Reaction of Stabilized Criegee Intermediates from Ozonolysis of Limonene with Water: Ab Initio and DFT Study

Abstract
:
1. Introduction
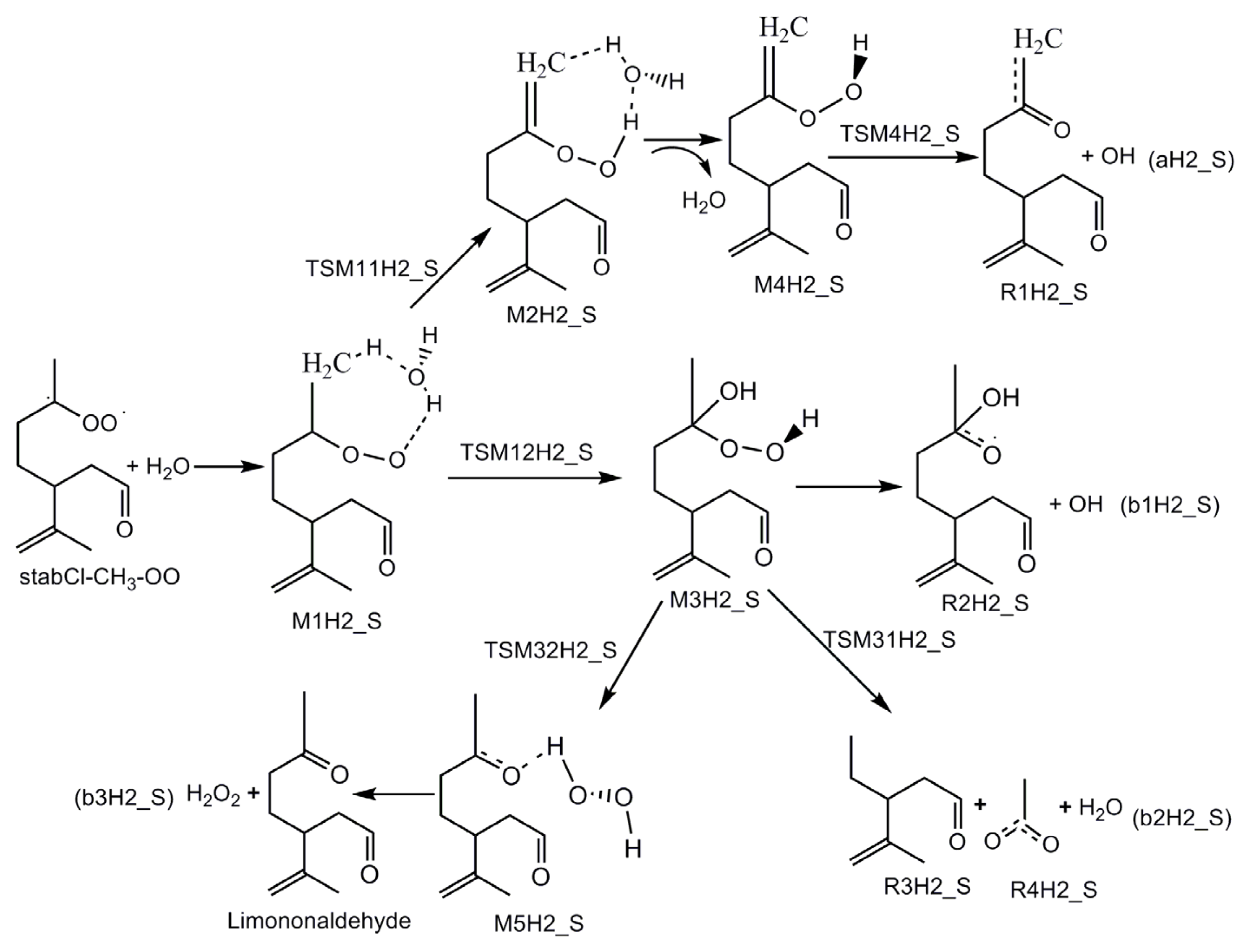
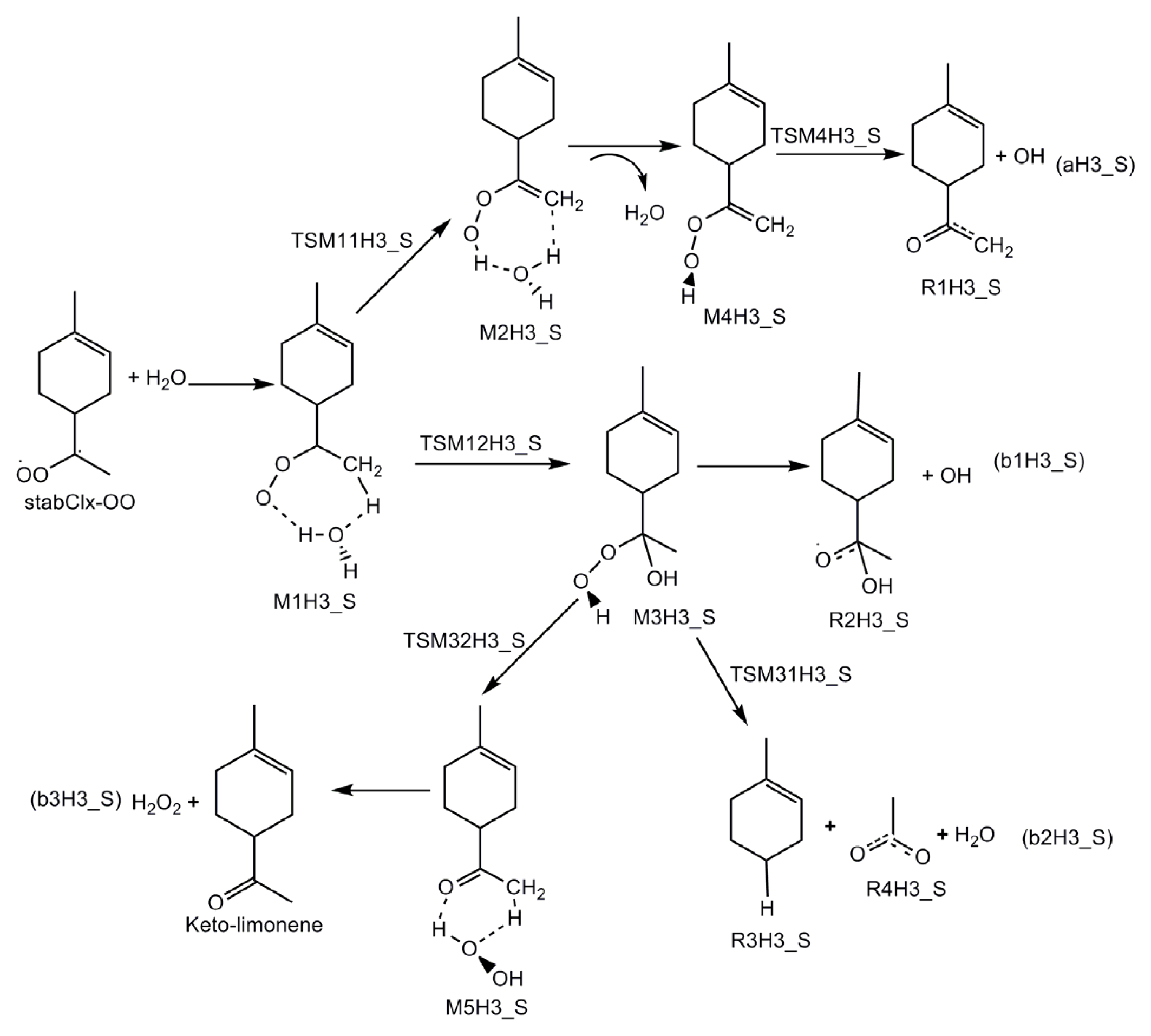
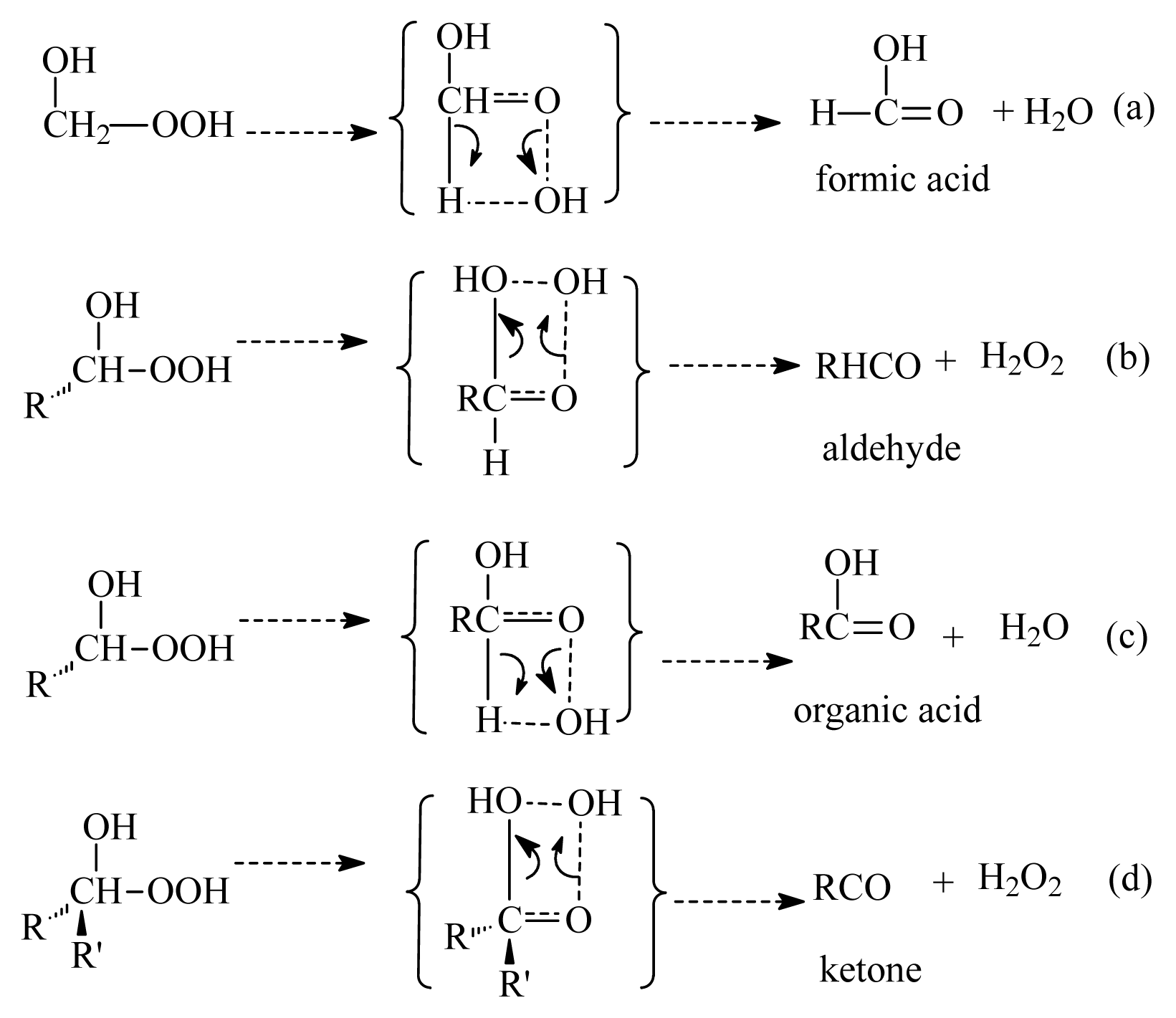
- Formation of OH radicals with water-catalyzed H migration. The water molecule assists the H migration toward the terminal oxygen of the COO moiety, which could lead to the formation of OH radicals.
- Formation of α-hydroxy hydroperoxide. Cycloaddition of H2O to stabilized Criegee intermediate, where the oxygen of water is linked to the carbon atom of the stabilized Criegee intermediate while a hydrogen atom of water is transferred to the terminal oxygen of the COO unit.
2. Computational Details
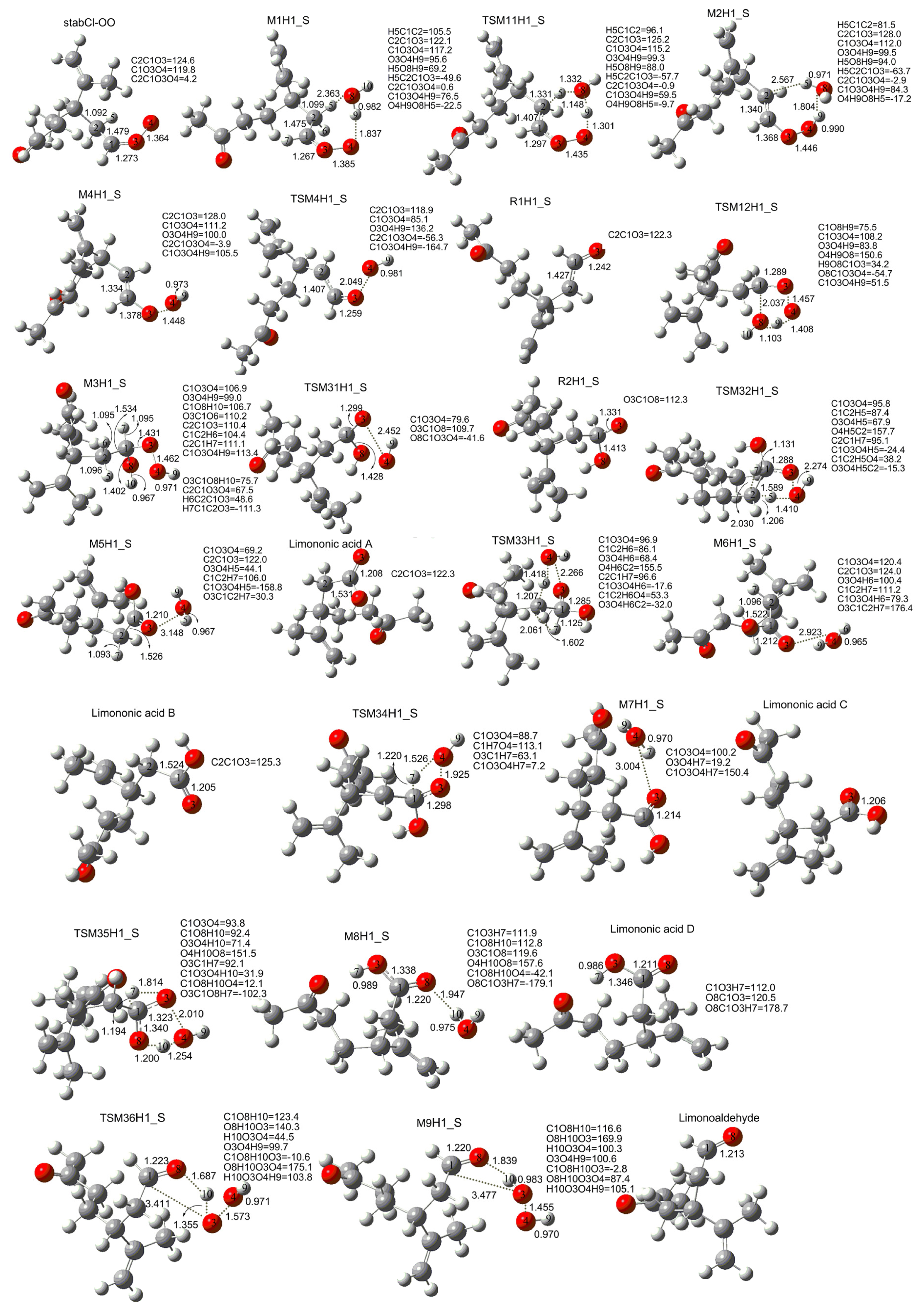
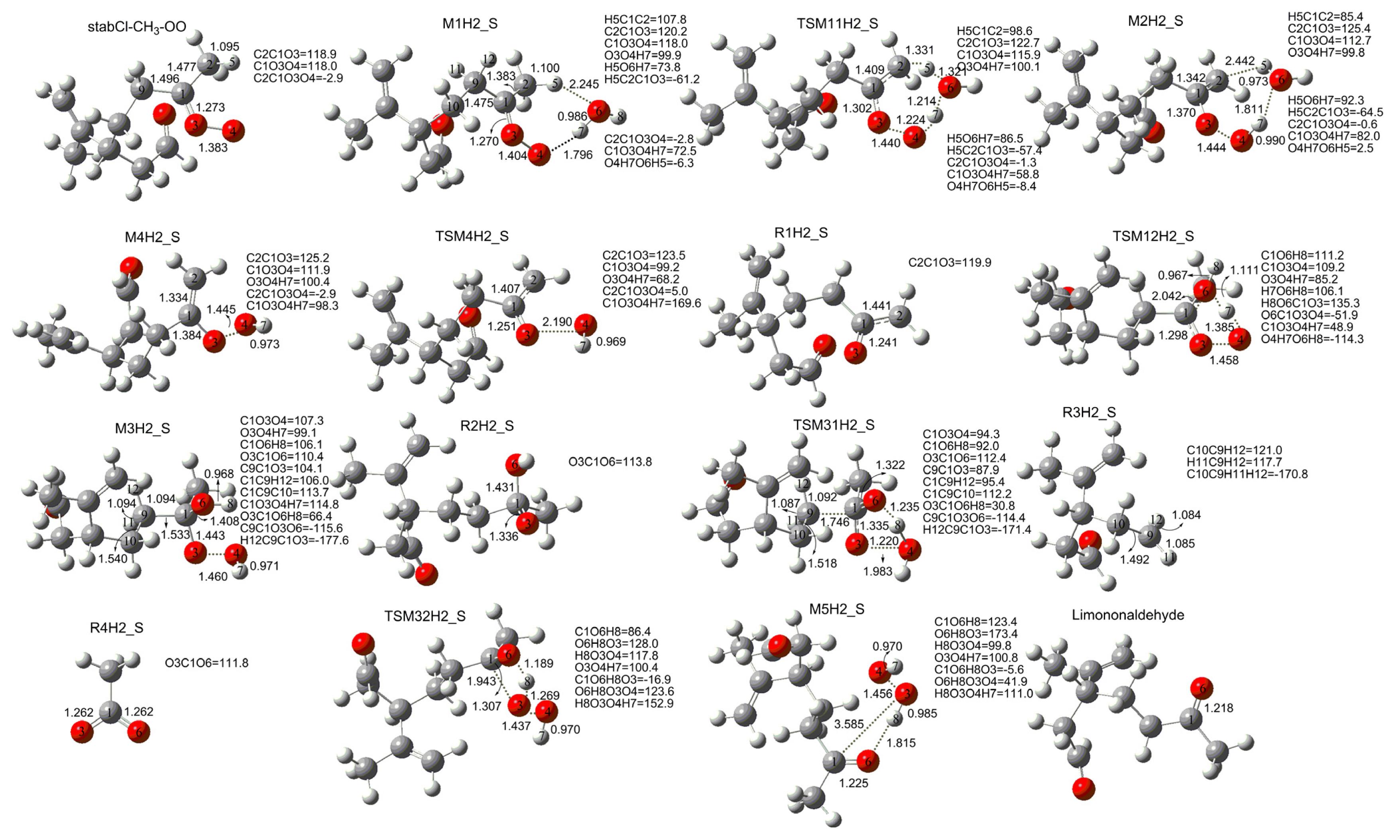
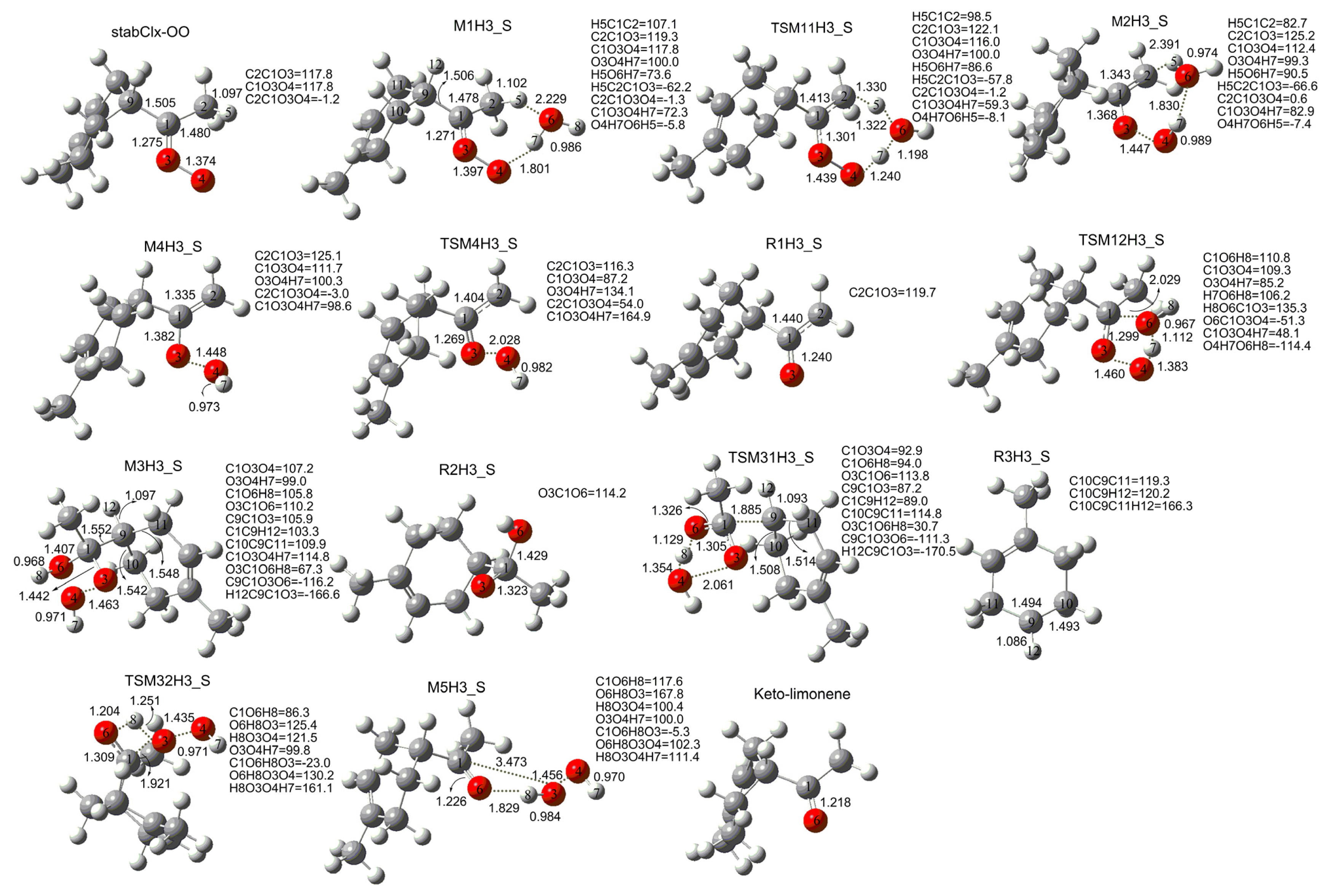
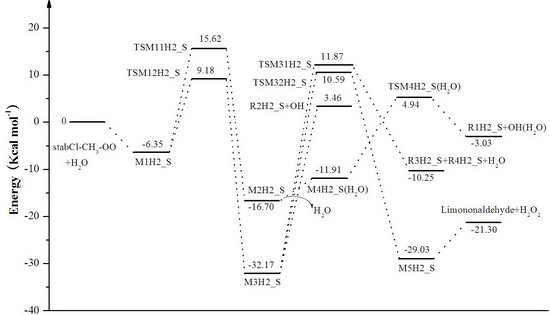
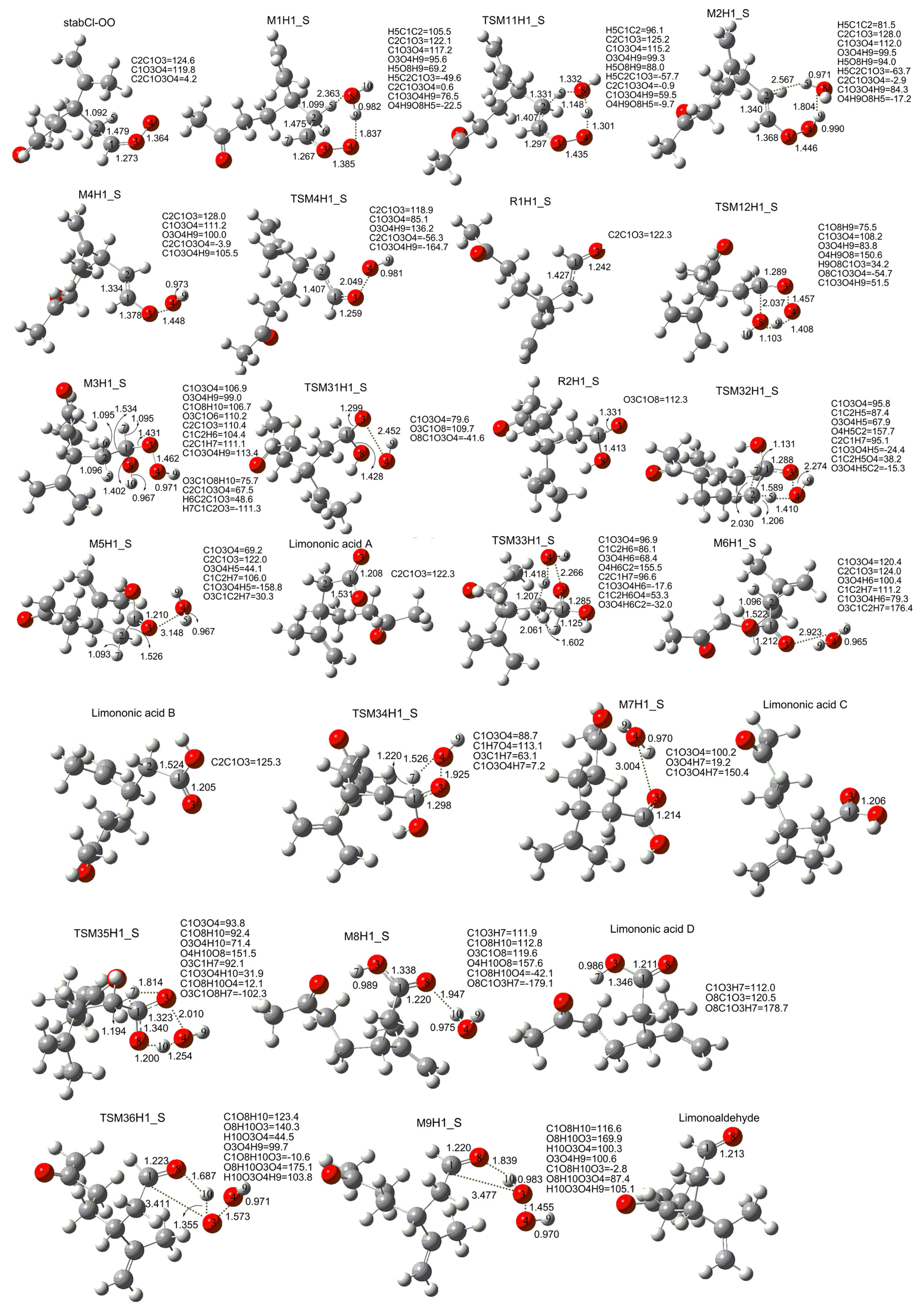
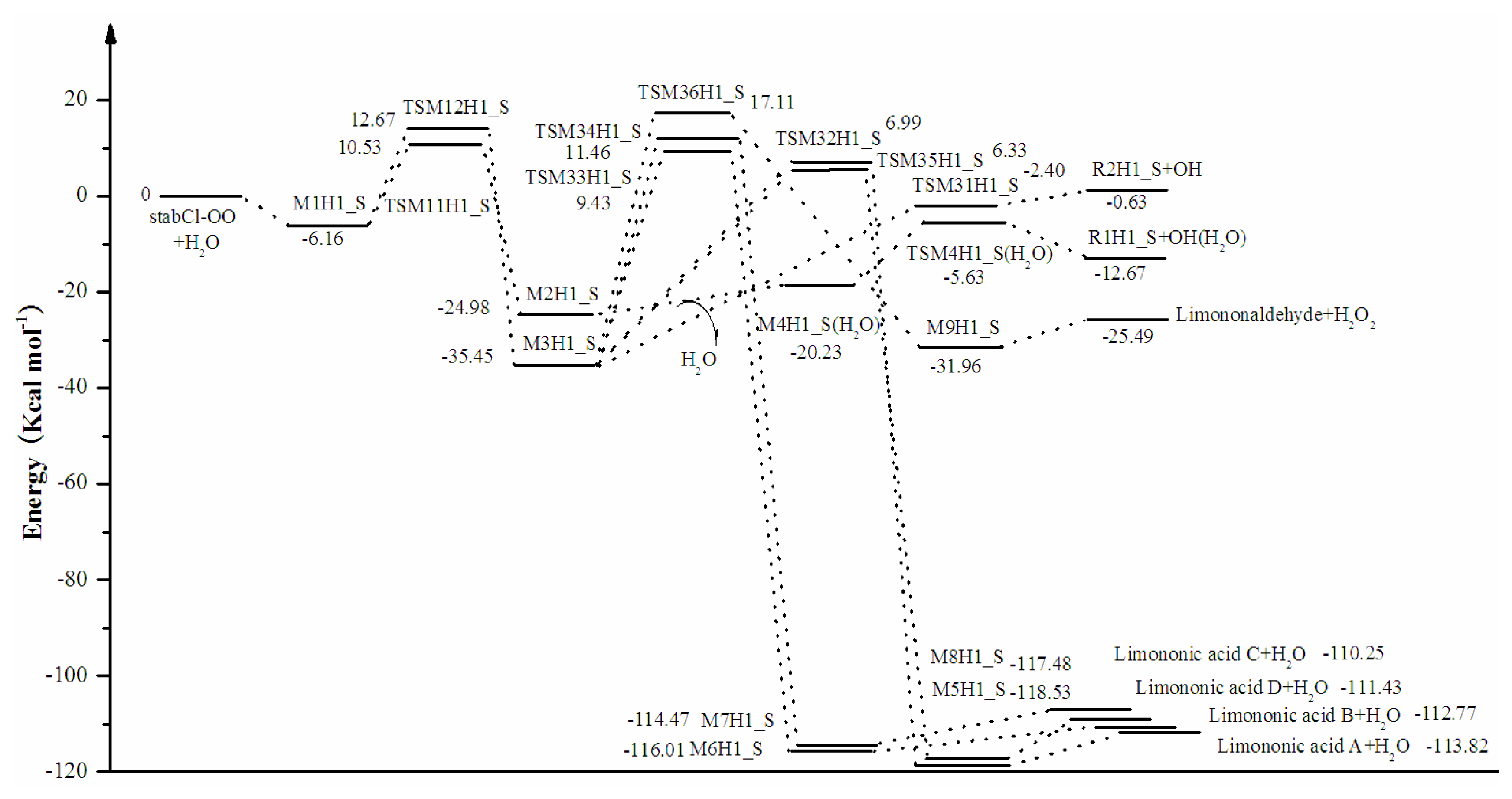
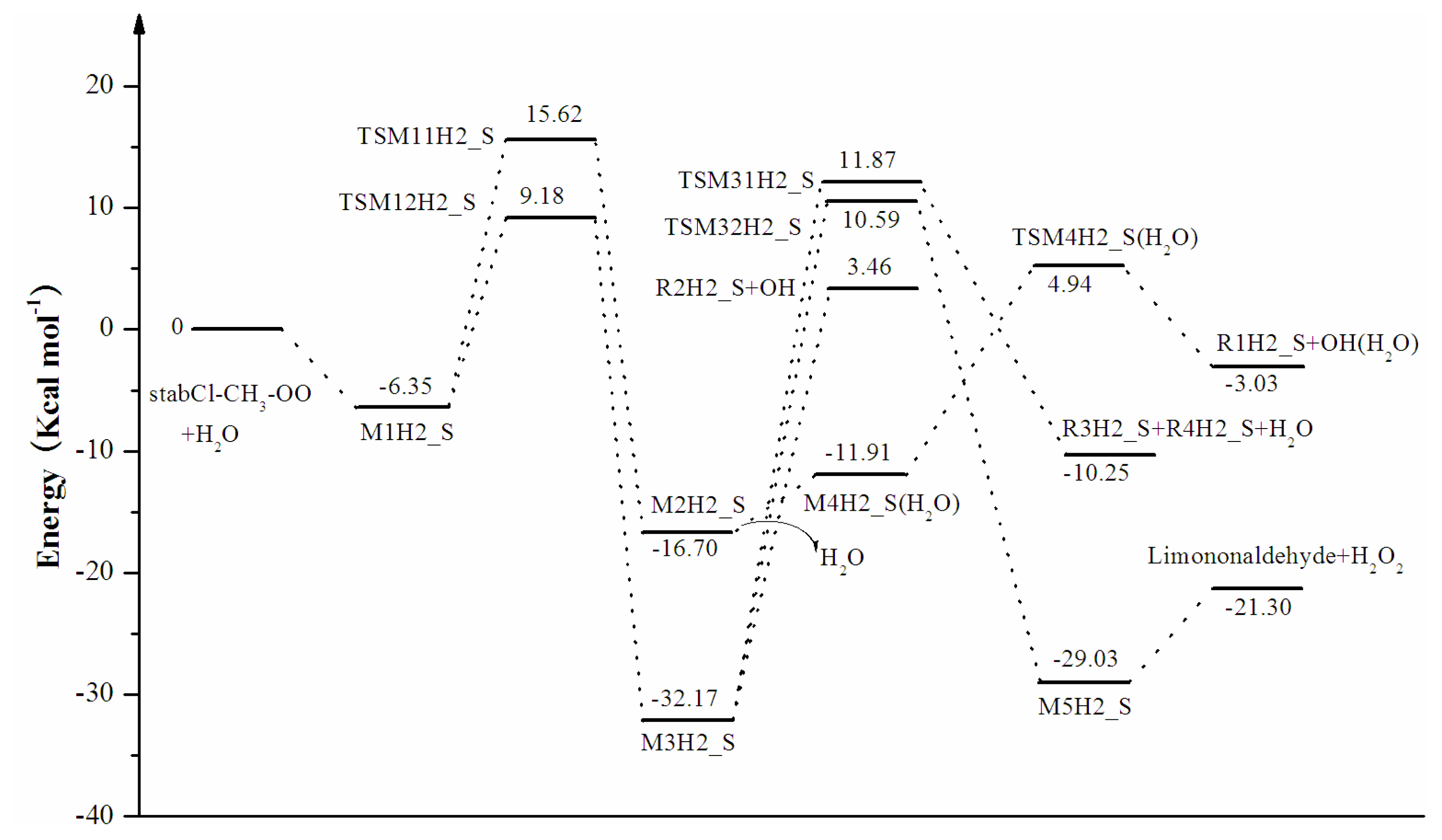
3. Results and Discussion
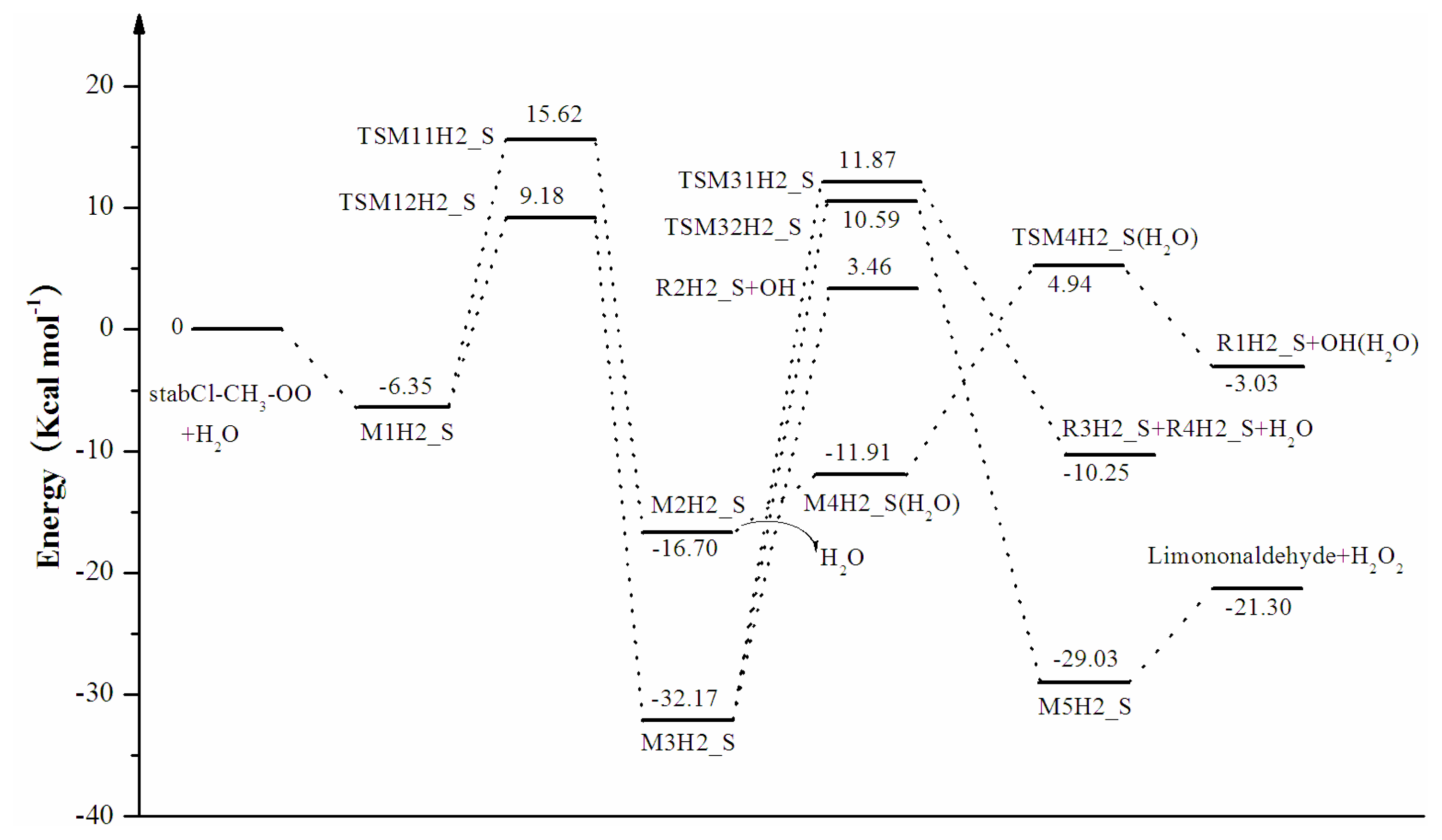
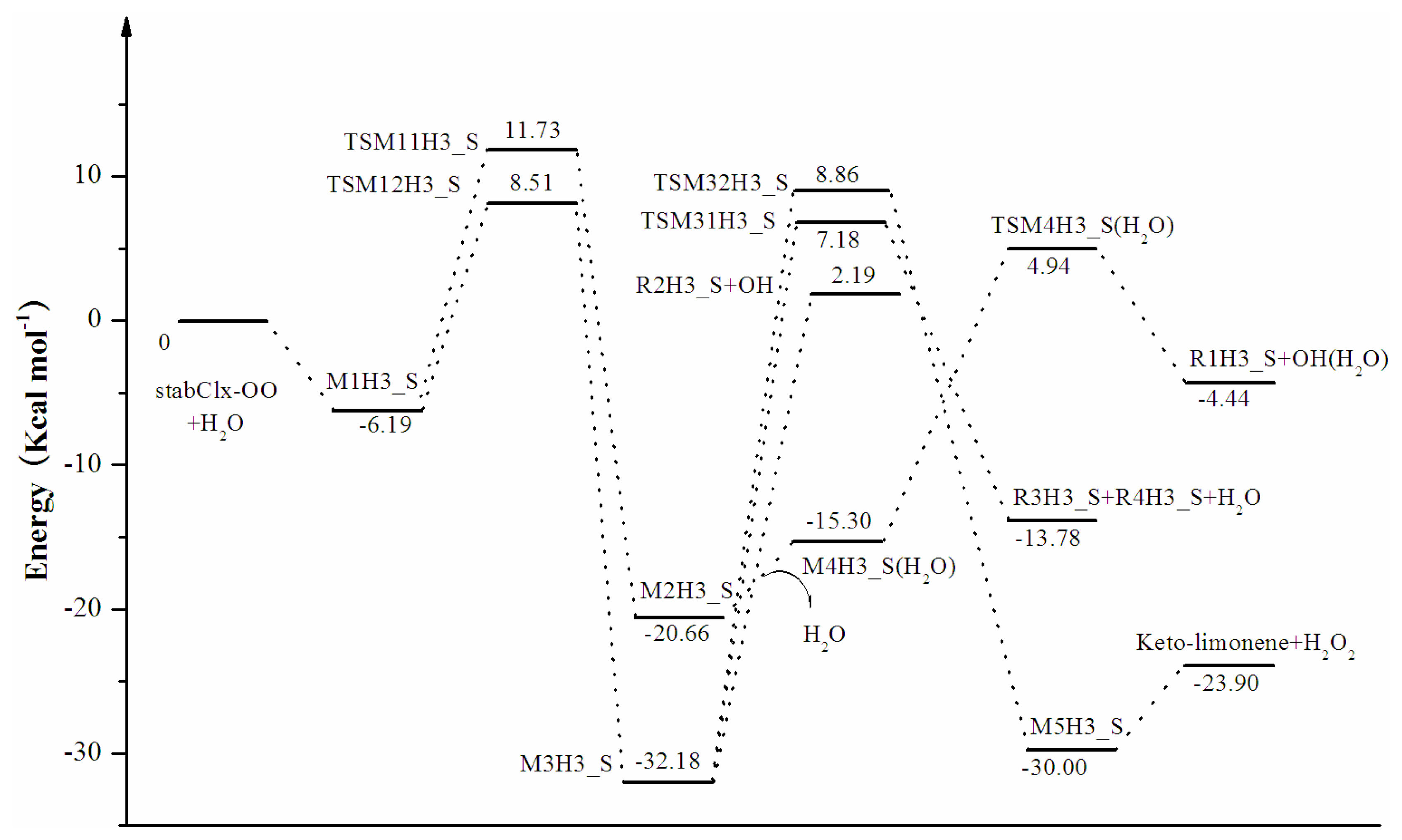
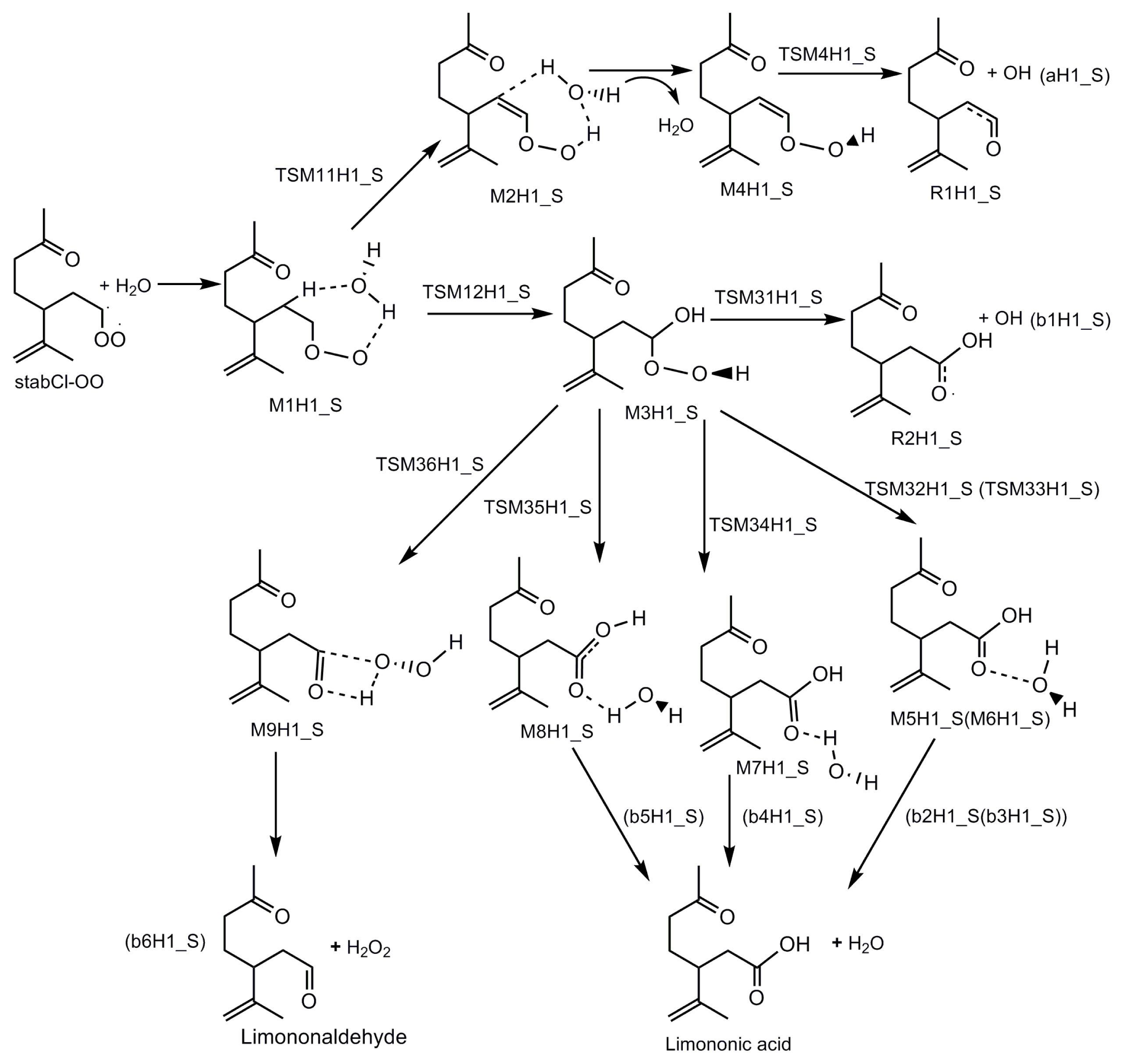
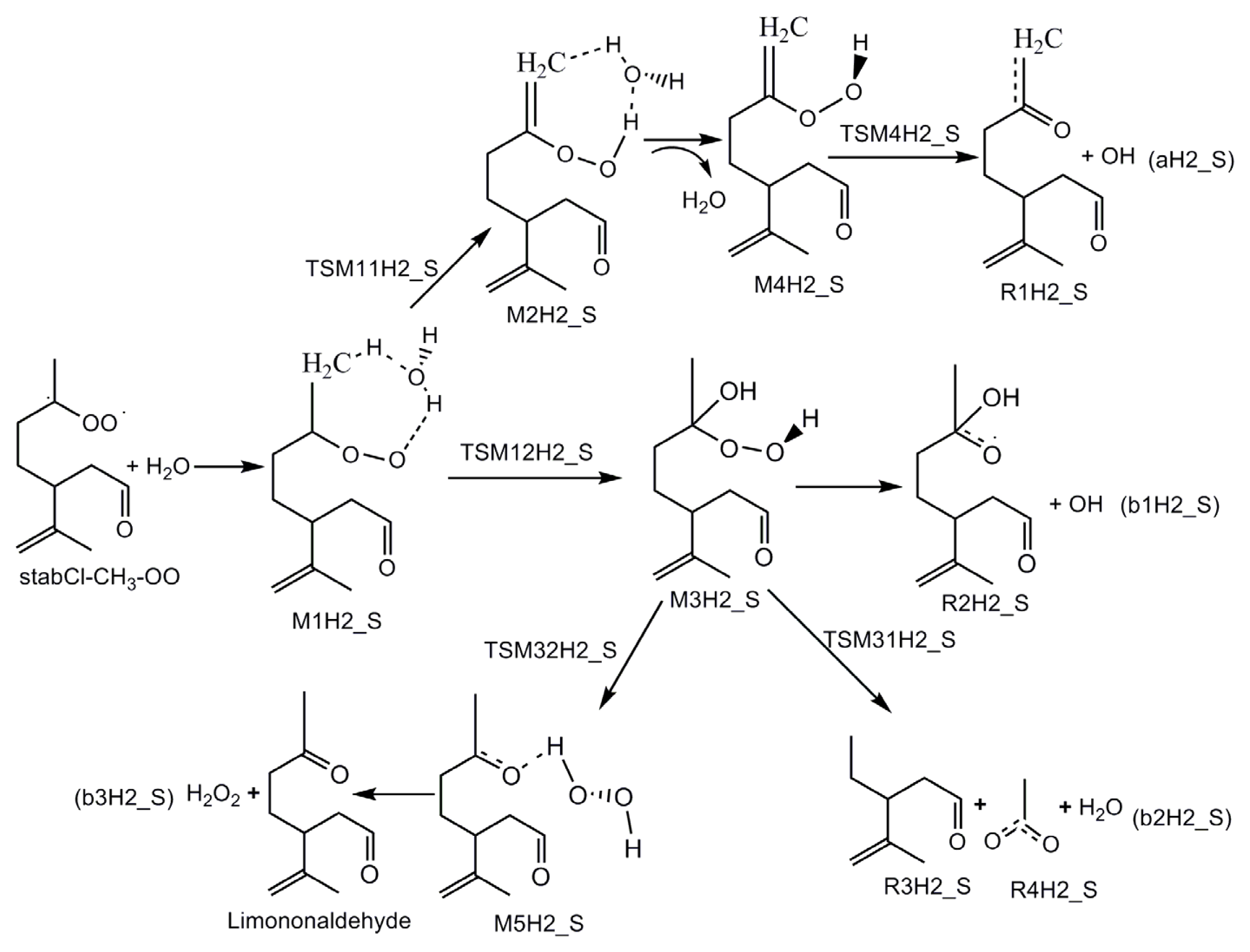
3.1. Reaction Mechanism
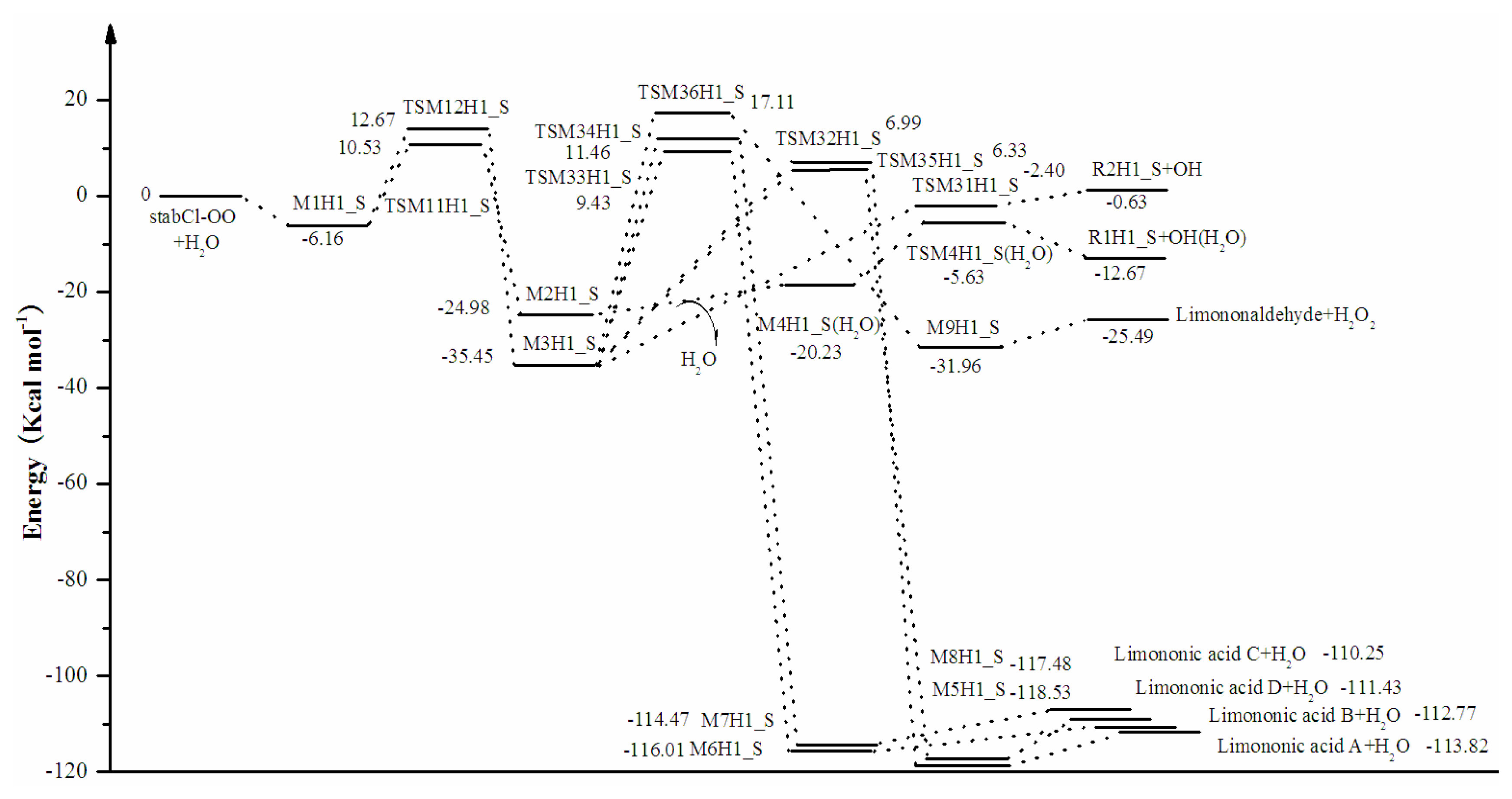
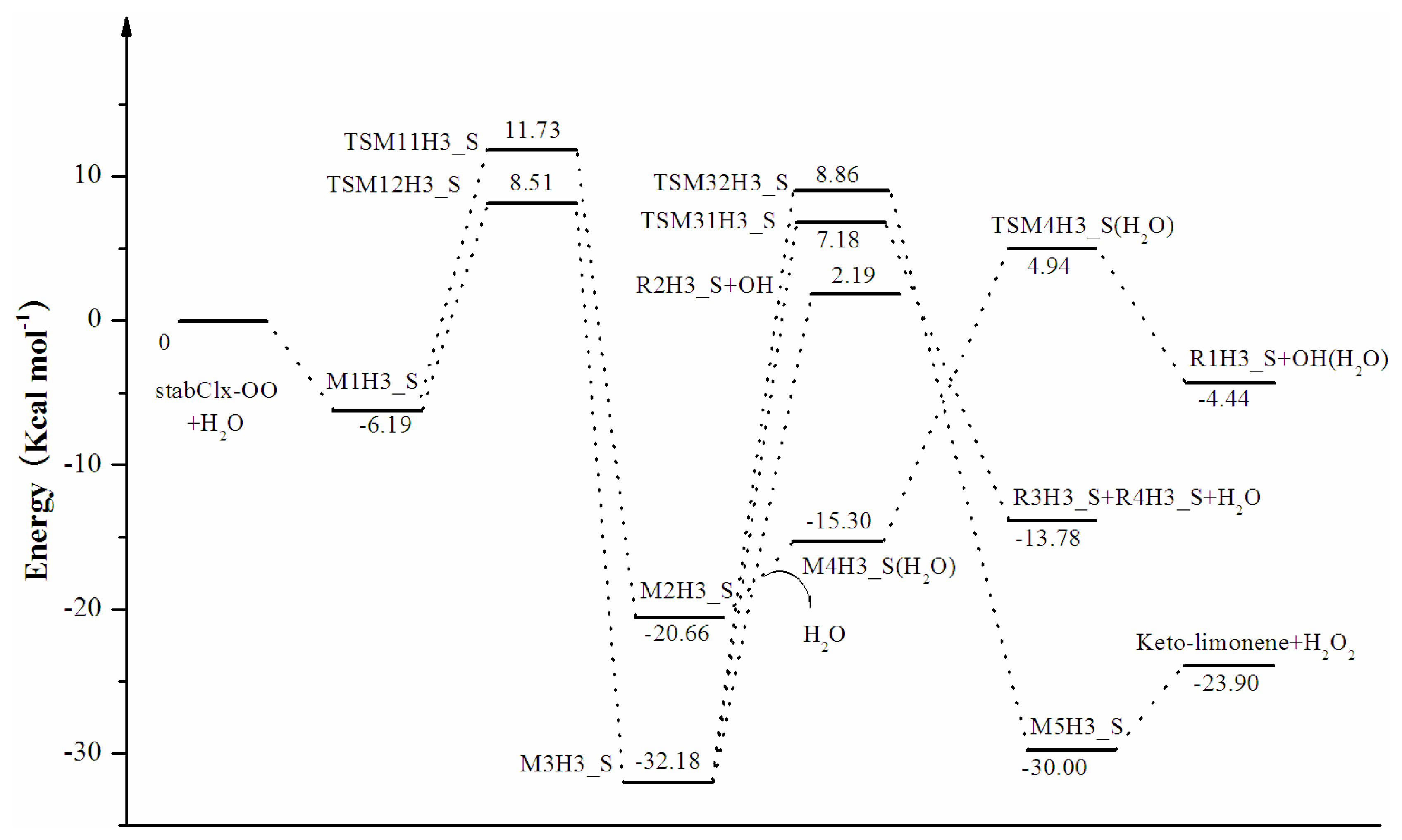
3.2. Thermochemical Analysis
4. Conclusions
- The reaction between the stabilized Criegee intermediates (stabCI-OO, stabCI-CH3-OO and stabCIx-OO) and H2O is the three-step reaction. At the first stage, the formation of a hydrogen-bonded complex occurs. At the second stage, the reaction can proceed via the following two reaction pathways: (1) formation of OH radicals with water-catalyzed H migration; and (2) formation of α-hydroxy hydroperoxide. The formation of α-hydroxy hydroperoxide (reaction (2)), the reaction of stabCI-OO and H2O occurs via six different degradation pathways, while the reaction of stabCI-CH3-OO and stabCIx-OO with H2O occurs via three different reaction pathways.
- The formation of OH radicals with water-catalyzed H migration path (aH1_S, aH2_S and aH3_S) and formation of α-hydroxy hydroperoxide (reaction pathways b1H1_S, b1H2_S and b1H3_S) may be considered as possible sources of OH radicals in the Earth’s atmosphere. The formation of α-hydroxy hydroperoxide (reaction pathways b6H1_S, b3H2_S and b3H3_S) can be considered as possible sources of atmospheric H2O2.
- The CCSD(T)/6-31G(d) + CF activation energies are in the range of 14.70–21.98 kcal mol−1 with respect to corresponding hydrogen-bond complexes (M1H1_S, M1H2_S, M1H3_S) between stabilized Criegee intermediates and H2O. The formation of α-hydroxy hydroperoxide (reaction (2)) for the reaction of stabCIx-OO and H2O is the most favorable pathway with the activation energy of 14.70 kcal mol−1.
- For the reaction of stabCI-OO and H2O, with the reaction between our optimized stabCI-OO (syn stabCI-OO) and water, the formation of OH radicals with water-catalyzed H migration path or reaction (1) is more favorable than the formation of α-hydroxy hydroperoxide or reaction (2). This conclusion is opposite to that obtained in the previous theoretical study of the reaction between isoprene stabilized Criegee intermediates and H2O by Anglada. For the reaction of stabCI-CH3-OO and stabCIx-OO with H2O, the formation of α-hydroxy hydroperoxide (reaction (2)) is more favorable than the formation of OH radicals with water-catalyzed H migration path or reaction (1). This conclusion is consistent with results of the previous theoretical study of the reaction between isoprene stabilized Criegee intermediates and H2O by Anglada.
Acknowledgments
Conflict of Interest
References
- Hatakeyama, S.; Akimoto, H. Reactions of Criegee intermediates in the gas phase. Res. Chem. Intermed 1994, 20, 503–524. [Google Scholar]
- Ryzhkov, A.B.; Ariya, P.A. A theoretical study of the reactions of carbonyl oxide with water in atmosphere: The role of water dimer. Chem. Phys. Lett 2003, 367, 423–429. [Google Scholar]
- Criegee, R. Mechanism of ozonolysis. Angew. Chem. Int. Ed. Engl 1975, 14, 745–751. [Google Scholar]
- Neeb, P.; Horie, O.; Moortgat, G.K. The nature of the transitory product in the gas-phase ozonolysis of ethane. Chem. Phys. Lett 1995, 246, 150–156. [Google Scholar]
- Hatakeyama, S.; Bandow, H.; Okuda, M.; Akimoto, H. Reactions of peroxymethylene and methylene (1A1) with water in the gas phase. J. Phys. Chem 1981, 85, 2249–2254. [Google Scholar]
- Crehuet, R.; Anglada, J.M.; Bofill, J.M. Tropospheric formation of hydroxymethyl hydroperoxide, formic acid, H2O2, and OH from carbonyl oxide in the presence of water vapor: A theoretical study of the reaction mechanism. Chem. Eur. J 2001, 7, 2227–2235. [Google Scholar]
- Jenkin, M.E.; Saunders, S.M.; Pilling, M.J. The tropospheric degradation of volatile organic compounds: A protocol for mechanism development. Atmos. Environ 1997, 31, 81–104. [Google Scholar]
- Gäb, S.; Hellpointner, E.; Turner, W.V.; Korte, F. Hydroxymethyl hydroperoxide and bis(hydroxymethy1) peroxide from gas-phase ozonolysis of naturally occurring alkenes. Nature 1985, 316, 535–536. [Google Scholar]
- Barnes, I.; Bastian, V.; Becker, K.H.; Zhu, T. Kinetics and products of the reactions of nitrate radical with monoalkenes, dialkenes, and monoterpenes. J. Phys.Chem 1990, 94, 2413–2419. [Google Scholar]
- Becke, A D. Density-functional thermochemistry. III. The role of exact exchange. J. Phys. Chem. 1993, 98, 5648–5652. [Google Scholar]
- Neeb, P.; Sauer, F.; Horie, O.; Moortgat, G.K. Formation of hydroxymethyl hydroperoxide and formic acid in alkene ozonolysis in the presence of water vapour. Atmos. Environ 1997, 31, 1417–1423. [Google Scholar]
- Hasson, A.S.; Orzechowska, G.; Paulson, S.E. Production of stabilized Criegee intermediates and peroxides in the gas phase ozonolysis of alkenes 1. ethene, trans-2-butene, and 2,3-dimethyl-2- butene. J. Geophys. Res 2001, 106, 34131–34142. [Google Scholar]
- Hasson, A.S.; Ho, A.W.; Kuwata, K.T.; Paulson, S.E. Production of stabilized Criegee intermediates and peroxides in the gas phase ozonolysis of alkenes 2. asymmetric and biogenic alkenes. J. Geophys. Res 2001, 106, 34143–34153. [Google Scholar]
- Sauer, F.; Schafer, C.; Neeb, P.; Horie, O.; Moortgat, G.K. Formation of hydrogen peroxide in the ozonolysis of isoprene and simple alkenes under humid conditions. Atmos. Environ 1999, 33, 229–241. [Google Scholar]
- Hewitt, C.N.; Kok, G.L. Formation and occurrence of organic hydroperoxides in the troposphere: Laboratory and field observations. J. Atmos. Chem 1991, 12, 181–194. [Google Scholar]
- Simonaitis, R.; Olsyna, K.J.; Meagher, J.F. Production of hydrogen peroxide and organic peroxides in the gas phase reactions of ozone with natural alkenes. Geophys. Res. Lett 1991, 18, 9–12. [Google Scholar]
- Martinez, R.I.; Herron, J.T. Gas-phase reaction of SO2 with a Criegee intermediate in the presence of water vapor. J. Environ. Sci. Health A 1981, 16, 623–636. [Google Scholar]
- Wolff, S.; Boddenberg, A.; Thamm, J.; Turner, W.V.; Gäb, S. Gas-phase ozonolysis of ethene in the presence of carbonyl-oxide scavengers. Atmos. Environ 1997, 31, 2965–2969. [Google Scholar]
- Winterhalter, R.; Neeb, P.; Grossmann, D.; Kolloff, A.; Horie, O.; Moortgat, G.K. Products and mechanism of the gas phase reaction of ozone with β-pinene. J. Atmos. Chem 2000, 35, 165–197. [Google Scholar]
- Puxbaum, H.; Rosenberg, C.; Gregori, M.; Lanzerstorfer, C.; Ober, E.; Winiwarter, W. Atmospheric concentrations of formic and acetic acid and related compounds in eastern and northern Austria. Atmos. Environ 1988, 22, 2841–2850. [Google Scholar]
- Hellpointner, E.; Gäb, S. Detection of Methyl, Hydroxymethyl and hydroxyethyl hydroperoxides in air and precipitation. Nature 1989, 107, 631–634. [Google Scholar]
- Grosjean, D. Organic acids in southern California air: Ambient concentrations, mobile source emissions, in situ formation and removal processes. Environ. Sci. Technol 1989, 23, 1506–1514. [Google Scholar]
- Sanhueza, E.; Santana, M.; Hermoso, M. Gas- and aqueous-phase formic and acetic acids at a tropical cloud forest site. Atmos. Environ 1992, 26A, 1421–1426. [Google Scholar]
- Lawrence, J.E.; Koutrakis, P. Measurement of atmospheric formic and acetic acids: Methods evaluation and results from field studies. Environ. Sci. Technol 1994, 28, 957–964. [Google Scholar]
- Granby, K.; Christensen, C.S.; Lohse, C. Urban and semi-rural observations of carboxylic acids and carbonyls. Atmos. Environ 1997, 31, 1403–1415. [Google Scholar]
- Marklund, S. Mechanisms of the irreversible inactivation of horseradish peroxidase caused by hydroxymethyl hydroperoxide. Arch. Biochem. Biophys 1973, 154, 614–622. [Google Scholar]
- Möller, D. The possible role of H2O2 in new-type forest decline. Atmos. Environ 1989, 23, 1625–1627. [Google Scholar]
- Hewitt, C.N.; Kok, G.L.; Fall, R. Hydroxyperoxides exposed to ozone mediate air pollution damage to alkene emitters. Nature 1990, 344, 56–58. [Google Scholar]
- Calvert, J.G.; Lazrus, A.; Kok, G.L.; Heikes, B.G.; Walega, J.G.; Lind, J.; Cantrell, C.A. Chemical mechanisms of acid generation in the troposphere. Nature 1985, 317, 27–35. [Google Scholar]
- Niki, H.; Maker, P.D.; Savage, C.M.; Breitenbach, L.P.; Hurley, M.D. FTIR spectroscopic study of the mechanism for the gas-phase reaction between ozone and tetramethylethylene. J. Phys. Chem 1987, 91, 941–946. [Google Scholar]
- Jonsson, Å.; Hallquist, M.; Ljungström, E. Influence of OH scavenger on the water effect on secondary organic aerosol formation from ozonolysisof limonene, Δ3-carene, and α-pinene. Environ. Sci. Technol. 2008, 42, 5938–5944. [Google Scholar]
- Leungsakul, S.; Jaoui, M.; Kamens, R.M. Kinetic Mechanism for predicting secondary organic aerosol formation from the reaction of D-limonene with ozone. Environ. Sci. Technol 2005, 39, 9583–9594. [Google Scholar]
- Dowideit, P.; von Sonntag, C. Reaction of ozone with ethene and its methyl- and chlorine-substituted derivatives in aqueous solution. Environ. Sci. Technol 1998, 32, 1112–1119. [Google Scholar]
- Anglada, J.M.; González, J.; Torrent-Sucarrat, M. Effects of the substituents on the reactivity of carbonyl oxides. A theoretical study on the reaction of substituted carbonyl oxides with water. Phys. Chem. Chem. Phys 2011, 13, 13034–13045. [Google Scholar]
- Aplincourt, P.; Ruiz-López, M.F. Theoretical study of formic acid anhydride formation from carbonyl oxide in the atmosphere. J. Phys. Chem. A 2000, 104, 380–388. [Google Scholar]
- Anglada, J.M.; Aplincourt, P.; Bofill, J.M.; Cremer, D. Atmospheric formation of OH radicals and H2O2 from alkene ozonolysis under humid conditions. Chemphyschem 2002, 3, 215–221. [Google Scholar]
- Aplincourt, P.; Anglada, J.M. Theoretical studies on isoprene ozonolysis under tropospheric conditions. 1. Reaction of substituted carbonyl oxides with water. J. Phys. Chem. A 2003, 107, 5798–5811. [Google Scholar]
- Mebel, A.M.; Morokuma, K.; Lin, M.C. Modification of the Gaussian-2 theoretical model: The use of coupled cluster energies, densityfunctional geometries, and frequencies. J. Chem. Phys 1995, 103, 7414–7421. [Google Scholar]
- Guenther, A.; Geron, C.; Pierce, T.; Lamb, B.; Harley, P.; Fall, R. Natural emissions of non-methane volatile organic compounds, carbon monoxide, and oxides of nitrogen from North America. Atmos. Environ 2000, 34, 2205–2230. [Google Scholar]
- Ramírez-Ramírez, V.M.; Nebot-Gil, I. Theoretical study of the OH addition to the endocyclic and exocyclic double bonds of the D-limonene. Chem. Phys. Lett 2005, 409, 23–28. [Google Scholar]
- Jiang, L.; Xu, Y.S.; Ding, A.Z. Reaction of stabilized Criegee intermediates from ozonolysis of limonene with sulfur dioxide: Ab initio and DFT study. 2010. J. Phys. Chem. A 2010, 114, 12452–12461. [Google Scholar]
- Wang, Y.D. Theory Study on Mechanism of O3-Initiated Atmospheric Photooxidation of Terpenes. Master’s Dissertation, Shandong University, Jinan, China, 2008. [Google Scholar]
- Leungsakul, S.; Jeffries, H.E.; Kamens, R.M. A kinetic mechanism for predicting secondary aerosol formation from the reactions of D-limonene in the presence of oxides of nitrogen and natural sunlight. Atmos. Environ 2005, 39, 7063–7082. [Google Scholar]
- Neeb, P.; Horie, O.; Moortgat, G.K. The ethene-ozone reaction in the gas phase. J. Phys. Chem. A 1998, 102, 6778–6785. [Google Scholar]
- Herron, J.T.; Huie, E. Stopped-flow studies of the mechanisms of ozone-alkene reactions in the gas phase. Ethylene. J. Am. Chem. Soc 1977, 99, 5430–5435. [Google Scholar]
- Martinez, R.I.; Herron, J.T. Stopped-flow studies of the mechanisms of ozone-alkene reactions in the gas phase: Trans-2-butene. J. Phys. Chem 1988, 92, 4644–4648. [Google Scholar]
- Gutbrod, R.; Kraka, E.; Schindler, R.N.; Cremer, D. Kinetic and theoretical investigation of the gas-phase ozonolysis of isoprene: Carbonyl oxides as an important source for OH radicals in the atmosphere. J. Am. Chem. Soc 1997, 119, 7330–7342. [Google Scholar]
- Anglada, J.M.; Bofill, J.M.; Olivella, S.; Sole, A. Theoretical investigation of the low-lying electronic states of dioxirane: Ring opening to dioxymethane and dissociation into CO2 and H2. J. Phys. Chem. A 1998, 102, 3398–3406. [Google Scholar]
- Chen, B.Z.; Anglada, J.M.; Huang, M.B.; Kong, F. The reaction of CH2 (X3B1) with O2 (X3 ): A theoretical CASSCF/CASPT2 investigation. J. Phys. Chem. A 2002, 106, 1877–1884. [Google Scholar]
- Paulson, S.E.; Chung, M.Y.; Hasson, A.S. OH radical formation from the gas-phase reaction of ozone with terminal alkenes and the relationship between structure and mechanism. J. Phys. Chem. A 1999, 103, 8125–8138. [Google Scholar]
- Aplincourt, P.; Ruiz-López, M.F. Theoretical investigation of reaction mechanisms for carboxylic acid formation in the atmosphere. J. Am. Chem. Soc 2000, 122, 8990–8997. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03, Revision E.01; Gaussian, Inc: Wallingford, CT, USA; p. 2004.
- Hariharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar]
- Gonzalez, C.; Schlegel, H.B. An improved algorithm for reaction path following. J. Chem. Phys 1989, 90, 2154–2161. [Google Scholar]
- Baptista, L.; Pfeifer, R.; da Silva, E.C.; Arbilla, G. Kinetics and thermodynamics of limonene ozonolysis. J. Phys. Chem. A 2011, 115, 10911–10919. [Google Scholar]
- Szabo, A.; Ostlund, N.S. Modern Quantum Chemistry: Introduction to Advanced Electronic Structure Theory; Macmillan: New York, NY, USA; p. 1986.
- Lei, W.; Derecskei-Kovacs, A.; Zhang, R. Ab initio study of OH addition reaction to isoprene. J. Chem. Phys 2000, 113, 5354–5360. [Google Scholar]
- Suh, I.; Lei, W.; Zhang, R. Experimental and theoretical studies of isoprene reaction with NO3. J. Phys. Chem. A 2001, 105, 6471–6478. [Google Scholar]
- Zhang, D.; Zhang, R. Mechanism of OH formation from ozonolysis of isoprene: A quantum-chemical study. J. Am. Chem. Soc 2002, 124, 2692–2703. [Google Scholar]
- Jiang, L.; Wang, W.; Xu, Y.S. Theoretical investigation of the NO3 radical addition to double bonds of limonene. Int. J. Mol. Sci 2009, 10, 3743–3754. [Google Scholar]
- Jiang, L.; Wang, W.; Xu, Y.S. Ab initio investigation of O3 addition to double bonds of limonene. Chem. Phys 2010, 368, 108–112. [Google Scholar]
- Gutbrod, R.; Schindler, R.N.; Kraka, E.; Cremer, D. Formation of OH radicals in the gas phase ozonolysis of alkenes: The unexpected role of carbonyl oxides. Chem. Phys. Lett 1996, 252, 221–229. [Google Scholar]
- Olzmann, M.; Kraka, E.; Cremer, D.; Gutbrod, R.; Andersson, S. Energetics, kinetics, and product distributions of the reactions of ozone with ethene and 2,3-dimethyl-2-butene. J. Phys. Chem. A 1997, 101, 9421–9429. [Google Scholar]
- Cremer, D.; Kraka, E.; Szalay, P.G. Decomposition modes of dioxirane, methyldioxirane and dimethyldioxirane—A CCSD(T), MR-AQCC and DFT investigation. Chem. Phys. Lett 1998, 292, 97–109. [Google Scholar]
- Xu, F.; Wang, H.; Zhang, Q.Z.; Zhang, R.X.; Qu, X.H.; Wang, W.X. Kinetic properties for the complete series reactions of chlorophenols with OH radicals—Relevance for dioxin formation. Environ. Sci. Technol 2010, 44, 1399–1404. [Google Scholar]
- Sun, X.Y.; He, M.X.; Zhang, Q.Z.; Wang, W.X.; Jalbout, A.F. Quantum chemical study on the atmospheric photooxidation of Methyl Vinyl Ether (MVE). J. Mol. Struc 2008, 868, 87–93. [Google Scholar]
- Drozd, G.T.; Kroll, J.; Donahue, N.M. 2,3-Dimethyl-2-butene (TME) ozonolysis: Pressure dependence of stabilized criegee intermediates and evidence of stabilized vinyl hydroperoxides. J. Phys. Chem. A 2011, 115, 161–166. [Google Scholar]
- Kurtén, T.; Donahue, N.M. MRCISD studies of the dissociation of vinylhydroperoxide, CH2CHOOH: There is a saddle point. J. Phys. Chem. A 2012, 116, 6823–6830. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Relative to | E(B3LYP/6-31G(d,p)) | E(MP2/6-31G(d)) | E(MP2/6-311++G(d,p)) | E(CCSD(T)/6-31G(d)) | E(CCSD(T)/6-31G(d) + CF) |
|---|---|---|---|---|---|---|
| stabCl-OO + H2O | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | |
| M1H1_S | stabCl-OO + H2O | −8.65 | −7.27 | −7.54 | −7.54 | −6.16 |
| TSM11H1_S | M1H1_S | 13.27 | 11.44 | 8.52 | 18.51 | 16.68 |
| M2H1_S | TSM11H1_S | −34.17 | −34.57 | −26.94 | −35.09 | −35.50 |
| M4H1_S + H2O | M2H1_S | 8.80 | 5.64 | 8.50 | 7.91 | 4.74 |
| TSM4H1_S | M4H1_S | 24.29 | 18.38 | 29.60 | 20.51 | 14.60 |
| R1H1_S+OH | TSM4H1_S | −10.64 | -8.29 | −17.17 | −9.39 | −7.04 |
| TSM12H1_S | M1H1_S | 14.96 | 17.01 | 13.62 | 16.78 | 18.83 |
| M3H1_S | TSM12H1_S | −50.15 | −50.35 | −41.65 | −47.92 | −48.12 |
| TSM31H1_S | M3H1_S | 56.79 | 54.26 | 60.18 | 35.57 | 33.04 |
| R2H1_S+OH | TSM31H1_S | −12.35 | −12.64 | −22.88 | 2.06 | 1.78 |
| TSM32H1_S | M3H1_S | 40.23 | 32.82 | 46.83 | 49.85 | 42.44 |
| M5H1_S | TSM32H1_S | −121.60 | −120.87 | −124.12 | −126.25 | −125.52 |
| Limononic acid A + H2O | M5H1_S | 7.41 | 5.01 | 6.31 | 7.12 | 4.71 |
| TSM33H1_S | M3H1_S | 44.84 | 35.62 | 51.29 | 54.09 | 44.87 |
| M6H1_S | TSM33H1_S | −121.88 | −120.57 | −126.09 | −126.76 | −125.44 |
| Limononic acid B + H2O | M6H1_S | 5.84 | 3.56 | 5.60 | 5.53 | 3.24 |
| TSM34H1_S | M3H1_S | 53.34 | 48.18 | 51.67 | 52.07 | 46.91 |
| M7H1_S | TSM34H1_S | −130.73 | −131.72 | −126.97 | −124.94 | −125.93 |
| Limononic acid C + H2O | M7H1_S | 8.06 | 4.72 | 7.79 | 7.56 | 4.22 |
| TSM35H1_S | M3H1_S | 44.65 | 42.72 | 34.16 | 43.70 | 41.78 |
| M8H1_S | TSM35H1_S | −125.34 | −129.75 | −111.65 | −119.40 | −123.81 |
| Limononic acid D + H2O | M8H1_S | 8.44 | 6.46 | 7.78 | 8.03 | 6.05 |
| TSM36H1_S | M3H1_S | 54.94 | 55.16 | 49.52 | 52.33 | 52.55 |
| M9H1_S | TSM36H1_S | −51.97 | −51.66 | −47.33 | −49.38 | −49.07 |
| Limononaldehyd e + H2O2 | M9H1_S | 8.58 | 6.79 | 8.12 | 8.26 | 6.47 |
| Compound | Relative to | E(B3LYP/6-31G(d,p)) | EG(MP2/6-31G(d)) | E(MP2/6-311++G(d,p)) | E(CCSD(T)/6-31G(d)) | E(CCSD(T)/6-31G(d) + CF) |
|---|---|---|---|---|---|---|
| stabCl-CH3-OO + H2O | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | |
| M1H2_S | stabCl-CH3- OO + H2O | −11.15 | −7.94 | −10.45 | −9.57 | −6.35 |
| TSM11H2_S | M1H2_S | 19.60 | 16.93 | 14.68 | 24.64 | 21.98 |
| M2H2_S | TSM11 | −30.19 | −31.48 | −22.41 | −31.04 | −32.32 |
| M4H2_S + H2O | M2H2_S | 8.74 | 5.66 | 8.71 | 7.86 | 4.79 |
| TSM4H2_S | M4H2_S | 29.25 | 25.80 | 41.29 | 20.30 | 16.86 |
| R1H2_S+OH | TSM4H2_S | −17.41 | −15.58 | −28.27 | −9.81 | −7.98 |
| TSM12H2_S | M1H2_S | 11.72 | 13.61 | 11.25 | 13.64 | 15.53 |
| M3H2_S | TSM12H2_S | −43.36 | −43.58 | −34.17 | −41.13 | −41.35 |
| R2H2_S + OH | M3H2_S | 46.53 | 43.33 | 38.15 | 38.84 | 35.63 |
| TSM31H2_S | M3H2_S | 45.47 | 43.40 | 37.90 | 46.11 | 44.04 |
| R3H2_S + R4H2_S + H2O | TSM31H2_S | −20.33 | −22.99 | −21.49 | −19.45 | −22.11 |
| TSM32H2_S | M3H2_S | 44.87 | 43.12 | 36.08 | 44.51 | 42.76 |
| M5H2_S | TSM32H2_S | −41.41 | −39.53 | −35.87 | −41.49 | −39.62 |
| Limononaldehyde + H2O2 | M5H2_S | 9.16 | 8.17 | 8.14 | 8.72 | 7.73 |
| Compound | Relative to | E(B3LYP/6-31G(d,p)) | E(MP2/6-31G(d)) | E(MP2/6-311++G(d,p)) | E(CCSD(T)/6-31G(d)) | E(CCSD(T)/6-31G(d) + CF) |
|---|---|---|---|---|---|---|
| stabClx-OO + H2O | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | |
| M1H3_S | stabClx-OO + H2O | −10.79 | −7.67 | −10.22 | −9.32 | −6.19 |
| TSM11H3_S | M1H3_S | 15.56 | 12.93 | 11.36 | 20.55 | 17.92 |
| M2H3_S | TSM11H3_S | −30.54 | −31.66 | −22.01 | −31.27 | −32.39 |
| M4H3_S + H2O | M2H3_S | 9.31 | 6.32 | 8.79 | 8.35 | 5.36 |
| TSM4H3_S | M4H3_S | 28.12 | 24.23 | 34.67 | 24.14 | 20.24 |
| R1H3_S+OH | TSM4H3_S | −12.80 | −11.39 | −18.84 | −10.79 | −9.38 |
| TSM12H3_S | M1H3_S | 11.50 | 12.81 | 12.09 | 13.39 | 14.70 |
| M3H3_S | TSM12H3_S | −42.89 | −42.86 | −33.40 | −40.72 | −40.69 |
| R2H3_S + OH | M3H3_S | 45.32 | 42.42 | 35.58 | 37.28 | 34.38 |
| TSM31H3_S | M3H3_S | 41.90 | 38.07 | 34.61 | 43.19 | 39.36 |
| R3H3_S + R4H3_S + H2O | TSM31H3_S | −19.76 | −20.40 | −24.55 | −20.32 | −20.95 |
| TSM32H3_S | M3H3_S | 42.47 | 41.24 | 34.04 | 42.27 | 41.04 |
| M5H3_S | TSM32H3_S | −40.78 | −38.49 | −36.13 | −41.14 | −38.86 |
| Keto-limonene + H2O2 | M5H3_S | 8.20 | 6.41 | 7.99 | 7.88 | 6.10 |
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jiang, L.; Lan, R.; Xu, Y.-S.; Zhang, W.-J.; Yang, W. Reaction of Stabilized Criegee Intermediates from Ozonolysis of Limonene with Water: Ab Initio and DFT Study. Int. J. Mol. Sci. 2013, 14, 5784-5805. https://doi.org/10.3390/ijms14035784
Jiang L, Lan R, Xu Y-S, Zhang W-J, Yang W. Reaction of Stabilized Criegee Intermediates from Ozonolysis of Limonene with Water: Ab Initio and DFT Study. International Journal of Molecular Sciences. 2013; 14(3):5784-5805. https://doi.org/10.3390/ijms14035784
Chicago/Turabian StyleJiang, Lei, Ru Lan, Yi-Sheng Xu, Wen-Jie Zhang, and Wen Yang. 2013. "Reaction of Stabilized Criegee Intermediates from Ozonolysis of Limonene with Water: Ab Initio and DFT Study" International Journal of Molecular Sciences 14, no. 3: 5784-5805. https://doi.org/10.3390/ijms14035784