Transcriptional Analysis of Drought-Induced Genes in the Roots of a Tolerant Genotype of the Common Bean (Phaseolus vulgaris L.)

Abstract
:1. Introduction
2. Results and Discussion
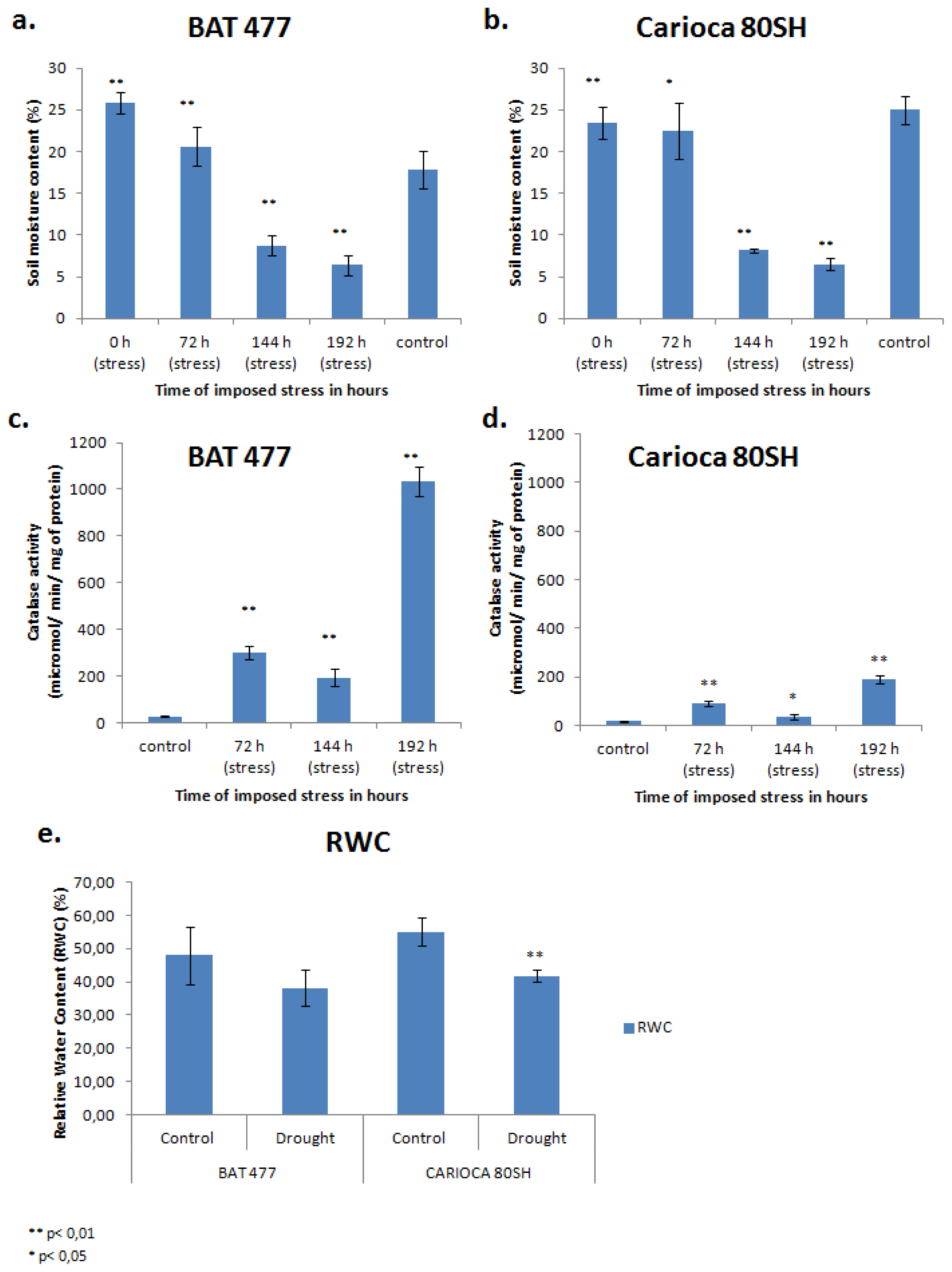
2.1. Soil Water Moisture
2.2. Biological Effects of Drought on Plants
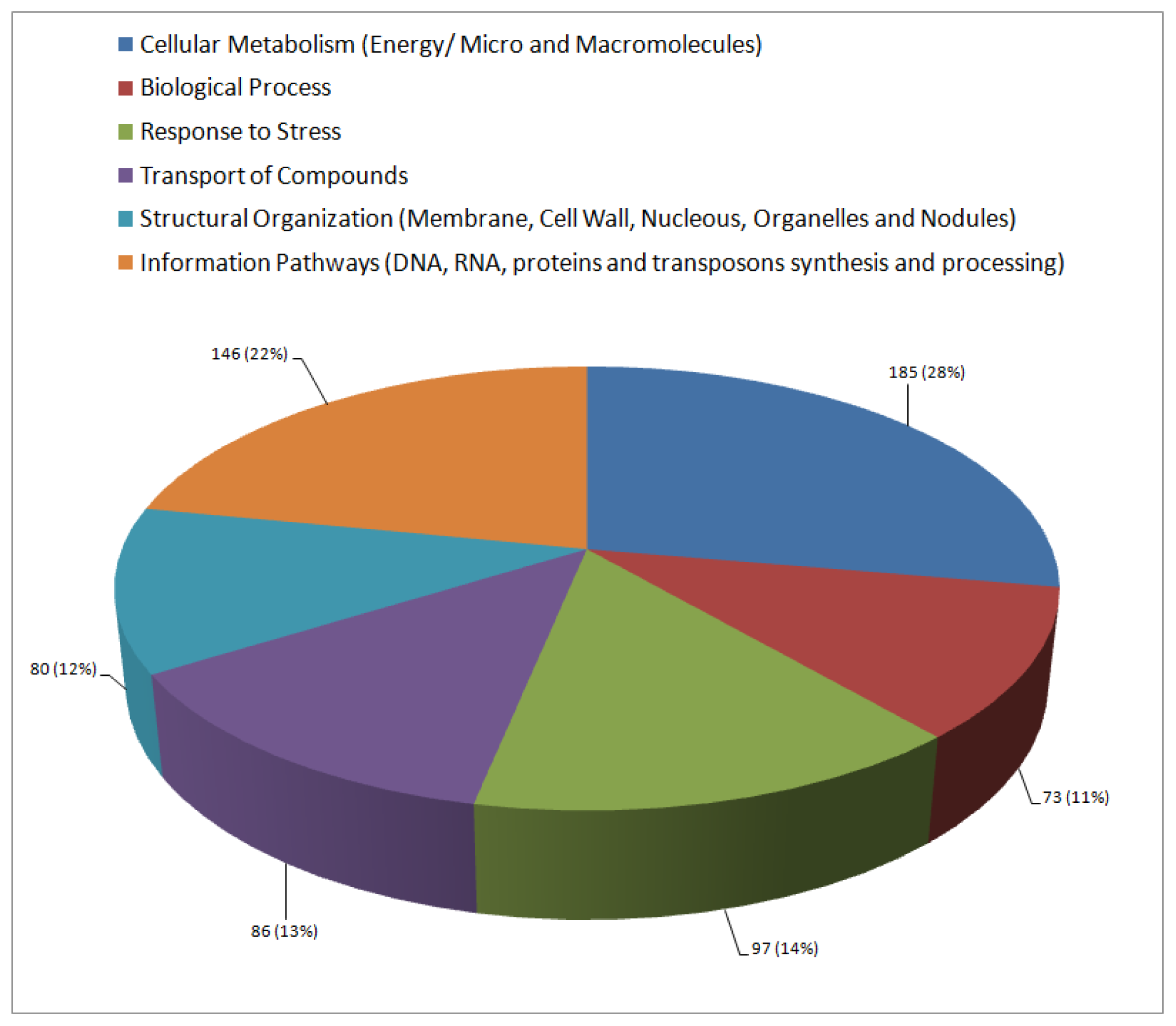
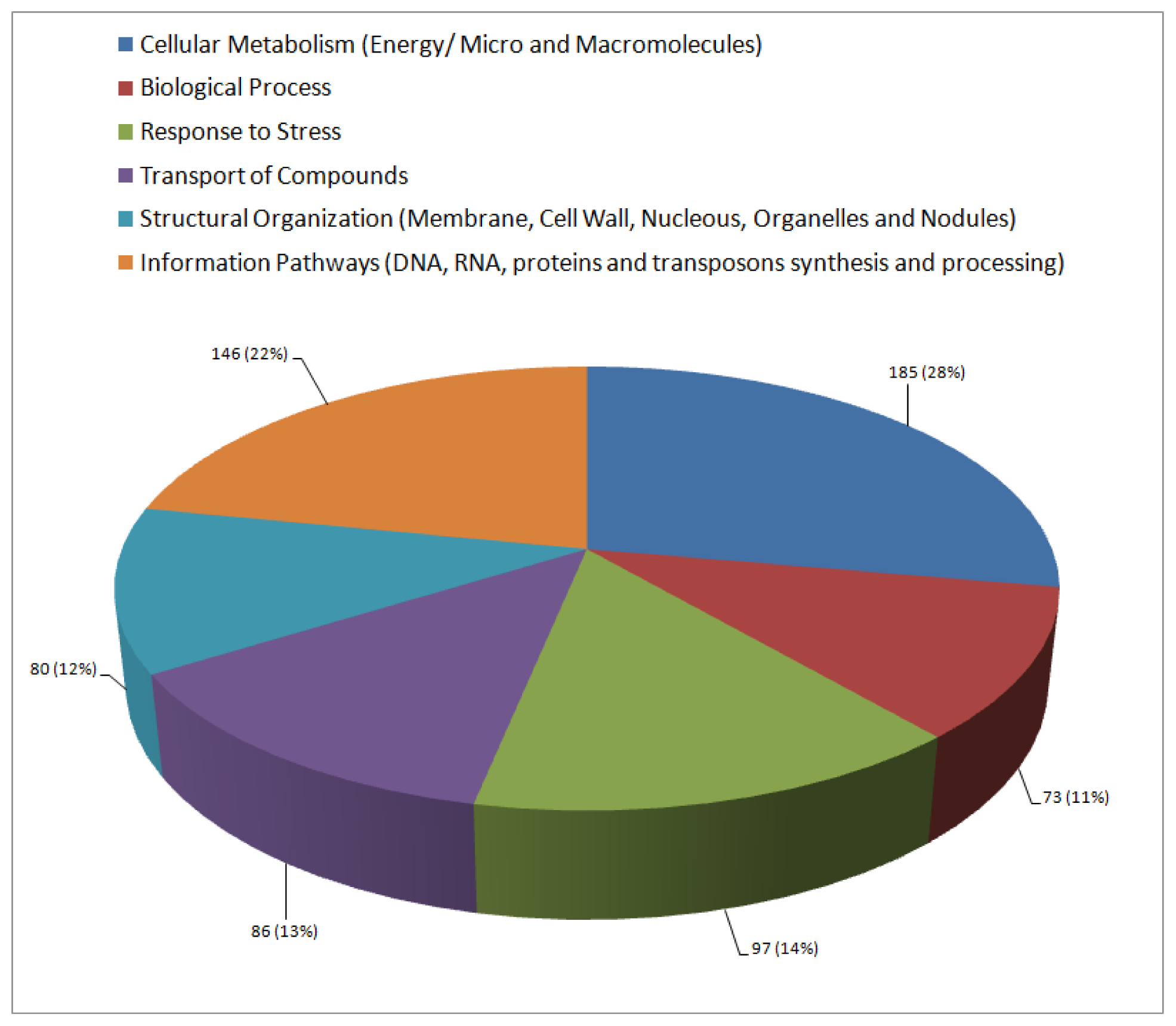
2.3. Functional Annotation and Classification of Transcripts
2.4. Major Response Mechanisms Activated
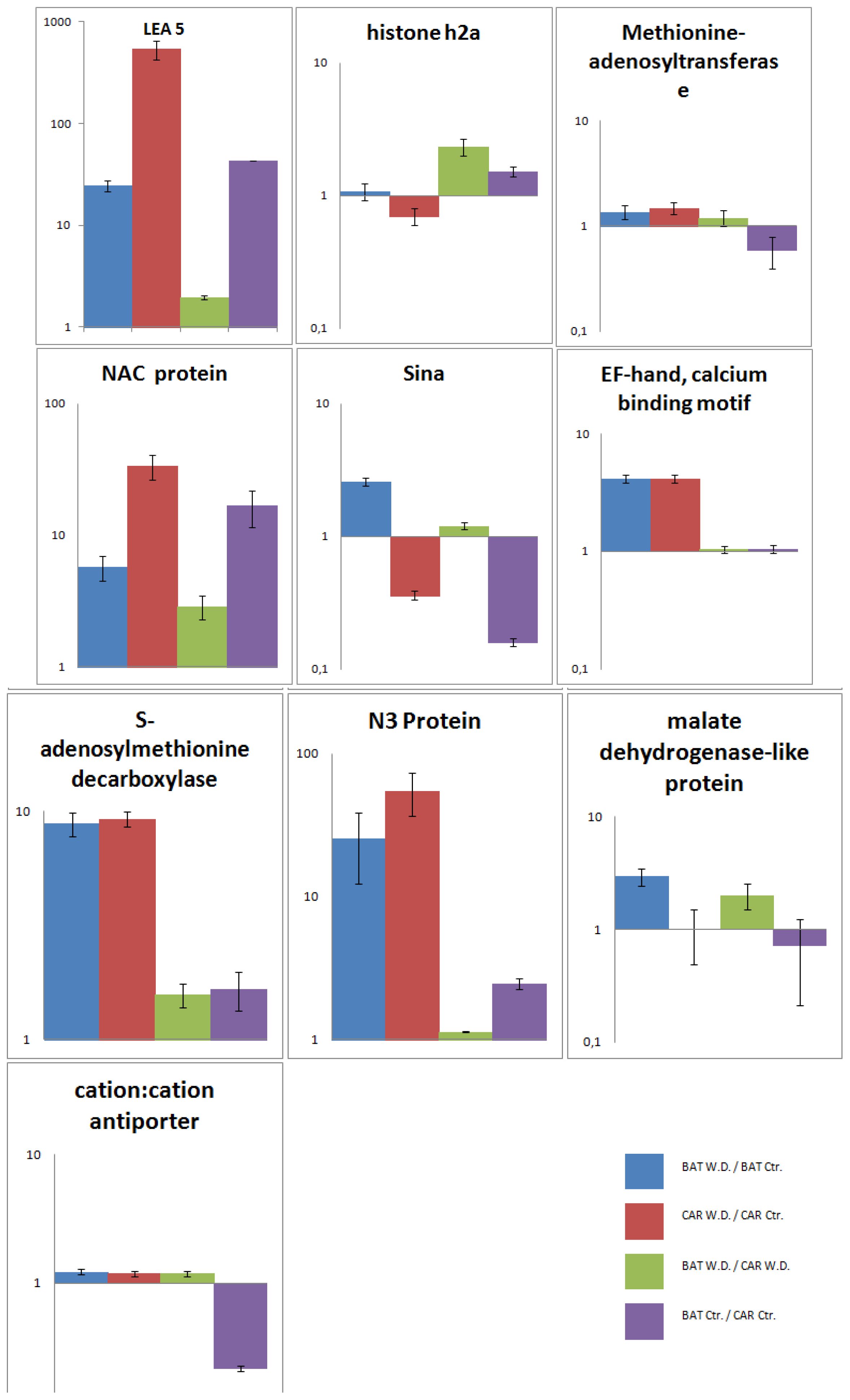
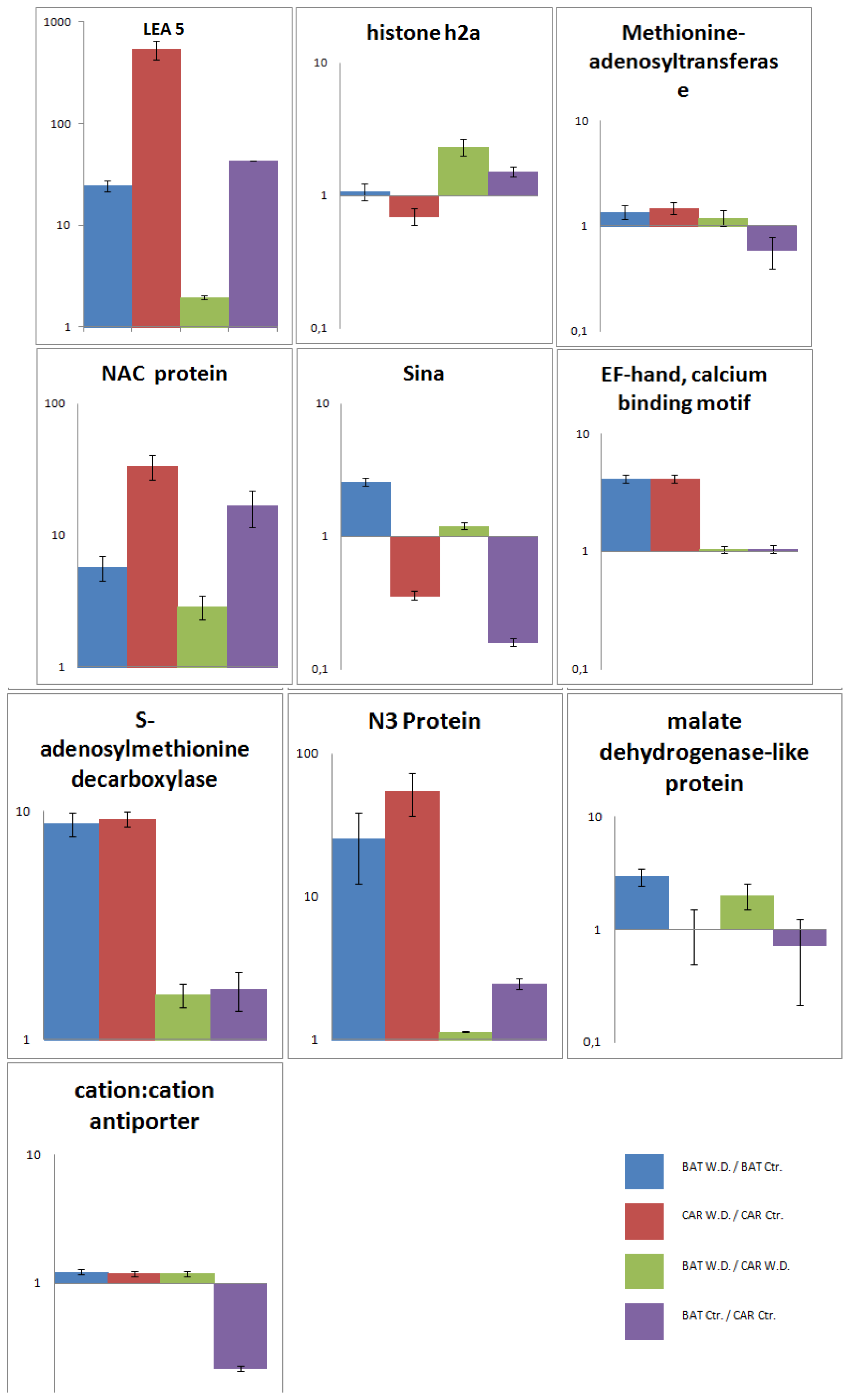
2.5. Validation of the SSH Library by RT-qPCR
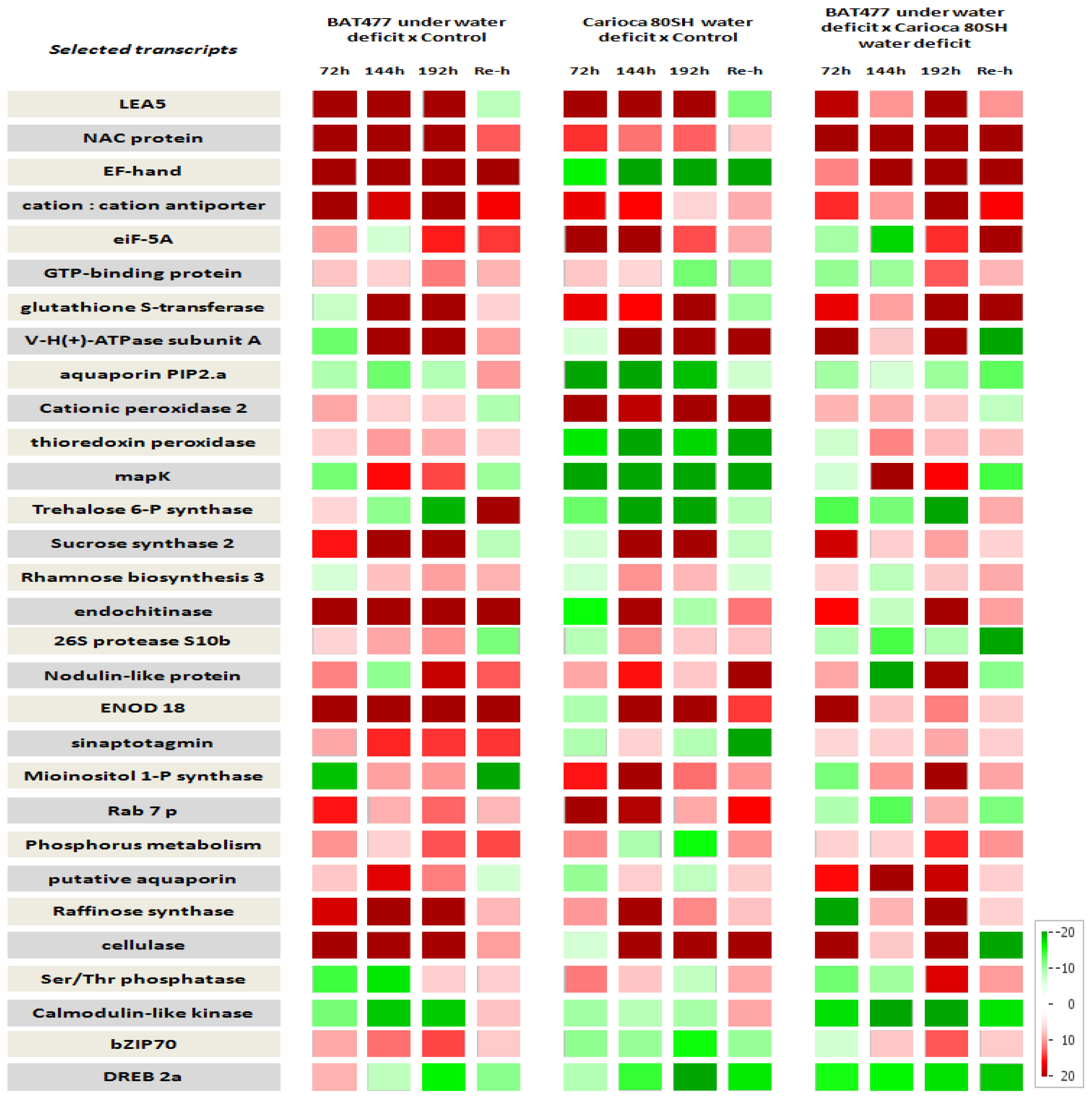
2.6. Time Scale Gene Expression Analysis
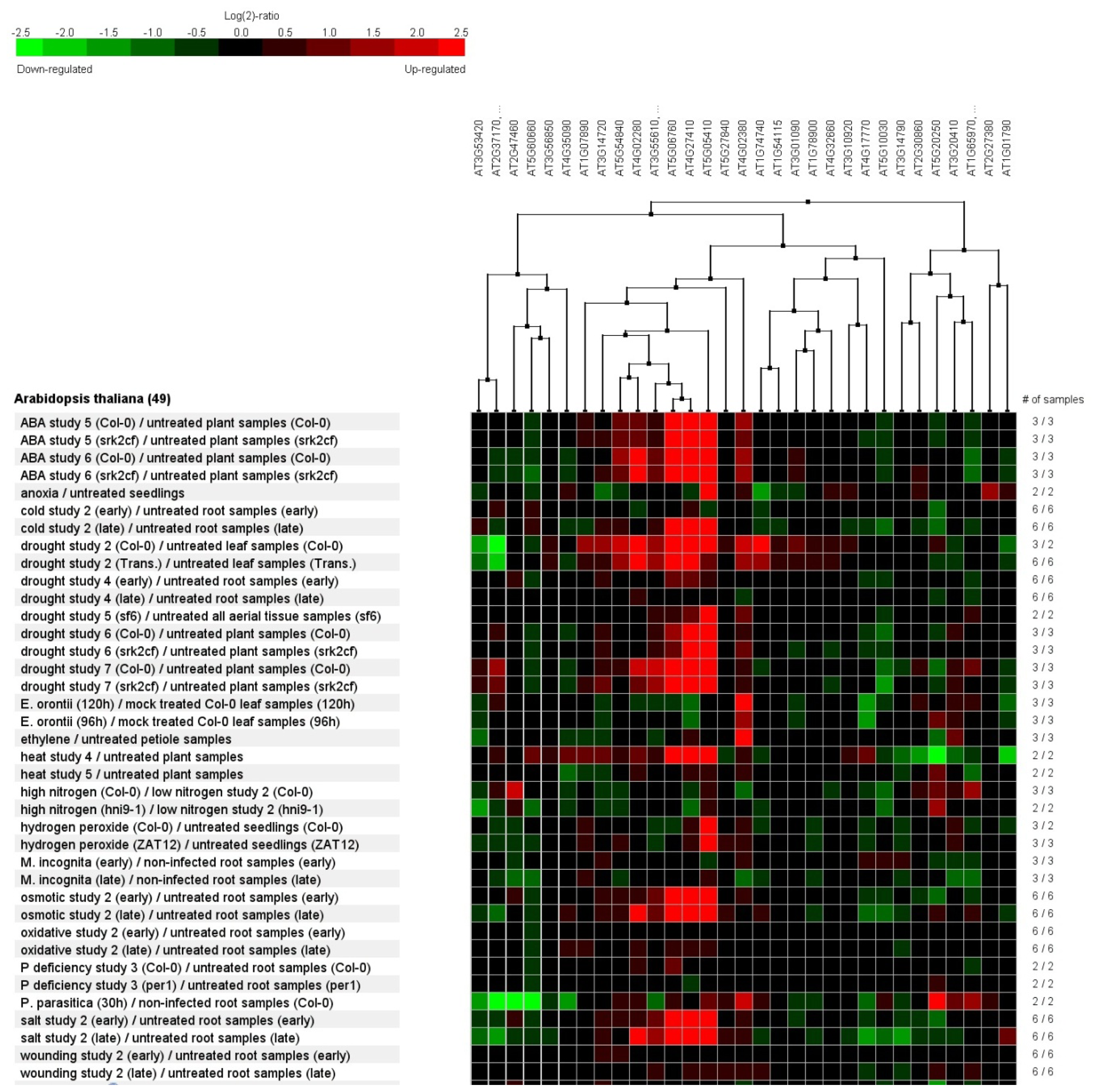
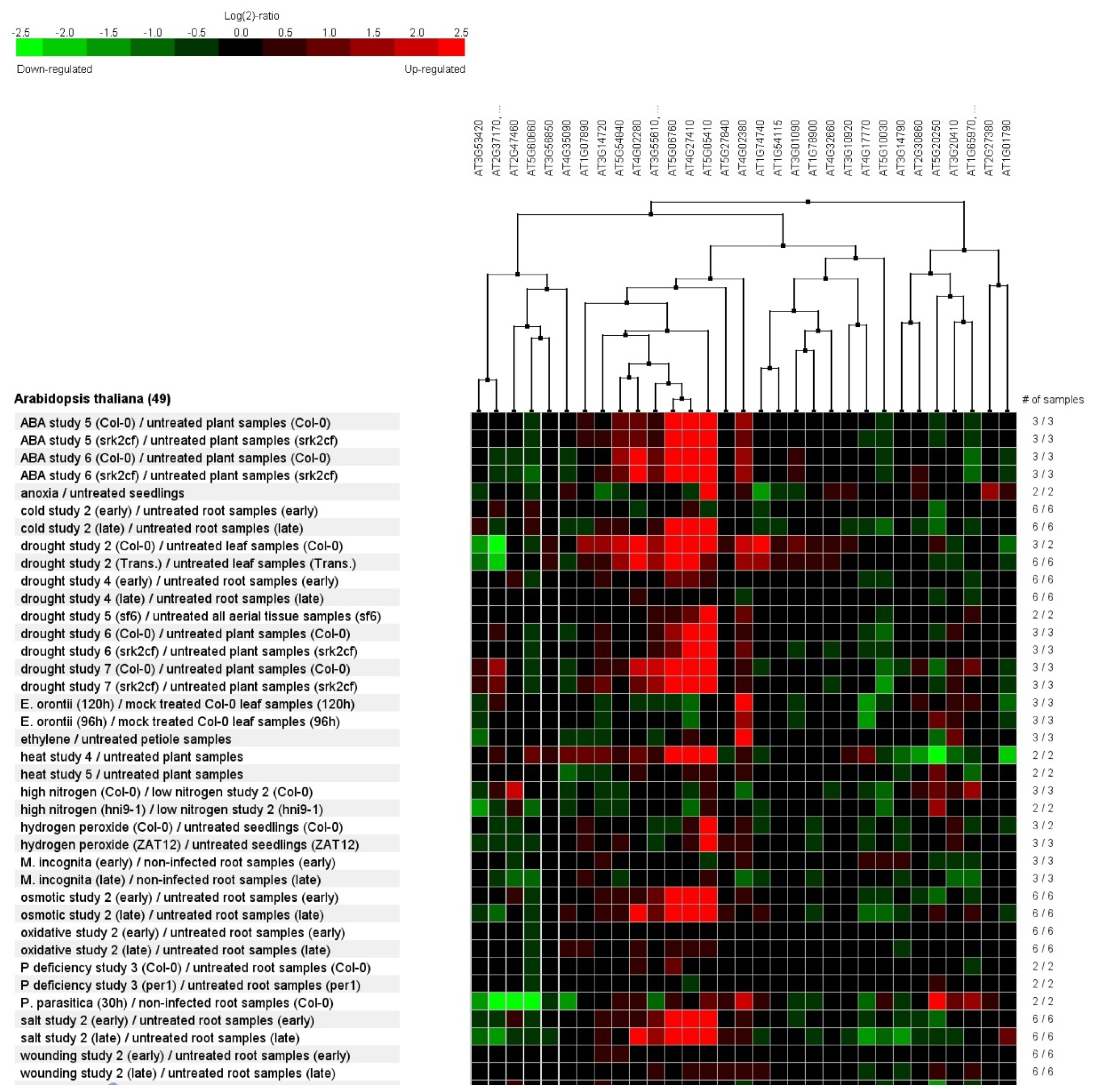
2.7. Genevestigator Cluster Analysis
3. Experimental Section
3.1. Plant Material and Induction of Drought Response
3.2. Catalase Enzymatic Activity Determination
3.3. SSH Library Construction
3.4. Bioinformatics Analyses
3.5. RT-qPCR for SSH Library Validation and Time Scale Gene Expression Analysis
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Hirayama, T.; Shinozaki, K. Research on plant abiotic stress responses in the post-genome era: Past, present and future. Plant J 2010, 61, 1041–1053. [Google Scholar]
- Bray, E.A.; Balley-Serres, J.; Weretilnik, E. Responses to Abiotic Stresses. In Biochemistry & Molecular Biology of Plants, 2nd ed.; Buchanan, B., Gruissen, W., Jones, R., Eds.; American Society of Plant Physiologists: Rockville, MD, USA, 2000; Volume 3, pp. 1158–1203. [Google Scholar]
- Singh, S.P.; Muñoz, C.G. Resistance to common bacterial blight among Phaseolus species and improvement. Crop Sci 1999, 39, 80–89. [Google Scholar]
- Pfeiffer, W.H.; McClafferty, B. Harvest plus: Breeding crops for better nutrition. Crop Sci 2007, 47, S88–S105. [Google Scholar]
- Beebe, S.; Idupulapati, R.; Blair, M.; Butare, L. Breeding for Abiotic Stress Tolerance in Common Bean: Present and Future Challenges. CIAT. Available online: http://km.fao.org/gipb/images/pdf_files/LiteratureViews/Beebe_PPT_apb09.pdf (accessed on 12 August 2011).
- Van Schoonhoven, A.; Voysest, O. Common Beans: Research for Crop Improvement, 1st ed.; CABI: Cali, Colombia, 1991; pp. 7–43. [Google Scholar]
- Grahan, P.H.; Ranalli, P. Common Bean (Phaseolus vulgaris L.). Field Crops Res 1997, 53, 131–146. [Google Scholar]
- Broughton, W.J.; Hernández, G.; Blair, M.; Beebe, S.; Gepts, P.; Vanderleyden, J. Beans (Phaseolus spp.)—Model food legumes. Plant Soil 2003, 252, 55–128. [Google Scholar]
- Ramírez-Vallejo, P.; Kelly, J.D. Traits related to drought resistance in common bean. Euphytica 1998, 99, 127–136. [Google Scholar]
- Pandey, R.K.; Herrera, W.A.T.; Pendleton, J.W. Drought response of grain legumes under irrigation gradient. Plant Growth Agron. J 1984, 76, 557–560. [Google Scholar]
- Guimarães, C.M.; Brunini, O.; Stone, L.F. Adaptação do feijoeiro (Phaseolus vulgaris L.) à seca. Rev. Bras. Eng. Agríc. Ambient 2006, 10, 70–75. [Google Scholar]
- Torres, G.A.M.; Pflieger, S.; Corre-Menguy, F.; Mazubert, C.; Hartmann, C.; Lelandais-Brière, C. Identification of novel drought-related mRNAs in common bean roots by differential display RT-PCR. Plant Sci 2006, 171, 300–307. [Google Scholar]
- Rodriguez-Uribe, L.; O’Connell, M.A. A root-specific bZIP transcription factor is responsive to water deficit stress in tepary bean (Phaseolus acutifolius) and common bean (P. vulgaris). J. Exp. Botany 2006, 57, 1391–1398. [Google Scholar]
- Montalvo-Henández, L.; Piedra-Ibarra, E.; Gómez-Silva, L.; Lira-Carmona, R.; Acosta-Gallegos, J.A.; Vasquez-Medrano, J.; Xoconostle-Cázares, B.; Ruíz-Medrano, R. Differential accumulation of mRNAs in drought-tolerant and susceptible common bean cultivars in response to water deficit. New Phytol 2008, 177, 102–113. [Google Scholar]
- Bui, L.C.; Leandri, R.D.; Renard, J.P.; Duranthon, V. SSH adequacy to pre-implantation mammalian development: Scarce specific transcripts cloning despite irregular normalization. BMC Genomics 2005, 6, 155–164. [Google Scholar]
- Gonzáles-Candelas, L.; Alamar, S.; Sánchez-Torres, P.; Zacarías, L.; Marcos, J.F. A transcriptomic approach highlights induction of secondary metabolism in citrus fruit in response to Penicillium digitatum infection. BMC Plant Biol 2010, 10, 194. [Google Scholar]
- Priyanka, B.; Sekhar, K.; Reddy, V.D.; Rao, K.V. Expression of pigeonpea high-proline-rich protein encoding gene (CcHyPRP) in yeast and Arabidopsis affords multiple abiotic stress tolerance. Plant Biotechnol 2010, 8, 76–87. [Google Scholar]
- Zhang, J.Z.; Li, Z.M.; Liu, L.; Mei, L.; Yao, J.L.; Hu, C.G. Identification of early-flower-related ESTs in an early-flowering mutant of trifoliate orange (Poncirus trifoliata) by suppression subtractive hybridization and macroarray analysis. Tree Physiol 2008, 28, 1449–1457. [Google Scholar]
- Zhang, Y.; Liu, X.; Zhang, W.; Han, R. Differential gene expression of the honey bees Apis mellifera and A. cerana induced by Varroa destructor infection. J. Insect Physiol 2010, 56, 1207–1218. [Google Scholar]
- Procópio, S.O.; Santos, J.B.; Silva, A.A.; Donagemma, G.K.; Mendonça, E.S. Permanent wilting point of soybean, bean and weeds. Planta Daninha 2004, 22, 35–41. [Google Scholar]
- Rodríguez, M.; Canales, E.; Borrás-Hidalgo, O. Molecular aspects of abiotic stress in plants. Biotecnol. Aplicada 2005, 22, 1–10. [Google Scholar]
- Cushman, J.; Bohnert, H.J. Genomic approaches to plant stress tolerance. Curr. Opin. Plant Biol 2000, 3, 117–124. [Google Scholar]
- Raheleh, R.; Ramazanali, K.-N.; Ali, G.; Abdolreza, B.; Farzaneh, N.; Masoud, R. Use of biochemical indices and antioxidant enzymes as a screening technique for drought tolerance in Chickpea genotypes (Cicer arietinum L.). Afr. J. Agric. Res 2012, 39, 5372–5380. [Google Scholar]
- Rodrigues, F.A.; Marcolino-Gomes, J.; Carvalho, J.F.C.; Nascimento, L.C.; Neumaier, N.; Farias, J.R.B.; Carazzolle, M.F.; Marcelino, F.C.; Nepomuceno, A.L. Subtractive libraries for prospecting differentially expressed genes in the soybean under water deficit. Genetic Mol. Biol 2012, 35, 304–314. [Google Scholar]
- Chaves, M.M.; Pereira, J.S.; Maroco, J.; Rodrigues, M.L.; Ricardo, C.P.P.; Osório, M.L.; Carvalho, I.; Faria, T.; Pinheiro, C. How plants cope with water stress in the field. Photosynthesis and Growth. Ann. Botany 2002, 89, 907–916. [Google Scholar]
- Pinheiro, C.; Chaves, M.M.; Ricardo, C.P.P. Plant Water Deficits in Mediterranean Ecosystems. In Plant Responses to Water Deficits—From Cell to Community, 1st ed.; Smith, J.A.C., Griffiths, H., Eds.; BIOS Scientific Publisher: London, UK, 2001; pp. 237–251. [Google Scholar]
- Alamillo, J.M.; Díaz-Leal, J.L.; Sánchez-Moran, M.A.V.; Pineda, M. Molecular analysis of ureide accumulation under drought stress in Phaseolus vulgaris L. Plant Cell Environ 2010, 33, 1828–1837. [Google Scholar]
- Reece, J.; Campbell, N. G Protein Signaling. In Biology, 1st ed.; Campbell, N., Reece, J., Eds.; Benjamin Cummings: San Francisco, CA, USA, 2002; pp. 26–27. [Google Scholar]
- Maurel, C.; Reizer, J.; Schroeder, J.I.; Chrispeels, M.J. The vacuolar membrane protein—TIP creates water specific channels in Xenopus oocytes. EMBO J 1993, 12, 2241–2247. [Google Scholar]
- Wang, W.; Vinocur, B.; Altman, A. Plant responses to drought, salinity and extreme temperatures: Towards genetic engineering for stress tolerance. Planta 2003, 218, 1–14. [Google Scholar]
- Davis, K.R.; Darvill, A.G.; Albersheim, P.; Dell, A. Host-pathogen interactions. Oligogalacturonides released from sodium polypectate by endopolygalacturonic acid lyase are elicitors of phytoalexines in soybean. Plant Physiol 1986, 80, 568–577. [Google Scholar]
- Dodd, A.N.; Kudla, J.; Sanders, D. The language of calcium signaling. Annu. Rev. Plant Biol 2010, 61, 593–620. [Google Scholar]
- Serrano, R.; Gaxiola, R.; Ríos, G.; Forment, J.; Vicente, O.; Ros, R. Salt stress proteins indentified by a functional approach in yeast. Monatsh Chem 2003, 134, 1445–1464. [Google Scholar]
- Xiong, L.; Zhu, J.K. Molecular and Genetic aspects of plant responses to osmotic stress. Plant Cell Environ 2002, 25, 131–139. [Google Scholar]
- Yamagushi-Shinozaki, K.; Kasuga, M.; Liu, Q.; Nakashima, K.; Sakuma, Y.; Abe, H.; Shinwari, Z.K.; Seki, M.; Shonozaki, K. Biological mechanisms of drought stress response. JIRCAS Work. Rep 2002, 23, 1–8. [Google Scholar]
- Hussain, S.S.; Kayani, M.A.; Amjad, M. Transcription factors tools to engineer enhanced drought stress tolerance in plants. Biotechnol. Prog 2011, 27, 297–306. [Google Scholar]
- Trujillo, L.; Menendez, C.; Ochogavia, M.E.; Hernandez, I.; Borras, O.; Rodriguez, R.; Coll, Y.; Arrieta, J.G.; Banguela, A.; Ramirez, R.; Hernandez, L. Engineering drought and salt tolerance in plants using SodERF3, a novel sugarcane ethylene responsive factor. Biotechnol. Apl 2009, 26, 168–171. [Google Scholar]
- Bohnert, H.J.; Ayoubi, P.; Borcherta, C.; Bressanc, R.A.; Burnapd, R.L.; Cushmane, J.C.; Cushmane, M.A.; Deyholosb, M.; Fischera, R.; Galbraithb, D.W.; et al. A genomics approach towards salt stress tolerance. Plant Physiol. Biochem 2001, 39, 295–311. [Google Scholar]
- Gutha, L.R.; Reddy, A.R. Rice DREB1B promoter shows distinct stress-specific responses, and the overexpression of cDNA in tobacco confers improved abiotic and biotic stress tolerance. Plant Mol. Biol 2008, 68, 533–555. [Google Scholar]
- Shinozaki, K.; Yamaguchi-Shinozaki, K. Molecular responses to dehydration and low temperature: Differences and cross-talk between two stress signaling pathways. Curr. Opin. Plant Biol 2000, 3, 217–223. [Google Scholar]
- Liu, Q.; Kasuga, M.; Xu, Y.; Chen, N.; Ma, Q.; Li, F.; Chong, K. Overexpression of OsCOIN, a putative cold inducible zinc finger protein, increased tolerance to chilling, salt and drought, and enhanced proline level in rice. Planta 2007, 226, 1007–1016. [Google Scholar]
- Xie, Q.; Frugis, G.; Colgan, D.; Chua, N.-H. Arabidopsis NAC1 transduces auxin signal downstream of TIR1 to promote lateral root development. Gene Dev 2000, 14, 3024–3036. [Google Scholar]
- Jakobi, M.; Weisshaar, B.; Droge-Laser, W.; Vicente-Carbajosa, J.; Tiedemann, J.; Kroj, T.; Parcy, F. bZIP transcription factors in Arabidopsis. Trends Plant Sci 2002, 7, 106–111. [Google Scholar]
- Fujita, Y.; Fujita, M.; Satoh, R.; Muruyama, K.; Parvez, M.; Seki, M.; Hiratsu, K.; Ohme-Takagi, M.; Shinozaki, K.; Yamagushi-Shinozaki, K. AREB1 is a transcriptional activator of novel ABRE dependent ABA signaling that enhances drought stress tolerance in Arabidopsis. Plant Cell 2005, 17, 3470–3488. [Google Scholar]
- Ausio, J.; Abbott, D.W. The many tails of a tail: Carboxyl-terminal tail heterogeneity specializes histone h2a variants for defined chromatin functions. Biochemistry 2002, 4, 5945–5949. [Google Scholar]
- Ascenzi, R.; Gantt, J.S. A drought-stress-inducible histone gene in Arabidopsis thaliana is a member of a distinct class of plant linker histone variants. Plant Mol. Biol 1997, 34, 629–641. [Google Scholar]
- Herder, G.D.; de Keyser, A.; de Rycke, R.; Rombauts, S.; van de Velde, W.; Clemente, M.R.; Verplancke, C.; Mergaert, P.; Kondorosi, E.; Holsters, M.; Goomarchtig, S. Seven in absentia proteins affect plant growth and nodulation in Medicago truncatula. Plant Physiol 2008, 148, 369–382. [Google Scholar]
- Tang, C.; Drevon, J.J.; Jaillard, B.; Souche, G.; Hinsinger, P. Proton release of two genotypes of bean (Phaseolus vulgaris L.) as affected by N nutrition and P deficiency. Plant Soil 2004, 260, 59–68. [Google Scholar]
- Lynch, J.; White, J.W. Shoot nitrogen dynamics in tropical common bean. Crop Sci 1992, 32, 392–397. [Google Scholar]
- Hruz, T.; Laule, O.; Szabo, G.; Wessendorp, F.; Bleuler, S.; Oertle, L.; Widmayer, P.; Gruissem, W.; Zimmermann, P. Genevestigator V3: A reference expression database for the meta-analysis of transcriptomes. Adv. Bioinforma. 2008. [Google Scholar] [CrossRef]
- Jebara, M.; Aouani, M.E.; Payre, H.; Drevon, J.J. Nodule conductance varied among common bean (Phaseolus vulgaris L.) genotypes under P deficiency. J. Plant Physiol 2005, 162, 309–315. [Google Scholar]
- Carvalho, M.H.C.; Laffray, D.; Louguet, P. Comparison of the physiological responses of Phaseolus vulgaris and Vigna unguiculata cultivars when submitted to drought conditions. Environ. Exp. Botany 1998, 40, 197–207. [Google Scholar]
- Azevedo, R.A.; Alas, R.M.; Smith, R.J.; Lea, P.J. Response of antioxidant enzymes to transfer from elevated carbon dioxide to air and ozone fumigation, in the leaves and roots of wild-type and a catalase-deficient mutant of barley. Physiol. Plant 1998, 104, 280–292. [Google Scholar]
- Green, P. Documentation for PHRAP and CROSSMATCH (Version 0.990319). Available online: http://www.phrap.org/phredphrapconsed.html (accessed on 10 December 2009).
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res 1997, 25, 3389–3402. [Google Scholar]
- Telles, G.P.; Silva, F.R. Trimming and clustering sugarcane ESTs. Genetic Mol. Biol 2001, 24, 17–23. [Google Scholar]
- Huang, X.; Maddan, A. CAP3: A DNA sequence assembly program. Genome Res 1999, 9, 868–877. [Google Scholar]
- Rison, S.C.G.; Hodgman, T.C.; Thornton, J.M. Comparison of functional annotation schemes for genomes. Funct. Integr. Genomics 2000, 1, 56–69. [Google Scholar]
- Ruepp, A.; Zollner, A.; Maier, D.; Albermann, K.; Hani, J.; Mokrejs, M.; Tetko, I.; Guldener, U.; Mannhaupt, G.; Munsterkotter, M.; Mewes, H.W. The FunCat, a functional annotation scheme for systematic classification of proteins from whole genomes. Nucleic Acids Res. 2004, 32, 5539–5545. [Google Scholar]
- The ArabidopsisInformation Resource (TAIR). Available online: http://arabidopsis.org/Blast/index.jsp (accessed on 14 October 2012).
- Rozen, S.; Skaletsky, H.J. Primer3 on the WWW for General Users and for Biologist Programmers. In Bioinformatics Methods and Protocols: Methods in Molecular Biology, 1st ed; Krawetz, S., Misener, S., Eds.; Humana Press: Totowa, NJ, USA, 2000; pp. 365–386. [Google Scholar]
- Netprimer. Available online: http://www.premierbiosoft.com/netprimer/index.html (accessed on 15 October 2010).
- Borges, A.; Tsai, S.M.; Caldas, D.G.G. Validation of reference genes for RT-qPCR normalization in common Bean during biotic and abiotic stresses. Plant Cell Rep 2011, 5, 827–838. [Google Scholar]
- Ramakers, C.; Ruijter, J.M.; Deprez, R.H.L.; Moorman, A.F.M. Assumption-free analysis of quantitative real-time PCR data. Neurosci. Lett 2003, 339, 62–66. [Google Scholar]
- Usadel, B.; Poree, F.; Nagel, A.; Lohse, M.; Czedik-Eysenberg, A.; Stitt, M. A guide to using MapMan to visualize and compare Omics data in plants: A case study in the crop species, Maize. Plant Cell Environ 2009, 9, 1–19. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Access code in library | Number of reads | GI number | Description/species | E-value |
|---|---|---|---|---|
| Cellular metabolism (Energy/micro and macromolecules) | ||||
| Contig147 | 3 | |255579310| | pyruvate decarboxylase, putative (Ricinus communis) | 4 × 10−80 |
| Contig7 | 3 | |83283965| | malate dehydrogenase-like protein (Solanum tuberosum) | 1 × 10−171 |
| Contig23 | 3 | |255638912| | glyceraldehyde-3-phosphate dehydrogenase (Glycine max) | 1 × 10−119 |
| Contig28 | 3 | |255540625| | glutaredoxin-1, grx1, putative (Ricinus communis) | 2 × 10−40 |
| Biological processes | ||||
| Contig123 | 3 | |224094081| | spliceosomal complex, (Populus trichocarpa) | 3 × 10−35 |
| Contig171 | 3 | |75304713| | Methionine adenosyltransferase, (Phaseolus lunatus) | 1 × 10−83 |
| Contig79 | 4 | |156181612| | S-adenosylmethionine decarboxylase (Phaseolus vulgaris) | 3 × 10−25 |
| Contig127 | 4 | |75304713| | Methionine adenosyltransferase, (Phaseolus lunatus) | 5 × 10−90 |
| Abiotic stress response | ||||
| Contig74 | 4 | |42571665| | interferon-related developmental regulator family protein (Arabidopsis thaliana) | 6 × 10−53 |
| Contig105 | 3 | |192910730| | light-inducible protein ATLS1, (Elaeis guineensis) | 2 × 10−30 |
| Contig14 | 3 | |75708857| | group 3 late embryogenesis abundant protein, (Phaseolus vulgaris) | 6 × 10−23 |
| Contig61 | 3 | |806310| | proline-rich protein, (Glycine max) | 7 × 10−18 |
| Contig37 | 4 | |1732556| | LEA5 (Glycine max) | 3 × 10−34 |
| Contig97 | 4 | |1350522| | LEA protein (Picea glauca) | 3 × 10−27 |
| Contig24 | 9 | |1732556| | LEA5 (Glycine max) | 3 × 10−34 |
| Biotic stress response | ||||
| Contig3 | 3 | |184202203| | isoflavone synthase 1 (Vigna unguiculata) | 1 × 10−85 |
| Contig3 | 3 | |184202203| | isoflavone synthase 1 (Vigna unguiculata) | 1 × 10−85 |
| Contig17 | 9 | |130835| | PvPR2 (Phaseolus vulgaris) | 1 × 10−79 |
| Transport | ||||
| Contig164 | 3 | |61651606| | plastidic phosphate translocator-like protein1 (Mesembryanthemum crystallinum) | 1 × 10−61 |
| Contig80 | 4 | |255587991| | cation:cation antiporter (Ricinus communis) | 1 × 10−39 |
| Contig2 | 3 | |255552798| | ATP binding protein, putative (Ricinus communis) | 8 × 10−30 |
| Contig64 | 4 | |255637247| | calcium ion binding (Glycine max) | 2 × 10−38 |
| Structural organization (Membrane, cell wall, nucleus, nodulation and organelle) | ||||
| Contig142 | 3 | |255549412| | Vesicle-associated membrane protein, putative (Ricinus communis) | 8 × 10−31 |
| Contig137 | 3 | |146233385| | abscisic acid ABA receptor (Populus trichocarpa) | 1 × 10−24 |
| Contig148 | 3 | |194466205| | putative L24 ribosomal protein (Arachis hypogaea) | 2 × 10−23 |
| Contig11 | 5 | |255584772| | histone h2a, putative (Ricinus communis) | 2 × 10−27 |
| Contig19 | 3 | |57013900| | NitaMp027 (Nicotiana tabacum) | 6 × 10−33 |
| Contig83 | 4 | |30682545| | ARF3 (ADP-Ribosylation factor 3) (Arabidopsis thaliana) | 1 × 10−59 |
| Information pathways (processing of DNA, RNA and proteins/transposons) | ||||
| Contig154 | 3 | |187940303| | NAC domain protein (Glycine max) | 8 × 10−84 |
| Contig51 | 4 | |20138704| | eIF-5A (Manihot esculenta) | 7 × 10−40 |
| Contig52 | 4 | |255646048| | transferase activity (Glycine max) | 2 × 10−58 |
| Contig162 | 3 | |155212489| | N3 protein (Glycine max) | 1 × 10−47 |
| Unclassified | ||||
| Contig72 | 3 | |255626205| | unknown (Glycine max) | 3 × 10−78 |
| Contig87 | 3 | |255639776| | unknown (Glycine max) | 3 × 10−71 |
| Contig98 | 3 | |255647862| | unknown (Glycine max) | 8 × 10−55 |
| Contig145 | 3 | |255646578| | unknown (Glycine max) | 5 × 10−47 |
| Contig6 | 4 | |224101339| | predicted protein (Populus trichocarpa) | 5 × 10−30 |
| Contig64 | 4 | |255637247| | unknown (Glycine max) | 2 × 10−38 |
| Contig77 | 4 | |255637264| | unknown (Glycine max) | 2 × 10−10 |
| Contig82 | 6 | |255629893| | unknown (Glycine max) | 7 × 10−27 |
| Transcription factor family | GenBank ID | GI/description/species |
|---|---|---|
| DREB | |75749717| | |292630743| Fe-S cluster assembly protein DRE2 homolog (Glycine max) |
| |75748469| | |32480821| DREB (Glycine max) | |
| ERF | |75749028| | |190361165| ethylene-responsive element binding factor 4 (Glycine max) |
| |75749407| | |18643339| transcription factor EIL2 (Vigna radiate) | |
| bHLH | |75749257| | |66947630| coiled-coil-helix-coiled-coil-helix domain containing protein (Medicago truncatula) |
| bZIP | |75749123| | |145652341| transcription factor bZIP70 (Glycine max) |
| |75748580| | |223452524| leucine-rich repeat protein (Glycine max) | |
| |75748298| | |15148922| TGA-type basic leucine zipper protein (Phaseolus vulgaris) | |
| |75748883| | |255558466| F-box/LRR-repeat protein, putative (Ricinus communis) | |
| NAC | |75749297| | |224088037| NAC domain protein, IPR003441 (Populus trichocarpa) |
| |75748424| | |62546189| NAC4 protein (Glycine max) | |
| |75749318| | |224088037| NAC domain protein (Populus trichocarpa) | |
| |75748418| | |187940303| NAC domain protein (Glycine max) | |
| MYB | |75748729| | |110931684| MYB transcription factor MYB185 (Glycine max) |
| GATA—factors | |75748743| | |255572876| GATA transcription factor, putative (Ricinus communis) |
| WRKY family | |75748775| | |151934195| WRKY36 (Glycine max) |
| Ubiquitous factors TFIIA e Sp1 | |75748702| | |255566898| transcription initiation factor ia, putative (Ricinus communis) |
| IAA (auxin-responsive) | |75748737| | |255552973| Auxin-responsive protein IAA1, putative (Ricinus communis) |
| |75748789| | |15226425| auxin-responsive family protein (Arabidopsis thaliana) | |
| GRAS | |75748648| | |224106445| GRAS family transcription factor (Populus trichocarpa) |
| |75749650| | |224106445| GRAS family transcription factor (Populus trichocarpa) | |
| Heteromeric Factors | |75748712| | |193237557| transcription factor CCAAT (Lotus japonicas) |
| eIF2—alpha family | |75748325| | |255544025| translation initiation factor eif-2b (Ricinus communis) |
| |75748617| | |20138704| Eukaryotic translation initiation factor 5A (Manihot esculenta) | |
| Zinc finger | |75749674| | |161087182| C2-H2 zinc finger protein (Glycine max) |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Recchia, G.H.; Caldas, D.G.G.; Beraldo, A.L.A.; Da Silva, M.J.; Tsai, S.M. Transcriptional Analysis of Drought-Induced Genes in the Roots of a Tolerant Genotype of the Common Bean (Phaseolus vulgaris L.). Int. J. Mol. Sci. 2013, 14, 7155-7179. https://doi.org/10.3390/ijms14047155
Recchia GH, Caldas DGG, Beraldo ALA, Da Silva MJ, Tsai SM. Transcriptional Analysis of Drought-Induced Genes in the Roots of a Tolerant Genotype of the Common Bean (Phaseolus vulgaris L.). International Journal of Molecular Sciences. 2013; 14(4):7155-7179. https://doi.org/10.3390/ijms14047155
Chicago/Turabian StyleRecchia, Gustavo Henrique, Danielle Gregorio Gomes Caldas, Ana Luiza Ahern Beraldo, Márcio José Da Silva, and Siu Mui Tsai. 2013. "Transcriptional Analysis of Drought-Induced Genes in the Roots of a Tolerant Genotype of the Common Bean (Phaseolus vulgaris L.)" International Journal of Molecular Sciences 14, no. 4: 7155-7179. https://doi.org/10.3390/ijms14047155