Kinetic Studies that Evaluate the Solvolytic Mechanisms of Allyl and Vinyl Chloroformate Esters

Abstract
:1. Introduction
2. Results and Discussion
3. Experimental Section
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Sartori, M. The War Gases. In Chemistry and Analysis; D. Van Nostrand Co., Inc.: New York, NY, USA, 1939. [Google Scholar]
- Wellings, R.E. Poison war gases. School Sci. Math 1942, 42, 331–339. [Google Scholar]
- Trumpener, U. The road to Ypres: The beginnings of gas warfare in World War I. J. Mod. Hist 1975, 47, 460–480. [Google Scholar]
- Hazardous Materials Advisory Committee, United States Environmental Agency Science Advisory Board, Herbicide Report. Chemistry and Analysis. Environmental Effects. Agricultural and Other Applied Uses; Washington DC, USA, May 1974.
- Kevill, D.N. Chloroformate Esters and Related Compounds. In Acyl Halides; Patai, S., Ed.; John Wiley & Sons, Ltd: Chichester, UK, 1972. [Google Scholar]
- Parrish, J.P.; Salvatore, R.N.; Jung, K.W. Perspectives of alkyl carbonates in organic synthesis. Tetrahedron 2000, 56, 8207–8237. [Google Scholar]
- Banerjee, S.S.; Aher, N.; Patel, R.; Khandare, J. Poly(ethylene glycol)-prodrug conjugates: Concepts, design, and application. J. Drug Deliv. 2012, 2012. [Google Scholar] [CrossRef]
- Winstein, S.; Grunwald, E.; Jones, H.W. The correlation of solvolyses rates and the classification of solvolysis reactions into mechanistic categories. J. Am. Chem. Soc 1951, 73, 2700–2707, , and references there in.. [Google Scholar]
- Bentley, T.W.; Garley, M.S. Correlations and predictions of solvent effects on reactivity: Some limitations of multi-parameter equations and comparisons with similarity models based on one solvent parameter. J. Phys. Org. Chem 2006, 19, 341–349. [Google Scholar]
- Kevill, D.N.; D’Souza, M.J. Sixty years of the Grunwald-Winstein equation: Development and recent applications. J. Chem. Res 2008, 2008, 61–66, , and references therein.. [Google Scholar]
- Bentley, T.W.; Carter, G.E. The SN2-SN1 spectrum. 4. Mechanism for solvolyses of tert-butyl chloride: A revised Y scale of solvent ionizing power based on solvolyses of 1-adamantyl chloride. J. Am. Chem. Soc 1982, 104, 5741–5747. [Google Scholar]
- Bentley, T.W.; Llewellyn, G. Yx scales of solvent ionizing power. Prog. Phys. Org. Chem 1990, 17, 121–158. [Google Scholar]
- Kevill, D.N.; D’Souza, M.J. Additional YClvalues and correlation of the specific rates of solvolysis of tert-butyl chloride in terms of NTand YClscales. J. Chem. Res. Synop 1993, 174–175. [Google Scholar]
- Lomas, J.S.; D’Souza, M.J.; Kevill, D.N. Extremely large acceleration of the solvolysis of 1-adamantyl chloride upon incorporation of a spiro adamantane substituent: Solvolysis of 1-chlorospiro[adamantane-2, 2′-adamantane]. J. Am. Chem. Soc 1995, 117, 5891–5892. [Google Scholar]
- Kevill, D.N.; Ryu, Z.H. Additional solvent ionizing power values for binary water-1,1,1,3,3,3,-hexafluoro-2-propanol solvents. Int. J. Mol. Sci 2006, 7, 451–455. [Google Scholar]
- Schadt, F.L.; Bentley, T.W.; Schleyer, P.R. The SN2-SN1 spectrum. 2. Quantitative treatments of nucleophilic solvent assistance. A scale of solvent nucleophilicities. J. Am. Chem. Soc 1976, 98, 7667–7674. [Google Scholar]
- Kevill, D.N.; Anderson, S.W. An improved scale of solvent nucleophilicity based on the solvolysis of the S-methyldibenzothiophenium Ion. J. Org. Chem 1991, 56, 1845–1850. [Google Scholar]
- Kevill, D.N. Development and Uses of Scales of Solvent Nucleophilicity. In Advances in Quantitative Structure-Property Relationships; Charton, M., Ed.; JAI Press: Greenwich, CT, USA, 1996; Volume 1, pp. 81–115. [Google Scholar]
- Queen, A. Kinetics of the hydrolysis of acyl chlorides in pure water. Can. J. Chem. 1967, 45, 1619–1629. [Google Scholar]
- Butler, A.R.; Robertson, I.H.; Bacaloglu, R. Kinetics and mechanism of the base-catalysed hydrolysis of some substituted phenyl chloroformates. J. Chem. Soc. Perkin Trans 2 1974, 1733–1736. [Google Scholar]
- Yew, K.H.; Koh, H.J.; Lee, H.W.; Lee, I. Nucleophilic substitution reactions of phenyl chloroformate. J. Chem. Soc. Perkin Trans 2 1995, 2263–2268. [Google Scholar]
- Koo, I.S.; Yang, K.; Kang, K.; Oh, H.K.; Lee, I. Stoichiometric solvation effects. Product-rate correlation for the solvolyses of phenyl chloroformate in alcohol-water mixtures. Bull. Korean Chem. Soc 1996, 17, 520–524. [Google Scholar]
- Kevill, D.N.; D’Souza, M.J. Correlation of the rates of solvolysis of phenyl chloroformate. J. Chem. Soc. Perkin Trans. 2 1997, 1721–1724. [Google Scholar]
- Koo, I.S.; Yang, K.; Koo, J.C.; Park, J.-K.; Lee, I. Stoichiometric solvation effects. Part 4. Product–rate correlations for solvolyses of p-methoxyphenyl chloroformate in alcohol–water mixtures. Bull. Korean Chem. Soc 1997, 18, 1017–1021. [Google Scholar]
- Koo, I.S.; Yang, K.; Kang, K.; Lee, I.; Bentley, T.W. Stoichiometric solvation effects. Part 3. Product–rate correlations for solvolyses of p-nitrophenyl chloroformate in alcohol–water mixtures. J. Chem. Soc. Perkin Trans 2 1998, 1179–1183. [Google Scholar]
- Koo, I.S.; Yang, K.; Kang, K.; Lee, I. Transition-state variation in the solvolyses of para-substituted phenyl chloroformates in alcohol-water mixtures. Bull. Korean Chem. Soc 1998, 19, 968–973. [Google Scholar]
- Kevill, D.N.; Koyoshi, F.; D’Souza, M.J. Correlation of the specific rates of solvolysis of aromatic carbamoyl chlorides, chloroformates, chlorothionoformates, and chlorodithioformates revisited. Int. J. Mol. Sci 2007, 8, 346–352. [Google Scholar]
- D’Souza, M.J.; Reed, D.; Koyoshi, F.; Kevill, D.N. Consideration of the factors influencing the specific rates of solvolysis of p-methoxyphenyl chloroformate. Int. J. Mol. Sci 2007, 8, 788–796. [Google Scholar]
- Bentley, T.W. Structural effects on the solvolytic reactivity of carboxylic and sulfonic acid chlorides. Comparisons with gas-phase data for cation formation. J. Org. Chem 2008, 73, 6251–6257. [Google Scholar]
- D’Souza, M.J.; Shuman, K.E.; Carter, S.E.; Kevill, D.N. Extended Grunwald-Winstein analysis—LFER used to gauge solvent effects in p-nitrophenyl chloroformate solvolysis. Int. J. Mol. Sci 2008, 9, 2231–2242. [Google Scholar]
- D’Souza, M.J.; Knapp, J.A.; Fernandez-Bueno, G.A.; Kevill, D.N. Use of LFERs to elucidate the mechanisms of reaction of a γ-methyl-β-alkynyl and an ortho-substituted aryl chloroformate ester. Int. J. Mol. Sci 2012, 13, 665–682. [Google Scholar]
- Koo, I.S.; Lee, J.S.; Yang, K.; Kang, K.; Lee, I. The studies on substituent and kinetic solvent isotope effect in solvolyses of phenyl chloroformates. Bull. Korean Chem. Soc 1999, 20, 573–576. [Google Scholar]
- Bentley, T.W.; Harris, H.C.; Ryu, Z.-H.; Lim, G.T.; Sung, D.D.; Szajda, S.R. Mechanisms of solvolyses of acid chlorides and chloroformates. Chloroacetyl and phenylacetyl chloride as similarity models. J. Org. Chem 2005, 70, 8963–8970. [Google Scholar]
- Castro, E.A.; Ruiz, M.G.; Salinas, S.; Santos, J.G. Kinetics and mechanism of the aminolysis of phenyl and 4-nitrophenyl chloroformates in aqueous solution. J. Org. Chem 1999, 64, 4817–4820. [Google Scholar]
- Castro, E.A.; Ruiz, M.G.; Santos, J.G. Structure-reactivity correlations in the aminolysis of aryl chloroformates. Int. J. Chem. Kinet. 2001, 281–287. [Google Scholar]
- Crugerias, J.; Leis, J.R.; Ríos, A. Micellar effects on the spontaneous hydrolysis of phenyl chloroformate. J. Chem. Educ 2001, 78, 1538–1540. [Google Scholar]
- Muňoz, M.; Rodríguez, A.; Graciani, M.M.; Moyá, M.L. Micellar medium effects on the hydrolysis of phenyl chloroformate in ionic, zwitterionic, nonionic, and mixed micellar solutions. Int. J. Chem. Kinet 2002, 34, 445–451. [Google Scholar]
- Graciani, M.d.M.; Rodríguez, A.; Muňoz, M.; Moyá, M. Water-ethylene glycol alkyltrimethylammonium bromide micellar solutions as reaction media: Study of spontaneous hydrolysis of phenyl chloroformate. Langmuir 2003, 19, 8685–8691. [Google Scholar]
- Moon, D.H.; Seong, M.H.; Kyong, J.B.; Lee, Y.; Lee, Y.-W. Correlation of the rates of solvolysis of 1- and 2-naphthyl chloroformates using the extended Grunwald-Winstein equation. Bull. Korean Chem. Soc 2011, 32, 2413–2417. [Google Scholar]
- Kevill, D.N.; Kim, J.C.; Kyong, J.B. Correlation of the rates of solvolysis of methyl chloroformate with solvent properties. J. Chem. Res. Synop. 1999, 150–151. [Google Scholar]
- Kevill, D.N.; D’Souza, M.J. Concerning the two reaction channels for the solvolyses of ethyl chloroformate and ethyl chlorothioformate. J. Org. Chem 1998, 63, 2120–2124. [Google Scholar]
- Kevill, D.N.; D’Souza, M.J. Correlation of the Rates of Solvolyses of n-Octyl Fluoroformate and a Comparison with n-Octyl Chloroformate Solvolyses. J. Chem. Soc. Perkin Trans. 2 2002, 240–243. [Google Scholar]
- Kyong, J.B.; Won, H.; Kevill, D.N. Application of the extended Grunwald-Winstein equation to solvolyses of n-propyl chloroformate. Int. J. Mol. Sci 2005, 6, 87–96. [Google Scholar]
- D’Souza, M.J.; McAneny, M.J.; Kevill, D.N.; Kyong, J.B.; Choi, S.H. Kinetic evaluation of the solvolysis of isobutyl chloro- and chlorothioformate esters. Beilstein J. Org. Chem 2011, 7, 543–552. [Google Scholar]
- D’Souza, M.J.; Carter, S.E.; Kevill, D.N. Correlation of the rates of solvolysis of neopentyl chloroformate—A recommended protecting agent. Int. J. Mol. Sci 2011, 12, 1161–1174. [Google Scholar]
- Seong, M.H.; Choi, S.H.; Lee, Y.W.; Kyong, J.B.; Kim, D.K.; Kevill, D.N. Correlation of the rates of solvolysis of methyl fluoroformate using the extended Grunwald-Winstein equation. Bull. Korean Chem. Soc 2009, 30, 2408–2412. [Google Scholar]
- Seong, M.H.; Kyong, J.B.; Kim, D.K.; Kevill, D.N. Application of the extended Grunwald-Winstein equation to solvolyses of n-propyl fluoroformate and a consideration of leaving group effects. Bull. Korean Chem. Soc 2008, 29, 1747–1751. [Google Scholar]
- Lee, S.H.; Rhu, C.J.; Kyong, J.B.; Kim, D.K.; Kevill, D.N. Correlation of the rates of solvolysis of isopropyl fluoroformate using the extended Grunwald-Winstein equation. Bull. Korean Chem. Soc 2007, 28, 657–661. [Google Scholar]
- Lee, Y.W.; Seong, M.H.; Kyong, J.B.; Kevill, D.N. Correlation of the specific rates of solvolysis of t-butyl fluoroformate using the extended Grunwald-Winstein equation. Bull. Korean Chem. Soc 2010, 31, 3366–3370. [Google Scholar]
- Lee, Y.; Park, K.-H.; Seong, M.H.; Kyong, J.B.; Kevill, D.N. Correlation of the rates of solvolysis of i-butyl fluoroformate and a consideration of leaving group effects. Int. J. Mol. Sci 2012, 12, 7806–7817. [Google Scholar]
- Kevill, D.N.; D’Souza, M.J. Correlation of the rates of solvolysis of benzoyl fluoride and a consideration of leaving-group effects. J. Org. Chem 2004, 69, 7044–7050. [Google Scholar]
- Bentley, T.W. Concurrent pathways to explain solvent and substituent effects for solvolyses of benzoyl chlorides in ethanol-trifluoroethanol mixtures. Arch. Org. Chem. 2012, 25–34. [Google Scholar]
- D’Souza, M.J.; Reed, D.N.; Erdman, K.J.; Kyong, J.B.; Kevill, D.N. Grunwald-Winstein analysis—Isopropyl chloroformate solvolysis revisited. Int. J. Mol. Sci 2009, 10, 862–879. [Google Scholar]
- Kyong, J.B.; Yoo, J.-S.; Kevill, D.N. Solvolysis-decomposition of 2-adamantyl chloroformate: Evidence for two reaction pathways. J. Org. Chem 2003, 68, 3425–3432. [Google Scholar]
- Kevill, D.N.; Kyong, J.B.; Weitl, F.L. Solvolysis-decomposition of 1-adamantyl chloroformate: Evidence for ion pair return in 1-adamantyl chloride solvolysis. J. Org. Chem 1990, 55, 4304–4311. [Google Scholar]
- Kyong, J.B.; Park, B.-C.; Kim, C.-B.; Kevill, D.N. Rate and product studies with benzyl and p-nitrobenzyl chloroformates under solvolytic conditions. J. Org. Chem 2000, 65, 8051–8058. [Google Scholar]
- D’Souza, M.J.; Shuman, K.E.; Omondi, A.O.; Kevill, D.N. Detailed analysis for the solvolysis of isopropenyl chloroformate. Eur. J. Chem 2011, 2, 130–135. [Google Scholar]
- D’Souza, M.J.; Darrington, A.M.; Kevill, D.N. A study of solvent effects in the solvolysis of propargyl chloroformate. ISRN Org. Chem 2011, 2011, 767141, :1–767141:6.. [Google Scholar]
- Hook, A.L; Chang, C.Y.; Yang, J.; Luckett, J.; Cockayne, A.; Atkinson, S.; Mei, Y.; Bayston, R.; Irvine, D.J.; Langer, R.; et al. Combinatorial discovery of polymers resistant to bacterial attachment. Nat. Biotechnol. 2012, 30, 868–875. [Google Scholar]
- Koh, H.J.; Kang, S.J. Rate and product studies on the solvolyses of allyl chloroformate. Bull. Korean Chem. Soc 2012, 33, 4117–4121. [Google Scholar]
- Li, C.J. Organic reactions in aqueous media with a focus on carbon-carbon bond formations: A decade update. Chem. Rev 2005, 105, 3095–3165. [Google Scholar]
- Kevill, D.N.; Rissman, T.J. Correlation of the rates of solvolysis of allyl and benzyl arenesulphonates. J. Chem. Soc. Perkin Trans 2 1984, 717–720. [Google Scholar]
- Kevill, D.N.; D'Souza, M.J. Correlation of the rates of solvolysis of benzoyl chloride and derivatives using extended forms of the Grunwald-Winstein equation. J. Phys. Org. Chem 2002, 15, 881–888. [Google Scholar]
- D’Souza, M.J.; Darrington, A.M.; Kevill, D.N. On the importance of the aromatic ring parameter in studies of the solvolyses of cinnamyl and cinnamoyl halides. Org. Chem. Int 2010, 2010, 13050621–13050629. [Google Scholar]
- Bentley, T.W.; Harris, H.C. Solvolyses of benzoyl chlorides in weakly nucleophilic media. Int. J. Mol. Sci 2011, 12, 4805–4818. [Google Scholar]
- Olah, G.; Mayr, H. Stable Carbocations. 198. Formation of allyl cations via protonation of alkenes in magic acid solution. Evidence of 1,2-alkyl shifts in the intermediate vinyl cations. J. Am. Chem. Soc 1976, 78, 7333–7340. [Google Scholar]
- Imhoff, M.A.; Summerville, R.H.; Schleyer, P.v.R.; Martinez, A.G.; Hanack, M.; Dueber, T.E.; Stang, P.J. Preparation and solvolysis of vinyl triflates. IV. Rearrangements involving simple vinyl cations generated by solvolysis. J. Am. Chem. Soc 1970, 92, 3802–3804. [Google Scholar]
- Summerville, R.H.; Senkler, C.A.; Schleyer, P.R.; Dueber, T.E.; Stang, P.J. Solvolysis of vinyl triflates. Effects of alkyl substituents, solvents, and added nucleophiles. J. Am. Chem. Soc 1974, 96, 1100–1110. [Google Scholar]
- Summerville, R.H.; Schleyer, P.R. Stereochemistry of vinyl cations and vinyl substitutions. J. Am. Chem. Soc 1974, 96, 1100–1120. [Google Scholar]
- Yates, K.; Mandrapilias, G. Vinyl cation intermediates in solvolytic and electrophilic reactions. 1. Solvolysis of α-arylvinyl derivatives. J. Org. Chem 1980, 45, 3892–3902. [Google Scholar]
- Jia, Z.S.; Ottosson, H.; Zeng, X.; Thibblin, A. The role of ion-molecule pairs in solvolysis reactions. Nucleophilic addition of water to a tertiary allylic carbocation. J. Org. Chem 2002, 67, 182–187. [Google Scholar]
- Okuyama, T. Solvolysis of vinyl iodonium salts. New insights into vinyl cation intermediates. Acc. Chem. Res 2002, 35, 12–18. [Google Scholar]
- Fujita, M.; Yamamoto, A.; Sugimura, T.; Okuyama, T. Solvolysis of chiral cyclohexylidenemethyl triflate. Evidence against a primary vinyl cation intermediate. J. Phys. Org. Chem 2002, 15, 550–555. [Google Scholar]
- D’Souza, M.J.; Ryu, Z.H.; Park, B.-C.; Kevill, D.N. Correlation of the rates of solvolysis of acetyl chloride and α-substituted derivatives. Can. J. Chem 2008, 86, 359–367. [Google Scholar]
- Kevill, D.N.; Casamassa, A.J.; D’Souza, M.J. Rates and product selectivities for the solvolyses of 4-(chloroformyl)morpholine. J. Chem. Res. (S) 1996, 472–473. [Google Scholar]
- Frost, A.A.; Pearson, R.G. Kinetics and Mechanism-a Study of Homogeneous Chemical Reactions, 2nd ed; Wiley: New York, NY, USA, 1961; pp. 49–50. [Google Scholar]
- Kevill, D.N.; Abduljaber, M.H. Correlation of the rates of solvolysis of cyclopropylcarbinyl and cyclobutyl bromides using the extended Grunwald-Winstein equation. J. Org. Chem 2000, 65, 2548–2554. [Google Scholar]
- D’Souza, M.J.; Dwyer, P.; Allison, B.E.; Miller, J.M.; Drohan, J. Wesley College ignites potential with undergraduate student research program. Counc. Undergrad Res 2011, 32, 41–45. [Google Scholar]
- D’Souza, M.J.; Wang, Q. Inter-institutional partnerships propel a successful undergraduate research program in chemistry. J. Coll. Teach. Learn 2012, 9, 245–252. [Google Scholar]




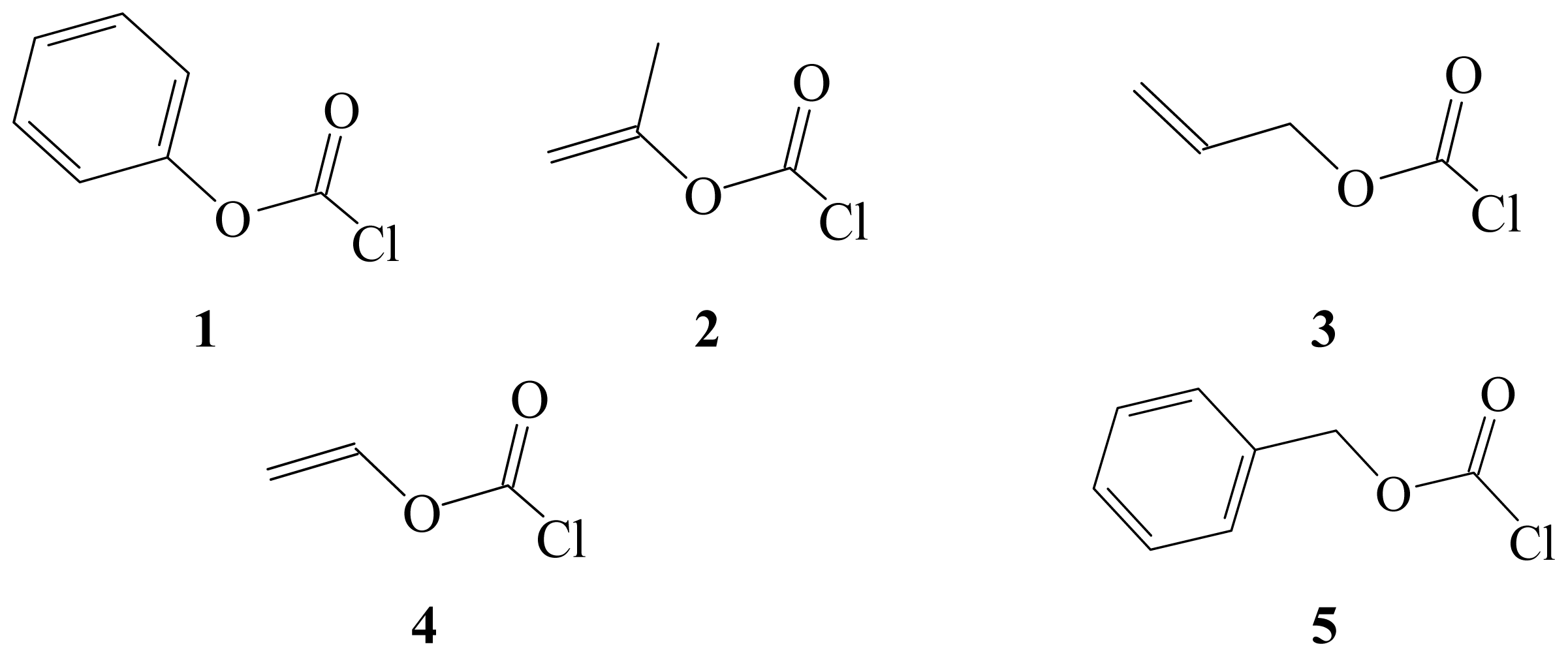{kind=link}
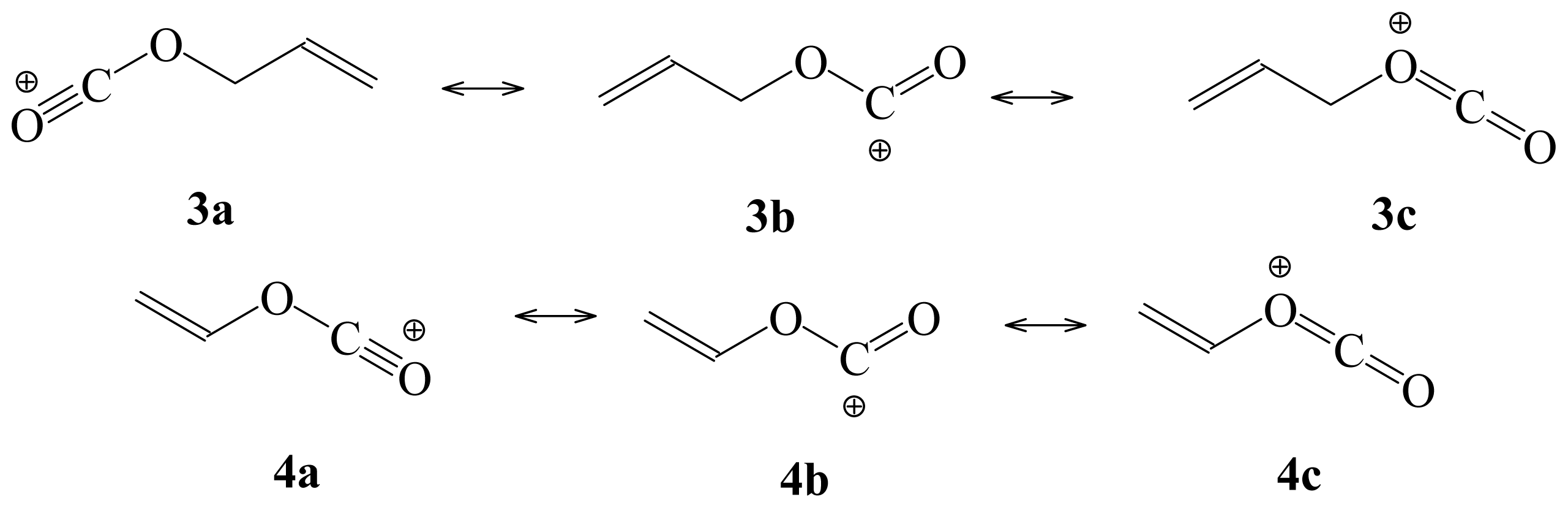{kind=link}
{kind=link}
{kind=link}
{kind=link}
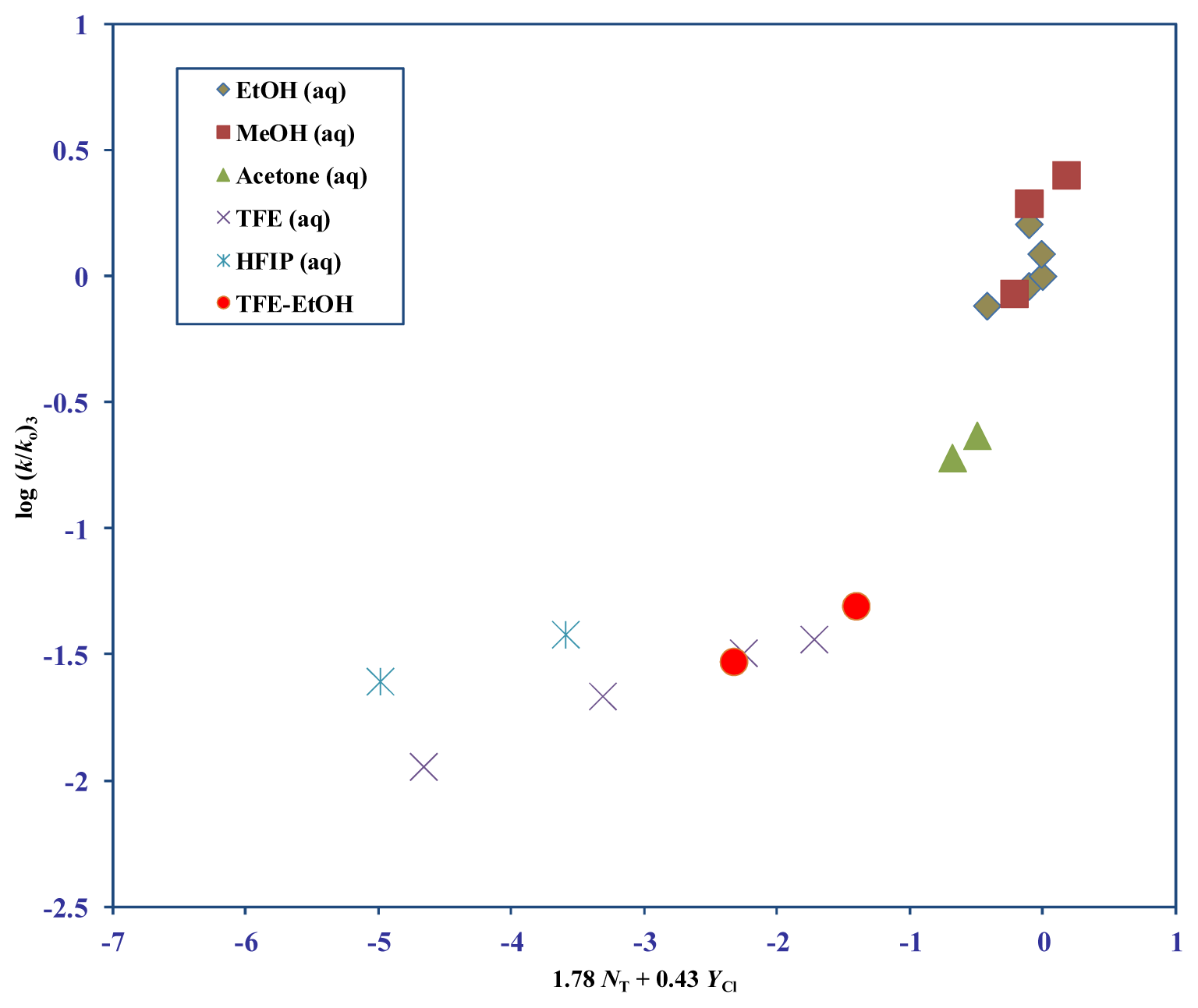
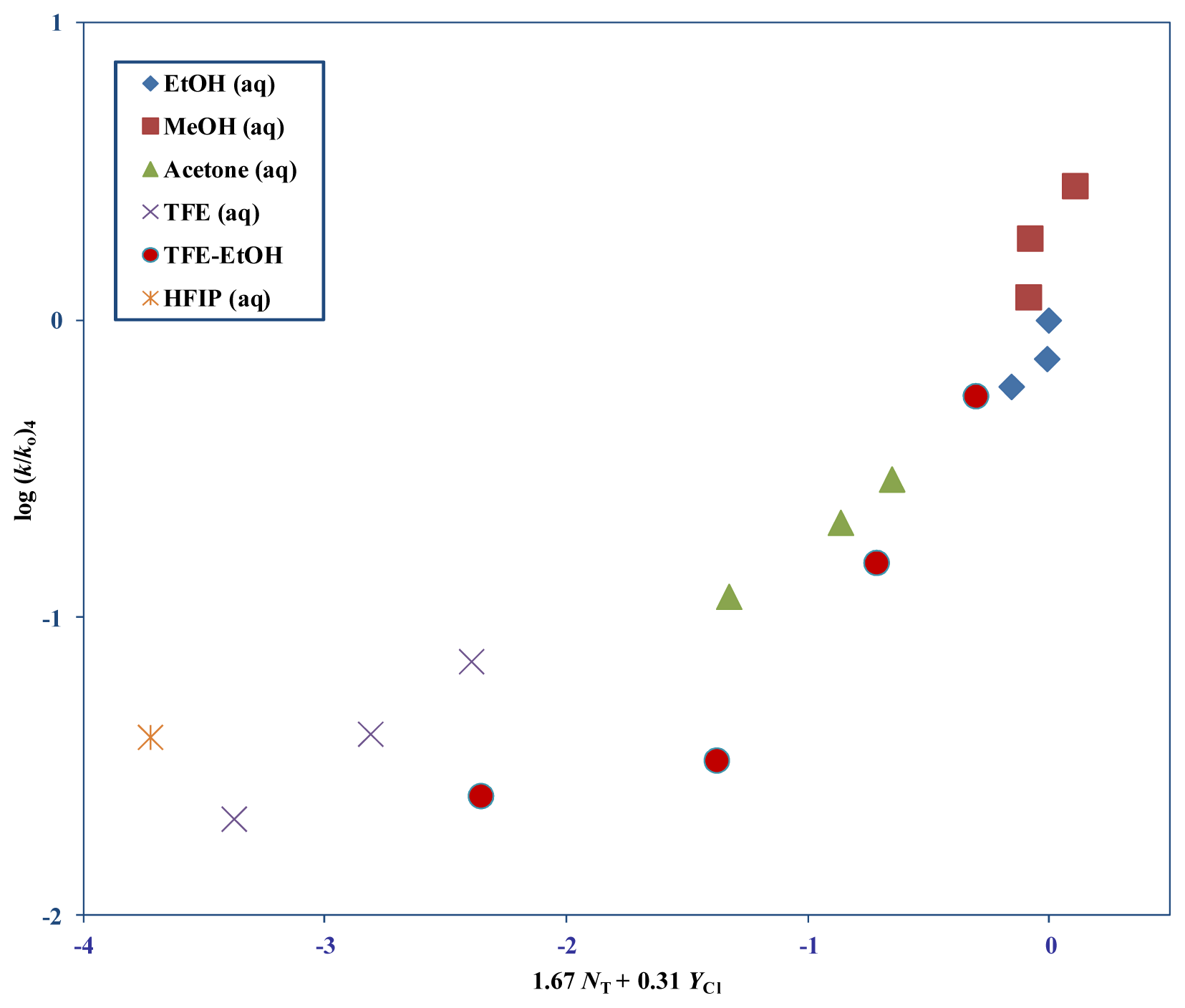
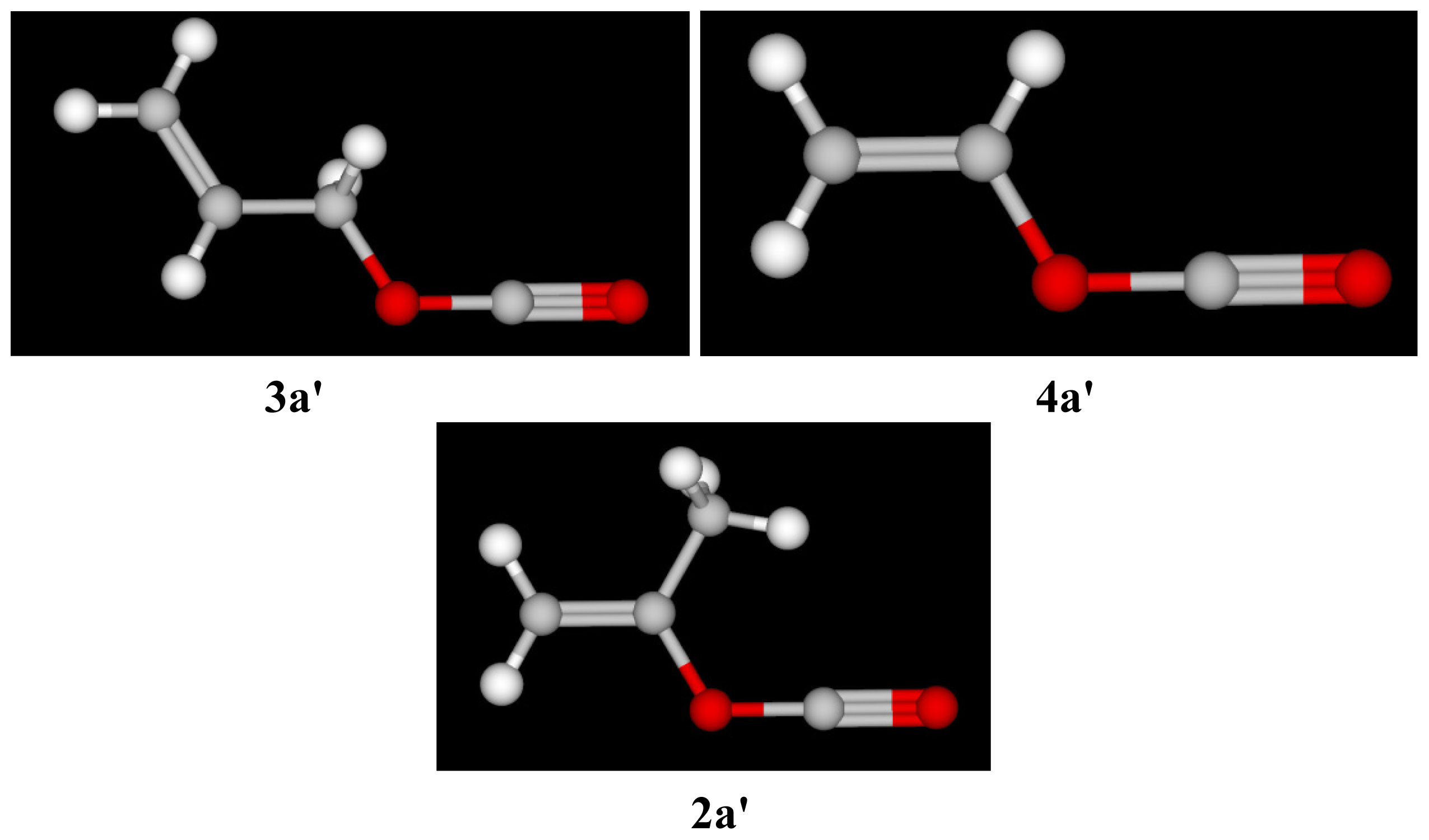
| Solvent (%) a | 1 b 105k, s−1 | 3 (T) 105k, s−1 c | 4 105k, s−1 c | 5 d 105k, s−1 | NTe | YClf |
|---|---|---|---|---|---|---|
| 100% EtOH | 260 | 11.1 ± 0.16 | 742 ± 1 | 5.16 | 0.37 | −2.50 |
| 90% EtOH | 389 | 13.4 ± 0.7 | 921 ± 3 | 12.9 | 0.16 | −0.90 |
| 80% EtOH | 503 | 14.7 ± 0.4 | 1252 ± 19 | 17.7 | 0.00 | 0.00 |
| 70% EtOH | 546 | 18.0 ± 0.6 | 21.5 | −0.20 | 0.78 | |
| 60% EtOH | 658 | 23.4 ± 0.9 | 25.6 | −0.38 | 1.38 | |
| 100% MeOH | 695 | 12.5 ± 0.7 | 1485 ± 13 | 18.8 | 0.17 | −1.20 |
| 90% MeOH | 1290 | 28.4 ± 1.5 | 2331 ± 42 | 38.4 | −0.01 | −0.20 |
| 80% MeOH | 1670 | 36.7 ± 2.0 | 3500 ± 55 | 55.4 | −0.06 | 0.67 |
| 60% MeOH | 2220 | 38.5 ± 0.8 | −0.54 | 2.07 | ||
| 90% Acetone | 23.8 | 146 ± 2 | −0.35 | −2.39 | ||
| 80% Acetone | 68.8 | 260 ± 4 | 2.13 | −0.37 | −0.80 | |
| 70% Acetone | 125 | 2.79 ± 0.14 | 365 ± 3 | 4.23 | −0.42 | 0.17 |
| 60% Acetone | 195 | 3.41 ± 0.14 | 7.62 | −0.52 | 1.00 | |
| 97% TFE (w/w) | 0.0570 | 0.166 ± 0.016 | 1.93 | −3.30 | 2.83 | |
| 90% TFE (w/w) | 1.15 | 0.317 ± 0.017 | 26.2 ± 0.3 (T) | 2.37 | −2.55 | 2.85 |
| 80% TFE (w/w) | 7.02 | 50.5 ± 1.2 | 3.44 | −2.19 | 2.90 | |
| 70% TFE (w/w) | 17.4 | 0.467 ± 0.021 | 88.6 ± 0.9 | 4.82 | −1.98 | 2.96 |
| 50% TFE (w/w) | 63.5 | 0.531 ± 0.022 | 9.39 | −1.73 | 3.16 | |
| 80T-20E | 2.43 | 0.434 ± 0.016 | 31.6 ± 0.1 (T) | 0.692 | −1.76 | 1.89 |
| 60T-40E | 17.5 | 0.723 ± 0.050 | 43.2 ± 0.1 (T) | 0.993 | −0.94 | 0.63 |
| 40T-60E | 57.7 | 178 ± 1 | 2.19 | −0.34 | −0.48 | |
| 20T-80E | 169 | 696 ± 1 | 3.90 | 0.08 | −1.42 | |
| 97% HFIP (w/w) | 14.8 × 10−4 | 1.80 ± 0.12 | 13.8 | −5.26 | 5.17 | |
| 90% HFIP (w/w) | 0.166 | 0.362 ± 0.023 | 11.5 | −3.84 | 4.41 | |
| 70% HFIP (w/w) | 10.5 | 0.553 ± 0.021 | 49.5 ± 0.2 | 11.3 | −2.94 | 3.83 |
| Substrate | na | lb | mb | cc | Rd | Fe |
|---|---|---|---|---|---|---|
| 3 f | 35 g | 0.98 ± 0.06 | 0.44 ± 0.03 | −0.04 ± 0.05 | 0.944 | 132 |
| 28 h | 1.43 ± 0.13 | 0.52 ± 0.03 | 0.10 ± 0.06 | 0.954 | 127 | |
| 7 i | 0.93 ± 0.12 | 0.66 ± 0.14 | −0.84 ± 0.30 | 0.974 | 36 | |
| 3 j | 12 k | 1.46 ± 0.19 | 0.37 ± 0.09 | 0.10 ± 0.08 | 0.943 | 37 |
| 12 l | 1.78 ± 0.18 | 0.43 ± 0.07 | 0.14 ± 0.06 | 0.965 | 61 | |
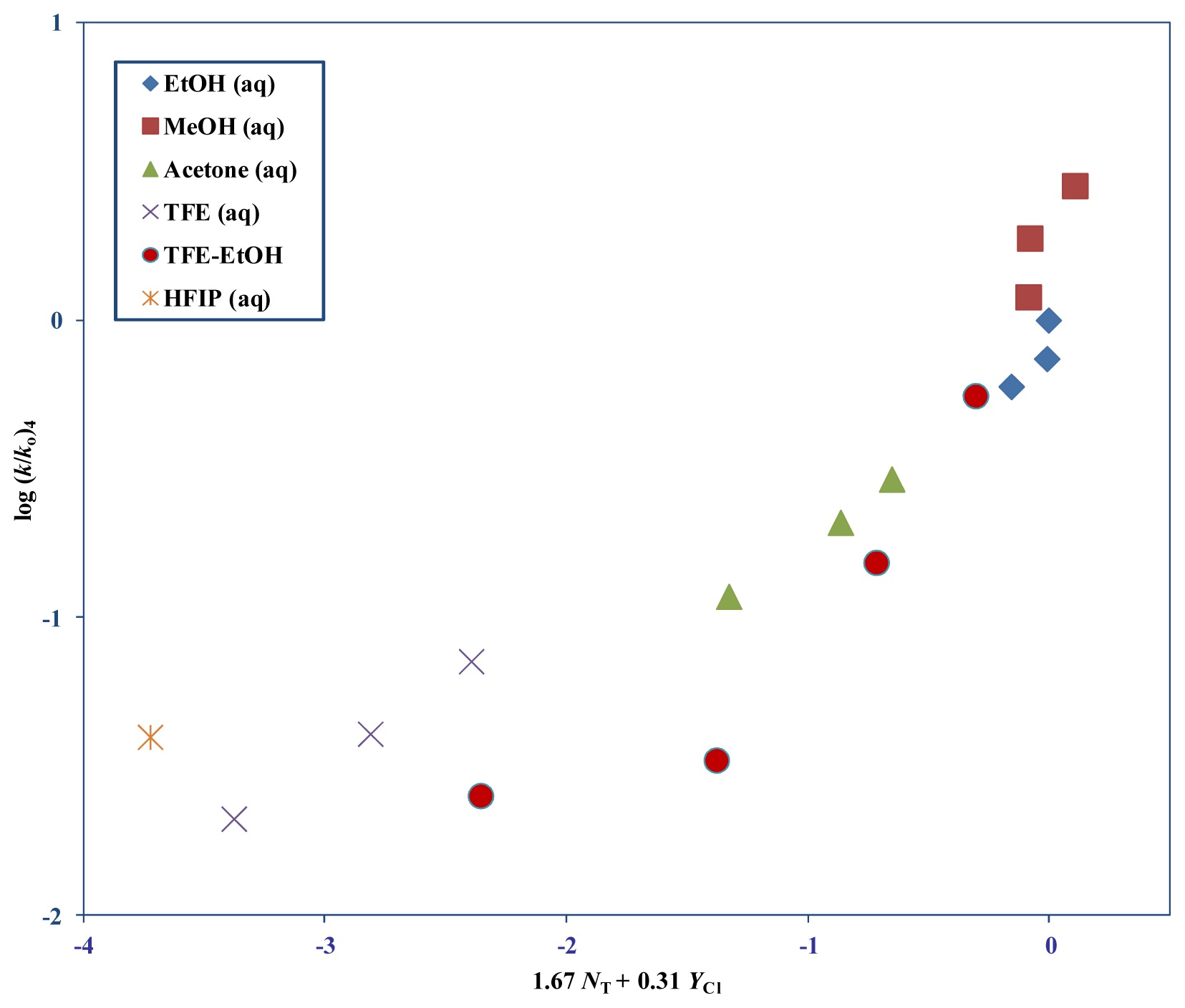
| 4 | 12 m | 1.67 ± 0.19 | 0.31 ± 0.07 | 0.10 ± 0.09 | 0.941 | 35 |
| 5 n | 0.80 ± 0.03 | 0.59± 0.01 | −1.31 ± 0.03 | 0.999 | 578 |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
D'Souza, M.J.; Givens, A.F.; Lorchak, P.A.; Greenwood, A.E.; Gottschall, S.L.; Carter, S.E.; Kevill, D.N. Kinetic Studies that Evaluate the Solvolytic Mechanisms of Allyl and Vinyl Chloroformate Esters. Int. J. Mol. Sci. 2013, 14, 7286-7301. https://doi.org/10.3390/ijms14047286
D'Souza MJ, Givens AF, Lorchak PA, Greenwood AE, Gottschall SL, Carter SE, Kevill DN. Kinetic Studies that Evaluate the Solvolytic Mechanisms of Allyl and Vinyl Chloroformate Esters. International Journal of Molecular Sciences. 2013; 14(4):7286-7301. https://doi.org/10.3390/ijms14047286
Chicago/Turabian StyleD'Souza, Malcolm J., Aaron F. Givens, Peter A. Lorchak, Abigail E. Greenwood, Stacey L. Gottschall, Shannon E. Carter, and Dennis N. Kevill. 2013. "Kinetic Studies that Evaluate the Solvolytic Mechanisms of Allyl and Vinyl Chloroformate Esters" International Journal of Molecular Sciences 14, no. 4: 7286-7301. https://doi.org/10.3390/ijms14047286