Synthesis of Thieno[2,3-b]thiophene Containing Bis-Heterocycles-Novel Pharmacophores

 ,
,  ,
,
Abstract
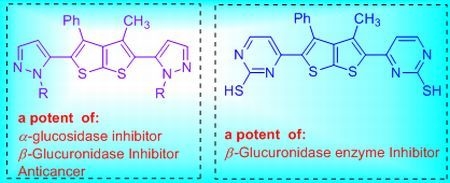
:
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Biological Activity Evaluation
3. Experimental Section
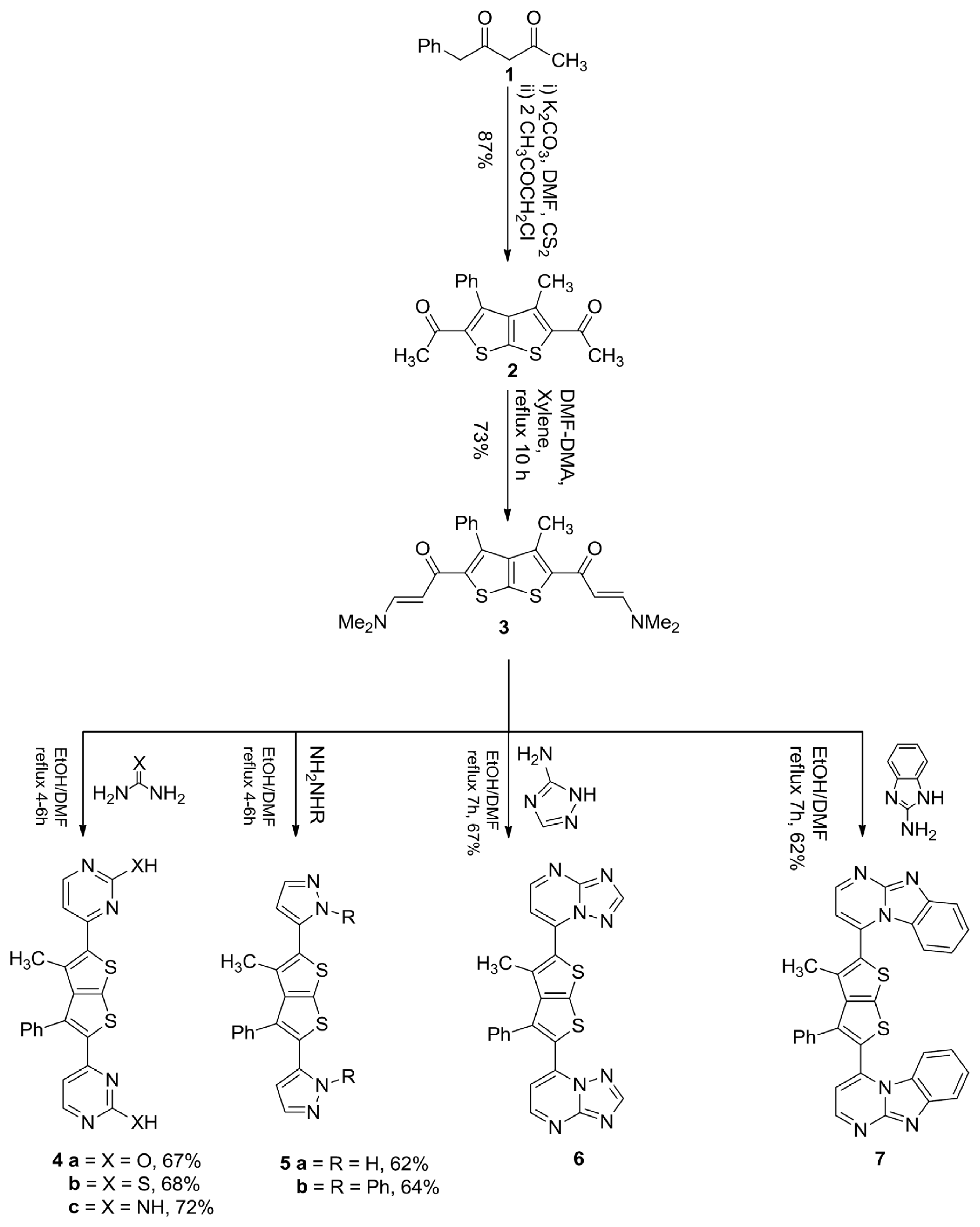
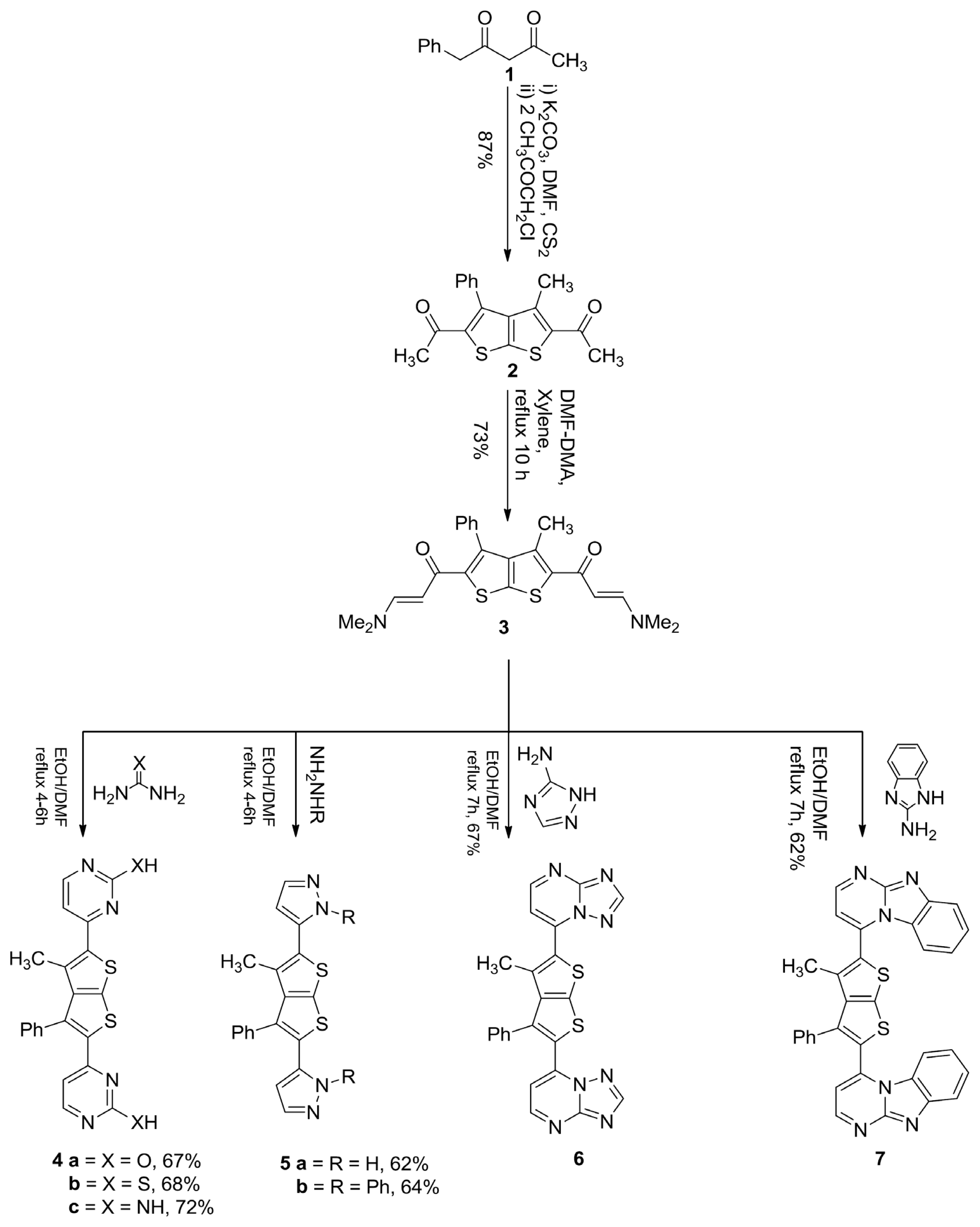
3.1. 1,1′-(3-Methyl-4-Phenylthieno[2,3-b]Thiophene-2,5-Diyl)Diethanone (2)
3.2. 1,1′-(3-Methyl-4-Phenylthieno[2,3-b]Thiophene-2,5-Diyl)Bis(3-(Dimethylamino)Prop-2-En-1-One) (3)
3.3. General Procedure for the Synthesis of Compounds 4a–c
3.3.1. 4,4′-(3-Methyl-4-Phenylthieno[2,3-b]Thiophene-2,5-Diyl)Dipyrimidin-2-Ol (4a)
3.3.2. 4,4′-(3-Methyl-4-Phenylthieno[2,3-b]Thiophene-2,5-Diyl)Dipyrimidin-2-Thiol (4b)
3.3.3. 4,4′-(3-Methyl-4-Phenylthieno[2,3-b]Thiophene-2,5-Diyl)Dipyrimidin-2-Amine (4c)
3.4. General Procedure for the Synthesis of Compounds 5a,b
3.4.1. 3,3′-(3-Methyl-4-Phenylthieno[2,3-b]Thiophene-2,5-Diyl)Bis(1H-Pyrazole) (5a)
3.4.2. 3,3′-(3-Methyl-4-Phenylthieno[2,3-b]Thiophene-2,5-Diyl)Bis(1-Phenyl-1H-Pyrazole) (5b)
3.5. General Procedure for the Synthesis of Compounds 6 and 7
3.5.1. 7,7′-(3-Methyl-4-Phenylthieno[2,3-b]Thiophene-2,5-Diyl)Di-[1,2,4]Triazolo[1,5-a]Pyrimidine (6)
3.5.2. 2,2′-(3-Methyl-4-Phenylthieno[2,3-b]Thiophene-2,5-Diyl)Bis(Benzo[4,5]Imidazo[1,2-a] Pyrimidine) (7)
3.6. Biological Activities
3.6.1. Anticancer Activity
3.6.2. In Vitro Antioxidant Activity
3.6.3. In Vitro β-Glucuronidase Inhibition Assay
3.6.4. In Vitro α-Glucosidase Inhibition Assay
3.6.5. Statistical Analysis
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Heeney, M.; Bailey, C.; Genevicius, K.; Shkunov, M.; Sparrowe, D.; Tierney, S.; Mculloch, I. Stable polythiophene semiconductors incorporating thieno[2,3-b]thiophene. J. Am. Chem. Soc 2005, 127, 1078–1079. [Google Scholar]
- Mashraqui, S.H.; Sangvikar, Y.S.; Meetsma, A. Synthesis and structures of thieno[2,3-b]thiophene incorporated [3,3]dithiacyclophanes. Enhanced first hyperpolarizability in an unsymmetrically polarized cyclophane. Tetrahedron Lett 2006, 47, 5599–5602. [Google Scholar]
- Mashraqui, S.H.; Sangvikar, Y.S.; Ashraf, M.; Kumar, S.; Daub, E. Dipyridyl/Pyridinium thieno[2,3-b]thiophenes as new atropisomeric systems. Synthesis, conformat-ional analysis and energy minimization. Tetrahedron 2005, 61, 3507–3513. [Google Scholar]
- Leriche, P.R.J.; Turbiez, M.M.; Monroche, V.; Allain, M.; Sauvage, F.X.; Roncali, J.; Frere, P.; Skabara, P.J. Linearly extended tetrathiafulvalene analogues with fused thiophene units as π conjugated spacers. J. Mater. Chem 2003, 13, 1324–1327. [Google Scholar]
- Lee, B.; Seshadri, V.; Palko, H.; Sotzing, G.A. Ring-Sulfonated poly(thienothiophene). J. Adv. Mater 2005, 17, 1792–1795. [Google Scholar]
- Lim, E.; Jung, B.J.; Lee, J.; Shim, H.K.; Lee, J.I.; Yang, Y.S.; Do, L.M. Thin-Film morphologies and solution-processable field-effect transistor behavior of a fluorine-thieno[3,2-b]thiophene-based conjugated copolymer. Macromolecules 2005, 38, 4531–4535. [Google Scholar]
- Kim, H.S.; Kim, Y.H.; Kim, T.H.; Noh, Y.Y.; Pyo, S.; Yi, M.H.; Kim, D.Y.; Kwon, S.K. Synthesis and studies on 2-hexylthieno[3,2-b]thiophene end-capped oligomers for OTFTs. Chem. Mater 2007, 19, 3561–3567. [Google Scholar]
- Jarak, I.; Kralj, M.; Piantanida, I.; Suman, L.; Zinic, M.; Pavelic, K.; Karminski-Zamola, G. Novel cyano- and amidino-substituted derivatives of thieno[2,3-b]- and thien-o[3,2-b]thiophene-2-carboxanilides and thieno[30,20:4,5]thieno- and thieno[20,30:4,5]thieno[2,3-c]quinolones: Synthesis, photochemical synthesis, DNA binding, and antitumor evaluation. Bioorg. Med. Chem 2006, 14, 2859–2868. [Google Scholar]
- Peters, D.; Hornfeldt, A.B.; Gronowitz, S. Synthesis of various 5-substituted uracils. J. Heterocycl. Chem 1990, 27, 2165–2173. [Google Scholar]
- Kukolja, S.; Draheim, S.E.; Graves, B.J.; Hunden, D.C.; Pfeil, J.L.; Cooper, R.D.G.; Ott, J.L.; Couter, F.T. Orally absorbable cephalosporin antibiotics. 2. Structure-Activity studies of bicyclic glycine derivatives of 7-aminodeacetoxycephalosporanic acid. J. Med. Chem 1985, 28, 1896–1903. [Google Scholar]
- Prugh, J.D.; Hartman, G.D.; Mallorga, P.J.; McKeever, B.M.; Michelson, S.R.; Murcko, M.A.; Schwam, H.; Smith, R.L.; Sondey, J.M.; Springer, J.P.; et al. New isomeric classes of topically active ocular hypotensive carbonic anhydrase inhibitors: 5-Substituted thieno[2,3-b]thiophene-2-sulfonamides and 5-substituted thieno[3,2-b]thiophene-2-sulfonamides. J. Med. Chem 1991, 34, 1805–1818. [Google Scholar]
- Egbertson, M.S.; Cook, J.J.; Bednar, B.; Prugh, J.D.; Bednar, R.A.; Gaul, S.L.; Gould, R.J.; Hartman, G.D.; Homnick, C.F.; Holahan, M.A.; et al. Centrally constrained thienothiophene α-sulfonamides are potent, long acting in vivo inhibitors of platelet aggregation. J. Med. Chem 1999, 42, 2409–2421. [Google Scholar]
- Allard, S.; Forster, M.; Souharce, B.; Thiem, H.; Scherf, U. Organic semiconductors for solution-processable field-effect transistors (OFETs). Angew. Chem. Int. Ed 2008, 47, 4070–4098. [Google Scholar]
- Wu, Z.-Q.; Ono, R.J.; Chen, Z.; Bielawski, C.W. Synthesis of poly(3-alkylthiophene)-block-poly( arylisocyanide): Two sequential, mechanistically distinct polymerizations using a single catalyst. J. Am. Chem. Soc 2010, 132, 14000–14001. [Google Scholar]
- Li, Z.; Ono, R.J.; Wu, Z.-Q.; Bielawski, C.W. Synthesis and self-assembly of poly(3-hexylthiophene)-block-poly(acrylic acid). Chem. Commun 2011, 47, 197–199. [Google Scholar]
- Wu, Z.-Q.; Ono, R.J.; Chen, Z.; Li, Z.; Bielawski, C.W. Polythiophene-block-poly(γ-benzyl l-glutamate): Synthesis and study of a new rod-rod block copolymer. Polym. Chem 2011, 2, 300–302. [Google Scholar]
- Sirringhaus, H.; Brown, P.J.; Friend, R.H.; Nielsen, M.M.; Bechgaard, K.; Langeveld-Voss, B.M.W.; Spiering, A.J.H.; Janssen, R.A.J.; Meijer, E.W.; Herwig, P.; et al. Two-Dimensional charge transport in self-organized, high-mobility conjugated polymers. Nature 1999, 401, 685–688. [Google Scholar]
- Schuett, W.; Raoort, H.S. The paralytic shellfish poison. Degradation to a pyrrolopyrimidine. J. Am. Chem. Soc 1962, 84, 2266–2267. [Google Scholar]
- Ho, Y.W. 5-(1-Pyrrolyl)-2-phenylthieno[2,3-d]pyrimidine as building block in heterocyclic synthesis: Novel synthesis of some pyrazoles, pyrimidines, imidazo[1,2-a]pyrimidines, pyrazolo[1,5-a]pyrimidines, pyrido-(pyrimido)pyrazolo[1,5-a]pyrimidines, 1,2,4-triazolo[1,5-a]pyrimidine and a 1,2,3,4-tetrazolo[1,5-a]pyrimidine derivative. J. Chin. Chem. Soc 2007, 54, 1075–1085. [Google Scholar]
- Alexander, S.K.; Smith, L. Novel one pot synthesis of polysubstituted pyrazolo[1,5-a] and imidazo[1,2-a]pyrimidines. Tetrahedron Lett 2006, 47, 2611–2614. [Google Scholar]
- Riyadh, S.M. Enaminones as building blocks for the synthesis of substituted pyrazoles with antitumor and antimicrobial activities. Molecules 2011, 16, 1834–1853. [Google Scholar]
- Mabkhot, Y.N.; Al-Majid, A.M.; Barakat, A.; Alshahrani, S.; Yamin, S. 1,1′-(3-Methyl-4-phenylthieno[2,3-b]thiophene-2,5-diyl)diethanone as a building block in heterocyclic synthesis. Novel synthesis of some pyrazole and pyrimidine derivatives. Molecules 2011, 16, 6502–6511. [Google Scholar]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Meth 1983, 65, 55–63. [Google Scholar]
- Khan, K.M.; Shah, Z.; Ahmad, V.U.; Khan, M.; Taha, M.; Rahim, F.; Ali, S.; Ambreen, N.; Perveen, S.; Choudhary, M.I.; Voelter, W. 2,4,6-Trichlorophenylhydrazine Schiff bases as DPPH radical and super oxide anion scavengers. Med. Chem 2012, 8, 452–461. [Google Scholar]
- Khan, K.M.; Rahim, F.; Halim, S.A.; Taha, M.; Khan, M.; Perveen, S.; Zaheer-ul-Haq; Mesaik, M.A.; Choudhary, M.I. Synthesis of novel inhibitors of β-glucuronidase based on benzothiazole skeleton and study of their binding affinity by molecular docking. Bioorg. Med. Chem. 2011, 19, 4286–4294. [Google Scholar]
- Choudhary, M.I.; Adhikari, A.; Rasheed, S.; Marasini, B.P.; Hussain, N.; Ahmad Kaleem, W.; Atta-ur-Rahman. Cyclopeptide alkaloids of Ziziphus oxyphylla Edgw as novel inhibitors of α-glucosidase enzyme and protein glycation. Phytochem. Lett. 2011, 4, 404–406. [Google Scholar]

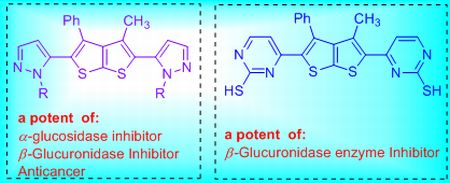{kind=link}
{kind=link}
| Compounds | IC50 ± SEM [μM] | |||
|---|---|---|---|---|
| Anticancer Activity (PC-3 Cell line) | DPPH Radical Scavenging Assay | β-Glucuronidase Inhibition | α-Glucosidase Inhibition | |
| 3 | >30 | NA | NA | NA |
| 4a | >30 | - | - | - |
| 4b | >30 | NA | 1.3 ± 0.2 | - |
| 4c | - | - | - | - |
| 5a | 22.5 ± 0.1 | NA | 2.3 ± 0.4 | 22.0 ± 0.3 |
| 5b | >30 | 165 ± 2.8 | 8.7 ± 0.1 | 58.4 ± 1.2 |
| 6 | >30 | - | - | - |
| 7 | >30 | - | NA | NA |
| Std. | Doxorubicin | N-Acetylcysteine | d-Saccharic acid | Acarbose |
| 0.91 ± 0.1 | 106 ± 1.1 | 1,4-lactone 45.8 ± 2.2 | 841 ± 1.7 | |
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mabkhot, Y.N.; Barakat, A.; Al-Majid, A.M.; Choudhary, M.I. Synthesis of Thieno[2,3-b]thiophene Containing Bis-Heterocycles-Novel Pharmacophores. Int. J. Mol. Sci. 2013, 14, 5712-5722. https://doi.org/10.3390/ijms14035712
Mabkhot YN, Barakat A, Al-Majid AM, Choudhary MI. Synthesis of Thieno[2,3-b]thiophene Containing Bis-Heterocycles-Novel Pharmacophores. International Journal of Molecular Sciences. 2013; 14(3):5712-5722. https://doi.org/10.3390/ijms14035712
Chicago/Turabian StyleMabkhot, Yahia Nasser, Assem Barakat, Abdullah Mohammed Al-Majid, and Muhammad Iqbal Choudhary. 2013. "Synthesis of Thieno[2,3-b]thiophene Containing Bis-Heterocycles-Novel Pharmacophores" International Journal of Molecular Sciences 14, no. 3: 5712-5722. https://doi.org/10.3390/ijms14035712