enoyl-Coenzyme A hydratase | A/T | A = 0.273 | 0.3819 | 0.402 | −0.208 |
| SmaSNP_261 | Sept2 protein | A/G | G = 0.197 | 1.0000 | 0.321 | −0.038 |
| SmaSNP_262 | DNA-directed RNA polymerase I subunit RPA34 | A/G | A = 0.078 | 1.0000 | 0.146 | −0.069 |
| SmaSNP_263 | Epithelial membrane protein 2 | A/G | G = 0.379 | 0.1336 | 0.480 | 0.306 |
| SmaSNP_264 | Retinol dehydrogenase 3 | C/G | G = 0.409 | 1.0000 | 0.491 | −0.050 |
| SmaSNP_265 | WD repeat-containing protein 54 | A/G | A = 0.076 | 1.0000 | 0.142 | −0.067 |
| SmaSNP_266 | tRNA pseudouridine synthase 3 | C/T | C = 0.136 | 0.4637 | 0.240 | 0.115 |
| SmaSNP_267 | Transmembrane protein 167 precursor | G/T | G = 0.258 | 0.6463 | 0.387 | −0.174 |
| SmaSNP_270 | Flotillin-1 | C/G | G = 0.438 | 0.1694 | 0.502 | 0.253 |
| SmaSNP_271 | NAD(P)H dehydrogenase quinone 1 | A/G | G = 0.359 | 0.0488 | 0.471 | 0.403 |
| SmaSNP_273 | Ubiquitin protein ligase E3 component | C/T | C = 0.484 | 0.7353 | 0.508 | 0.077 |
| SmaSNP_274 | K13213 matrin 3 | C/G | G = 0.106 | 0.2983 | 0.193 | 0.216 |
| SmaSNP_275 | Dolichol-phosphate mannosyltransferase | A/G | A = 0.091 | 0.2209 | 0.169 | 0.281 |
| SmaSNP_276 | DNA-directed RNA polymerases i II and III subunit rpabc1 | A/G | A = 0.197 | 0.5728 | 0.322 | 0.153 |
| SmaSNP_277 | Syndecan 2 | A/C | A = 0.429 | 1.0000 | 0.505 | −0.130 |
| SmaSNP_278 | Peptide methionine sulfoxide reductase | C/G | C = 0.078 | 1.0000 | 0.146 | −0.069 |
| SmaSNP_279 | Methyltransferase-like protein 21D | G/T | G = 0.470 | 0.0129 | 0.509 | 0.465 |
| SmaSNP_281 | Phosphatidylinositol transfer protein beta isoform-like isoform 2 | C/T | T = 0.318 | 0.4333 | 0.439 | −0.172 |
| SmaSNP_282 | Zona pellucida protein C | A/T | T = 0.121 | 1.0000 | 0.216 | −0.123 |
| SmaSNP_283 | AP-2 complex subunit alpha-2-like | G/T | G = 0.333 | 0.2669 | 0.450 | −0.213 |
| SmaSNP_284 | Apoptosis regulator BAX | A/G | G = 0.409 | 0.0780 | 0.493 | 0.324 |
| SmaSNP_285 | Borealin | G/T | T = 0.032 | 1.0000 | 0.063 | −0.017 |
| SmaSNP_286 | Brain protein 44 | C/T | C = 0.394 | 0.2691 | 0.483 | −0.255 |
| SmaSNP_287 | Exosome component 8 | A/G | G =1.000 | - | 0.000 | NA |
| SmaSNP_288 | Atrophin-1 domain containing protein | G/T | T = 0.439 | 1.0000 | 0.500 | −0.030 |
| SmaSNP_289 | similar to connectin/titin | A/T | A = 0.303 | 0.0018 | 0.433 | 0.580 |
| SmaSNP_290 | Ubiquitin carboxyl-terminal hydrolase L5 | C/T | T = 0.469 | 1.0000 | 0.506 | 0.012 |
| SmaSNP_292 | Histone deacetylase complex subunit SAP18 | C/T | C = 0.188 | 0.5568 | 0.308 | −0.216 |
| SmaSNP_293 | Replication protein A 14 kDa subunit | C/G | G = 0.182 | 1.0000 | 0.302 | −0.003 |
| SmaSNP_296 | Carbonic anhydrase | G/T | G = 0.076 | 1.0000 | 0.142 | −0.067 |
| SmaSNP_297 | UPF0414 transmembrane protein | C/T | C = 0.212 | 0.6080 | 0.340 | 0.109 |
| SmaSNP_298 | Queuine tRNA-ribosyltransferase | C/T | T = 0.061 | 1.0000 | 0.116 | −0.049 |
| SmaSNP_299 | NHP2-like protein 1 | C/G | C = 0.379 | 0.1358 | 0.480 | 0.306 |
| SmaSNP_304 | Microsomal glutathione S-transferase 3 | A/G | A = 0.091 | 1.0000 | 0.168 | −0.085 |
| SmaSNP_305 | Actin related protein 2/3 complex subunit 4 | C/T | T = 0.030 | 1.0000 | 0.060 | −0.016 |
| SmaSNP_306 | Cyclophilin B | C/G | C = 0.061 | 1.0000 | 0.116 | −0.049 |
| SmaSNP_307 | Dynein light chain Tctex-type 3 | C/G | C = 0.061 | 1.0000 | 0.116 | −0.049 |
| SmaSNP_308 | Ependymin-1 | A/G | A = 0.234 | 0.3135 | 0.366 | 0.231 |
| SmaSNP_309 | C-4 methylsterol oxidase | A/G | A = 0.297 | 1.0000 | 0.424 | 0.043 |
| SmaSNP_310 | Dynein light chain LC8-type | G/T | T = 0.045 | 1.0000 | 0.088 | −0.032 |
| SmaSNP_311 | Rho-related GTP-binding protein RhoF | A/T | T = 0.394 | 0.2669 | 0.483 | −0.255 |
| SmaSNP_312 | Golgi SNAP receptor complex member 1 | A/T | A = 0.188 | 0.5587 | 0.308 | −0.216 |
| SmaSNP_314 | Ribosomal L1 domain-containing protein 1 | A/G | A = 0.203 | 1.0000 | 0.329 | −0.046 |
| SmaSNP_315 | N-alpha-acetyltransferase 50 | A/T | A = 0.242 | 1.0000 | 0.373 | 0.025 |
| SmaSNP_316 | Oncogene DJ-1 isoform 1 | C/T | C = 0.453 | 1.0000 | 0.503 | −0.056 |
| SmaSNP_317 | Wu:fj40d12 protein n = 7 Tax = Euteleostomi RepID = A3KP21_DANRE | A/G | A = 0.438 | 1.0000 | 0.500 | 0.000 |
| SmaSNP_318 | Mucin multi-domain protein | C/G | C = 0.167 | 0.5617 | 0.281 | −0.185 |
| SmaSNP_319 | Adenosine kinase | A/G | A = 0.182 | 0.5575 | 0.301 | −0.208 |
| SmaSNP_320 | No homology found | A/G | A = 0.394 | 0.4901 | 0.486 | 0.127 |
| SmaSNP_321 | Zymogen granule membrane protein 16 | A/G | G = 0.333 | 1.0000 | 0.451 | −0.076 |
| SmaSNP_322 | 6-Pyruvoyl tetrahydrobiopterin synthase | C/T | C = 0.031 | 1.0000 | 0.061 | −0.016 |
| SmaSNP_323 | Proteasome subunit beta | C/T | T = 0.125 | 1.0000 | 0.222 | −0.127 |
| SmaSNP_324 | RING finger protein 4 | A/G | A = 0.394 | 0.0652 | 0.488 | 0.379 |
| SmaSNP_325 | Lipocalin | C/G | C = 0.136 | 1.0000 | 0.239 | −0.143 |
| SmaSNP_326 | Choline transporter-like protein 2 | A/G | G = 0.455 | 0.0311 | 0.507 | 0.402 |
| SmaSNP_328 | RNA-binding proteins (RRM domain) | C/T | C = 0.106 | 1.0000 | 0.192 | −0.103 |
| SmaSNP_329 | Type II keratin | C/G | G = 0.061 | 1.0000 | 0.116 | −0.049 |
| SmaSNP_330 | Novel protein similar to vertebrate thyroid hormone receptor interactor 12 (TRIP12) | C/T | T = 0.094 | 1.0000 | 0.172 | −0.088 |
| SmaSNP_332 | Ribosomal protein S6 kinase | A/C | A = 0.470 | 0.7287 | 0.507 | 0.103 |
| SmaSNP_333 | Transmembrane 6 superfamily member 2 | A/T | T = 0.288 | 0.0796 | 0.419 | 0.348 |
| SmaSNP_334 | PREDICTED: hypothetical protein LOC100712283 [Oreochromis niloticus] | C/T | T =1.000 | - | 0.000 | NA |
| SmaSNP_337 | 1-Alkyl-2-acetylglycerophosphocholine esterase | C/T | C = 0.234 | 1.0000 | 0.365 | 0.058 |
| SmaSNP_338 | CD151 antigen | C/T | T = 0.266 | 0.3909 | 0.395 | −0.186 |
| SmaSNP_339 | Arsenite methyltransferase 1 | A/T | A = 0.313 | 1.0000 | 0.436 | −0.002 |
| SmaSNP_340 | Receptor expression-enhancing protein 5 | C/T | T = 0.234 | 0.6507 | 0.364 | −0.116 |
| SmaSNP_341 | Cathepsin S | C/G | G = 0.333 | 0.1119 | 0.454 | 0.332 |
| SmaSNP_342 | Trans-1,2-dihydrobenzene-1,2-diol dehydrogenase | A/G | A = 0.424 | 0.2818 | 0.494 | −0.226 |
| SmaSNP_343 | High mobility group protein 2 | G/T | G = 0.470 | 0.2980 | 0.508 | 0.224 |
| SmaSNP_346 | ATP-binding cassette, sub-family A (ABC1) | C/T | T = 0.288 | 1.0000 | 0.417 | 0.055 |
| SmaSNP_347 | Myomesin 1a (skelemin) | C/T | T = 0.091 | 1.0000 | 0.168 | −0.085 |
| SmaSNP_348 | Retinoic acid receptor responder protein 3 | A/G | G = 0.439 | 1.0000 | 0.500 | −0.030 |
| SmaSNP_349 | Nucleophosmin 1 | A/C | A = 0.258 | 0.6466 | 0.387 | −0.174 |
| SNP Name | SNP location/effect | GO term |
|---|---|---|
| SmaSNP_211 | 3′ UTR | phosphorylation (GO:0016310) |
| SmaSNP_212 | Non synonymous | reproduction (GO:0000003) |
| SmaSNP_215 | Synonymous | mitotic cell cycle (GO:0000278) |
| SmaSNP_216 | 3′ UTR | protein localization to cell division site (GO:0072741) |
| SmaSNP_217 | Non synonymous | binding of sperm to zona pellucida ( GO:0007339) |
| SmaSNP_218 | Non synonymous | protein import into mitochondrial matrix (GO:0030150) |
| SmaSNP_219 | 3′ UTR | ribonucleoprotein complex biogenesis (GO:0022613) |
| SmaSNP_220 | Synonymous | ribosomal large subunit assembly (GO:0000027) |
| SmaSNP_222 | 5′ UTR | regulation of peptidoglycan recognition protein signaling pathway (GO:0061058) |
| SmaSNP_223 | Synonymous | cell adhesion (GO:0007155) |
| SmaSNP_224 | Synonymous | DNA-dependent transcription, initiation (GO:0006352) |
| SmaSNP_225 | 3′ UTR | ribosomal large subunit assembly (GO:0000027) |
| SmaSNP_226 | Synonymous | cellular alcohol metabolic process (GO:0044107) |
| SmaSNP_227 | Synonymous | thioredoxin biosynthetic process (GO:0042964) |
| SmaSNP_228 | 5′ UTR | regulation of nucleotide-binding oligomerization domain containing signaling pathway (GO:0070424 ) |
| SmaSNP_229 | 3′ UTR | immune response to tumor cell (GO:0002418) |
| SmaSNP_230 | 3′ UTR | reproduction (GO:0000003) |
| SmaSNP_231 | Non synonymous | phosphorylation of RNA polymerase II C-terminal domain (GO:0070816) |
| SmaSNP_232 | Synonymous | resolution of meiotic recombination intermediates (GO:0000712) |
| SmaSNP_233 | Synonymous | ubiquitin-dependent protein catabolic process (GO:0006511) |
| SmaSNP_234 | 3′ UTR | regulation of macrophage inflammatory protein 1 alpha production (GO:0071640) |
| SmaSNP_235 | Synonymous | protein localization to nucleolar rDNA repeats (GO:0034503) |
| SmaSNP_236 | Synonymous | T-helper 1 cell activation (GO:0035711) |
| SmaSNP_237 | 5′ UTR | termination of G-protein coupled receptor signaling pathway (GO:0038032) |
| SmaSNP_238 | Non synonymous | transcription initiation from RNA polymerase III type 2 promoter (GO:0001023) |
| SmaSNP_239 | 5′ UTR | Not found |
| SmaSNP_240 | Synonymous | MHC class I protein complex assembly (GO:0002397) |
| SmaSNP_241 | 3′ UTR | reproduction (GO:0000003) |
| SmaSNP_243 | Non synonymous | neuronal stem cell maintenance (GO:0097150) |
| SmaSNP_244 | Unknown | Not found |
| SmaSNP_245 | 5′ UTR | reproduction (GO:0000003) |
| SmaSNP_246 | Synonymous | intracellular protein transmembrane transport (GO:0065002) |
| SmaSNP_247 | Non synonymous | ribosomal protein import into nucleus (GO:0006610) |
| SmaSNP_248 | 3′ UTR | regulation of mitotic recombination (0000019) |
| SmaSNP_249 | 3′ UTR | alpha-l-fucosidase activity (GO:0004560) |
| SmaSNP_250 | 3′ UTR | modulation by virus of host protein serine/threonine phosphatase activity (GO:0039517) |
| SmaSNP_252 | 3′ UTR | regulation of macrophage inflammatory protein 1 alpha production (GO:0071640) |
| SmaSNP_253 | Synonymous | regulation of cytokinesis (GO:0032465) |
| SmaSNP_256 | Synonymous | regulation of ribonuclease activity (GO:0060700) |
| SmaSNP_257 | Synonymous | centromere complex assembly (GO:0034508) |
| SmaSNP_259 | 3′ UTR | complement activation, lectin pathway (GO:0001867) |
| SmaSNP_260 | 3′ UTR | amitosis (GO:0051337) |
| SmaSNP_261 | 3′ UTR | protein processing (GO:0016485) |
| SmaSNP_262 | Synonymous | RNA polymerase I transcriptional preinitiation complex assembly (GO:0001188) |
| SmaSNP_263 | Synonymous | membrane protein proteolysis (GO:0033619) |
| SmaSNP_264 | Non synonymous | reproduction (GO:0000003) |
| SmaSNP_265 | 5′ UTR | ribosomal subunit export from nucleus (GO:0000054) |
| SmaSNP_266 | Synonymous | reproduction (GO:0000003) |
| SmaSNP_267 | 3′ UTR | smoothened signaling pathway involved in regulation of cerebellar granule cell precursor cell proliferation (GO:0021938) |
| SmaSNP_270 | 3′ UTR | flotillin complex (GO:0016600) |
| SmaSNP_271 | Synonymous | NAD(P)H dehydrogenase complex assembly (GO:0010275) |
| SmaSNP_273 | 3′ UTR | regulation of ubiquitin-protein ligase activity involved in mitotic cell cycle (GO:0051439) |
| SmaSNP_274 | Unknown | reproduction (GO:0000003) |
| SmaSNP_275 | Synonymous | dolichyl-phosphate beta-d-mannosyltransferase activity (GO:0004582) |
| SmaSNP_276 | Synonymous | transcription from RNA polymerase III type 2 promoter (GO:0001009) |
| SmaSNP_277 | 3′ UTR | T-helper 2 cell activation (GO:0035712) |
| SmaSNP_278 | 3′ UTR | cellular response to methionine (GO:0061431) |
| SmaSNP_279 | Non synonymous | protein import (GO:0017038) |
| SmaSNP_281 | 5′ UTR | regulation of beta 2 integrin biosynthetic process (GO:0045115) |
| SmaSNP_282 | Non synonymous | regulation of binding of sperm to zona pellucida (GO:2000359) |
| SmaSNP_283 | 3′ UTR | cellular macromolecular complex subunit organization (GO:0034621) |
| SmaSNP_284 | Non synonymous | regulation of apoptotic process (GO:0042981) |
| SmaSNP_285 | Synonymous | chromosome passenger complex localization to kinetochore (GO:0072356) |
| SmaSNP_286 | 5′ UTR | brain development (GO:0007420) |
| SmaSNP_287 | Non synonymous | extracellular vesicular exosome assembly (GO:0071971) |
| SmaSNP_288 | Unknown | Not found |
| SmaSNP_289 | Unknown | Not found |
| SmaSNP_290 | Synonymous | regulation of ubiquitin-specific protease activity (GO:2000152) |
| SmaSNP_292 | 3′ UTR | suppression by virus of host TAP complex (GO:0039589) |
| SmaSNP_293 | 3′ UTR | DNA replication preinitiation complex assembly (GO:0071163) |
| SmaSNP_296 | Synonymous | carbon utilization (GO:0015976) |
| SmaSNP_297 | 3′ UTR | membrane protein proteolysis (GO:0033619) |
| SmaSNP_298 | Non synonymous | queuine tRNA-ribosyltransferase activity (GO:0008479) |
| SmaSNP_299 | Synonymous | Not found |
| SmaSNP_304 | Synonymous | reproduction (GO:0000003) |
| SmaSNP_305 | 3′ UTR | protein-DNA complex subunit organization (GO:0071824) |
| SmaSNP_306 | 3′ UTR | behavioral response to stimulus (GO:0007610) |
| SmaSNP_307 | Synonymous | reproduction (GO:0000003) |
| SmaSNP_308 | Non synonymous | Not found |
| SmaSNP_309 | Synonymous | testosterone secretion (GO:0035936) |
| SmaSNP_310 | 3′ UTR | dynein-driven meiotic oscillatory nuclear movement (GO:0030989) |
| SmaSNP_311 | 3′ UTR | suppression by virus of host tapasin activity (GO:0039591) |
| SmaSNP_312 | 5′ UTR | Not found |
| SmaSNP_314 | Synonymous | regulation of macrophage inflammatory protein 1 alpha production (GO:0071640) |
| SmaSNP_315 | 3′ UTR | menopause (GO:0042697) |
| SmaSNP_316 | 3′ UTR | T-helper 1 cell activation (GO:0035711) |
| SmaSNP_317 | 5′ UTR | Not found |
| SNP Name | SNP location/effect | GO term |
| SmaSNP_318 | Unknown | Not found |
| SmaSNP_319 | 5′ UTR | phosphorylation (GO:0016310) |
| SmaSNP_320 | Unknown | Not found |
| SmaSNP_321 | 3′ UTR | Golgi to plasma membrane protein transport (GO:0043001) |
| SmaSNP_322 | 3′ UTR | regulation of ATP citrate synthase activity (GO:2000983) |
| SmaSNP_323 | 3′ UTR | regulation of G-protein beta subunit-mediated signal transduction in response to host (GO:0075162) |
| SmaSNP_324 | Non synonymous | cytokinesis, actomyosin contractile ring assembly (GO:0000915) |
| SmaSNP_325 | Non synonymous | tear secretion (GO:0070075) |
| SmaSNP_326 | 5′ UTR | Not found |
| SmaSNP_328 | Unknown | Not found |
| SmaSNP_329 | 5′ UTR | regulation of type II hypersensitivity (GO:0002892) |
| SmaSNP_330 | Unknown | Not found |
| SmaSNP_332 | 5′ UTR | phosphorylation (GO:0016310) |
| SmaSNP_333 | 5′ UTR | Not found |
| SmaSNP_334 | 5′ UTR | Not found |
| SmaSNP_337 | 3′ UTR | juvenile-hormone esterase activity (GO:0004453) |
| SmaSNP_338 | 3′ UTR | inflammatory response to antigenic stimulus (GO:0002437) |
| SmaSNP_339 | Synonymous | T-helper 1 cell activation (GO:0035711) |
| SmaSNP_340 | Synonymous | regulation of G-protein coupled receptor protein signaling pathway (GO:0008277) |
| SmaSNP_341 | Synonymous | sperm entry (GO:0035037) |
| SmaSNP_342 | Unknown | Not found |
| SmaSNP_343 | Synonymous | collagen metabolic process (GO:0032963) |
| SmaSNP_346 | 3′ UTR | chromatin silencing at silent mating-type cassette (GO:0030466) |
| SmaSNP_347 | 3′ UTR | nucleoside oxidase activity (GO:0033715) |
| SmaSNP_348 | 5′ UTR | retinoic acid receptor signaling pathway (GO:0048384) |
| SmaSNP_349 | 3′ UTR | T-helper 1 cell activation (GO:0035711) |
| Inmune 1 | Hypothalamic pituitary-gonad axis 2 | |
|---|---|---|
| Samples | ||
| Number of individuals | 52 | 30 |
| Origin | Commercial fish farm | Commercial fish farm |
| Data | ||
| Number of reads | 915,782 | 1,191,866 |
| Total megabases (Mb) | 291.04 | 341.20 |
| Average read length | 317.8 | 286.0 |
| Assembly | ||
| Number of contigs | 55,504 | 65,472 |
| Mean length (bp) | 671.3 | 625.9 |
| Average contig coverage | 4.4 | 4.6 |
Supplementary Files
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Vera, M.; Alvarez-Dios, J.-A.; Fernandez, C.; Bouza, C.; Vilas, R.; Martinez, P. Development and Validation of Single Nucleotide Polymorphisms (SNPs) Markers from Two Transcriptome 454-Runs of Turbot (Scophthalmus maximus) Using High-Throughput Genotyping. Int. J. Mol. Sci. 2013, 14, 5694-5711. https://doi.org/10.3390/ijms14035694
Vera M, Alvarez-Dios J-A, Fernandez C, Bouza C, Vilas R, Martinez P. Development and Validation of Single Nucleotide Polymorphisms (SNPs) Markers from Two Transcriptome 454-Runs of Turbot (Scophthalmus maximus) Using High-Throughput Genotyping. International Journal of Molecular Sciences. 2013; 14(3):5694-5711. https://doi.org/10.3390/ijms14035694
Chicago/Turabian StyleVera, Manuel, Jose-Antonio Alvarez-Dios, Carlos Fernandez, Carmen Bouza, Roman Vilas, and Paulino Martinez. 2013. "Development and Validation of Single Nucleotide Polymorphisms (SNPs) Markers from Two Transcriptome 454-Runs of Turbot (Scophthalmus maximus) Using High-Throughput Genotyping" International Journal of Molecular Sciences 14, no. 3: 5694-5711. https://doi.org/10.3390/ijms14035694