A Role for Protein Phosphatase 2A in Regulating p38 Mitogen Activated Protein Kinase Activation and Tumor Necrosis Factor-Alpha Expression during Influenza Virus Infection

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
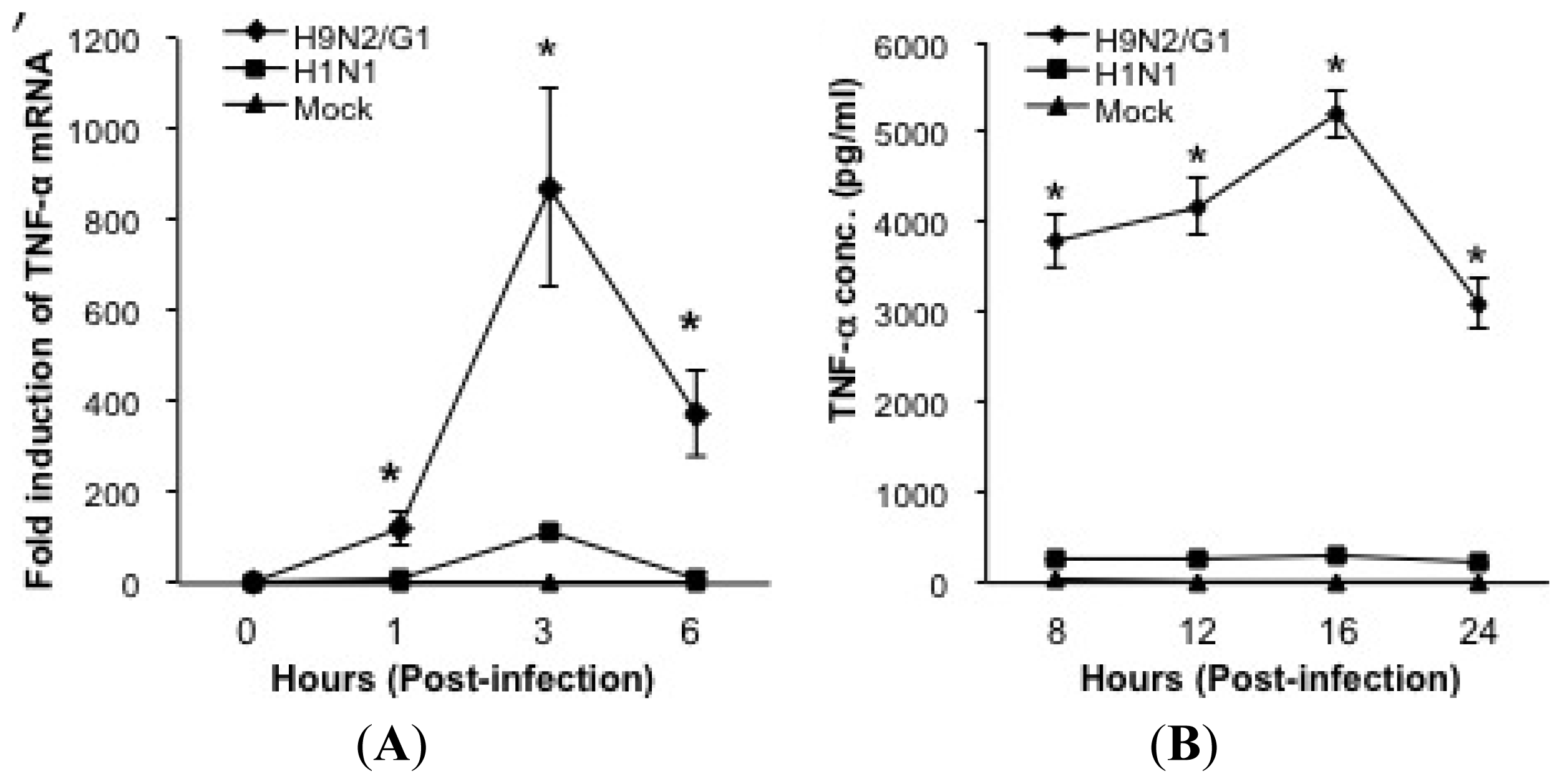
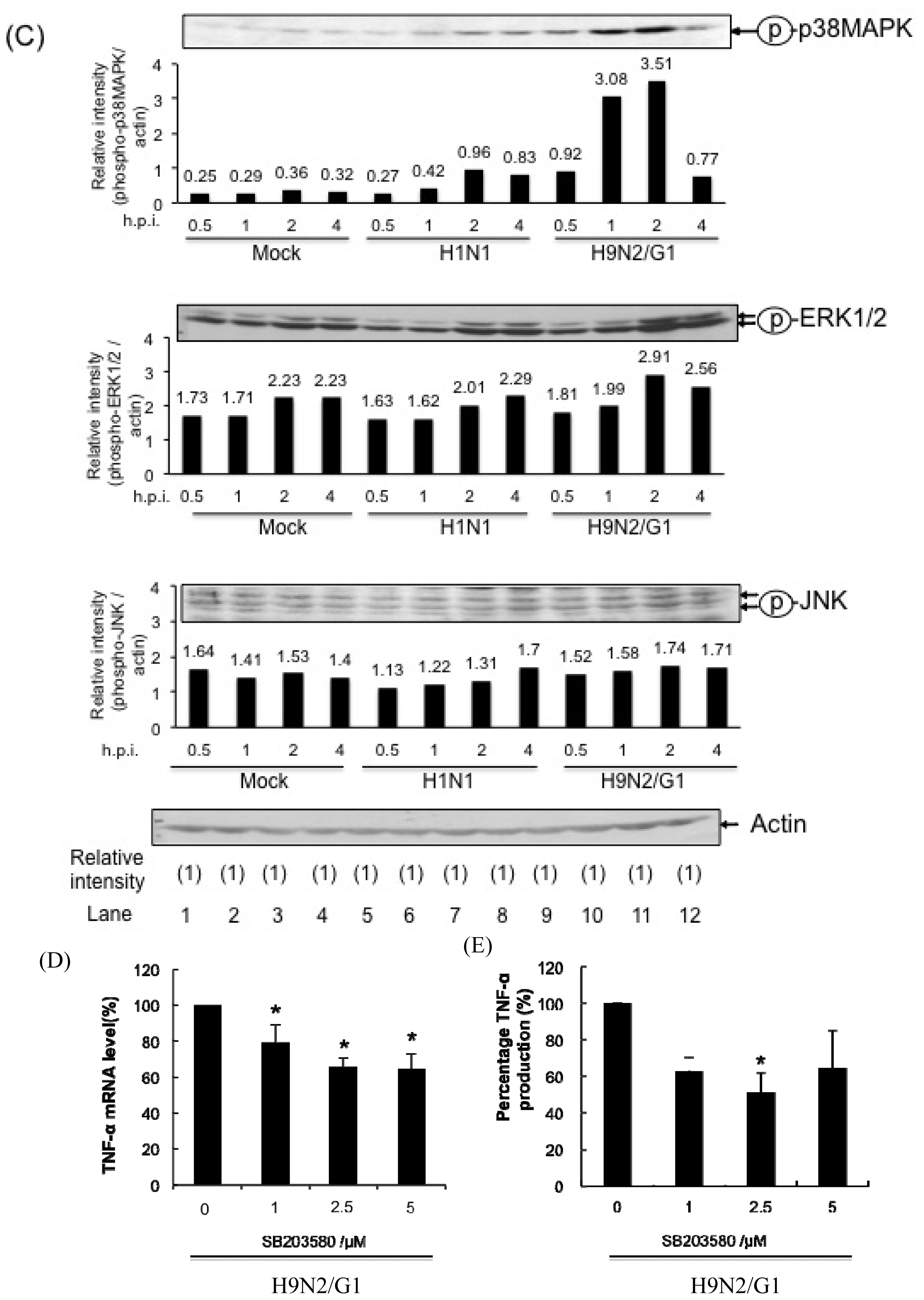
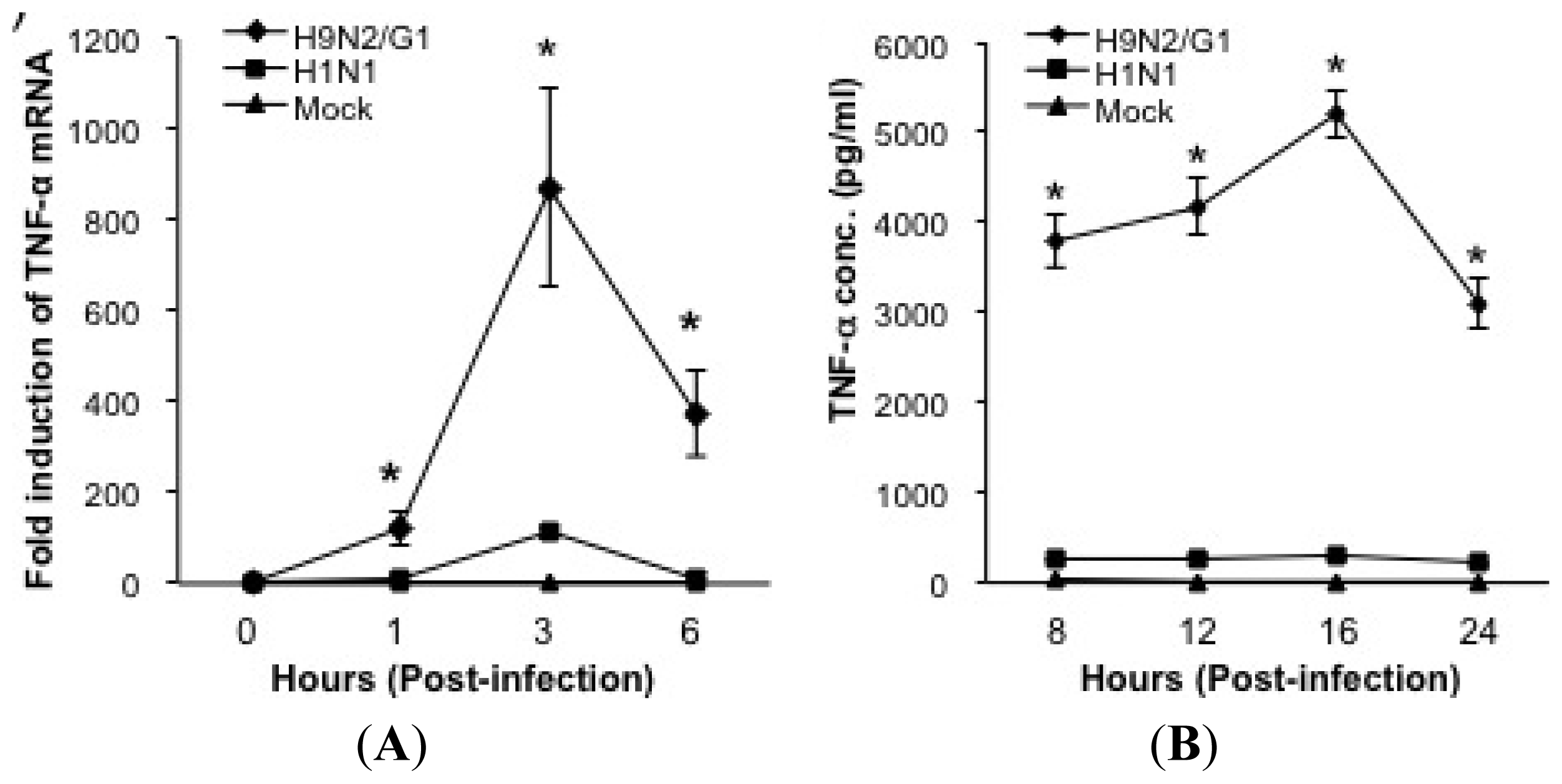
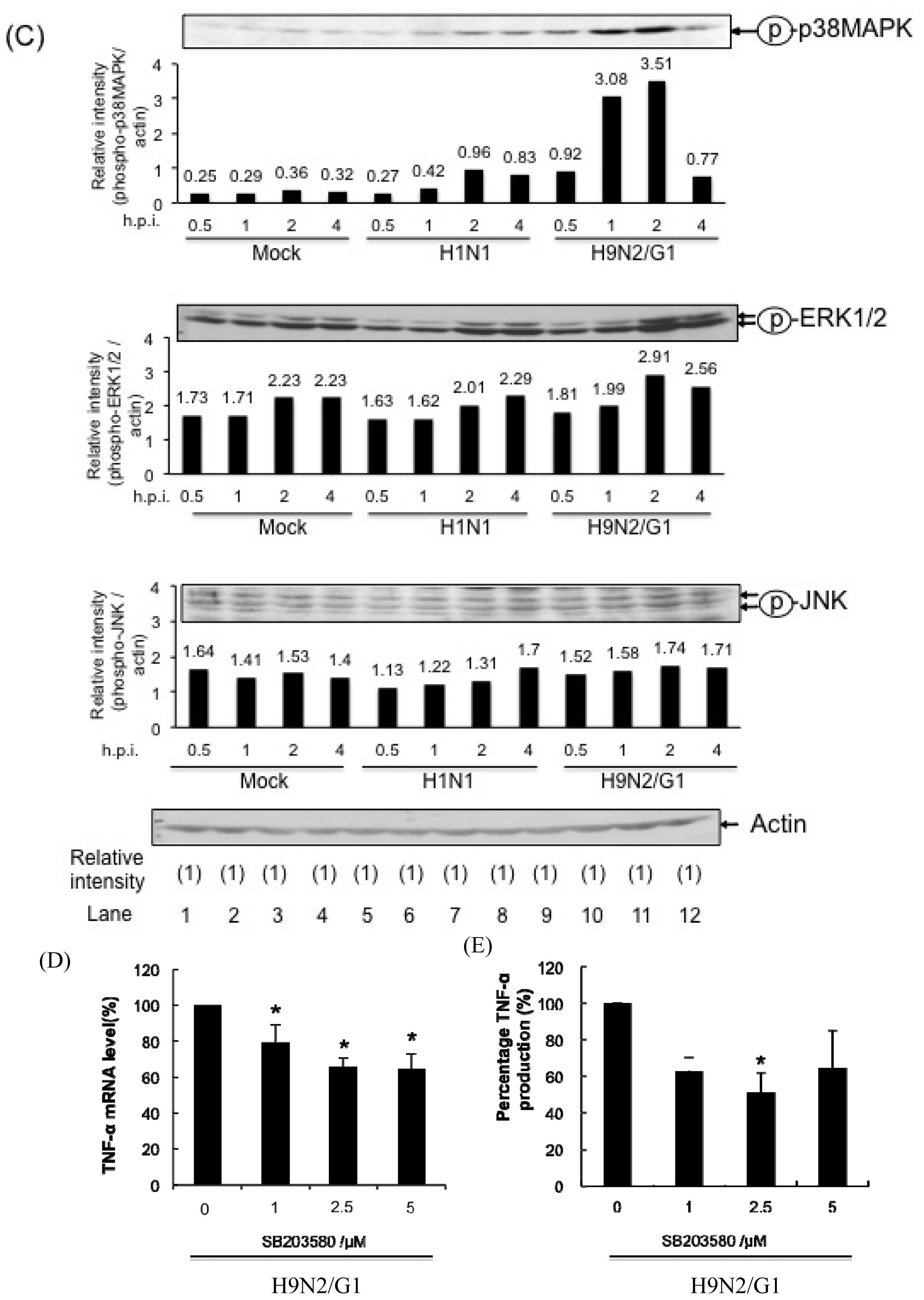
2.1. H9N2/G1 Hyperinduced TNF-alpha Expression through the Activation of p38MAPK in Primary Human Blood Macrophages
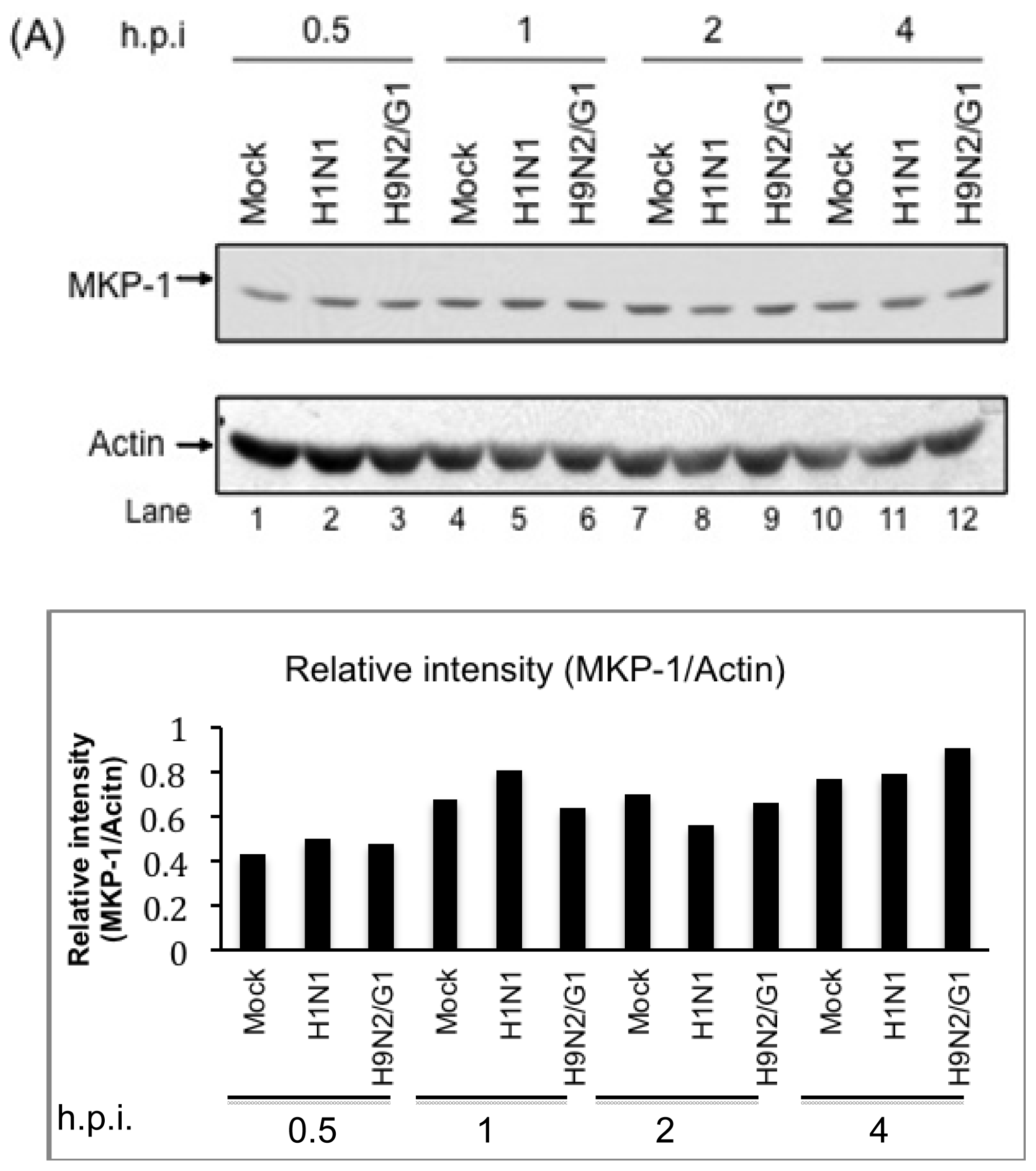
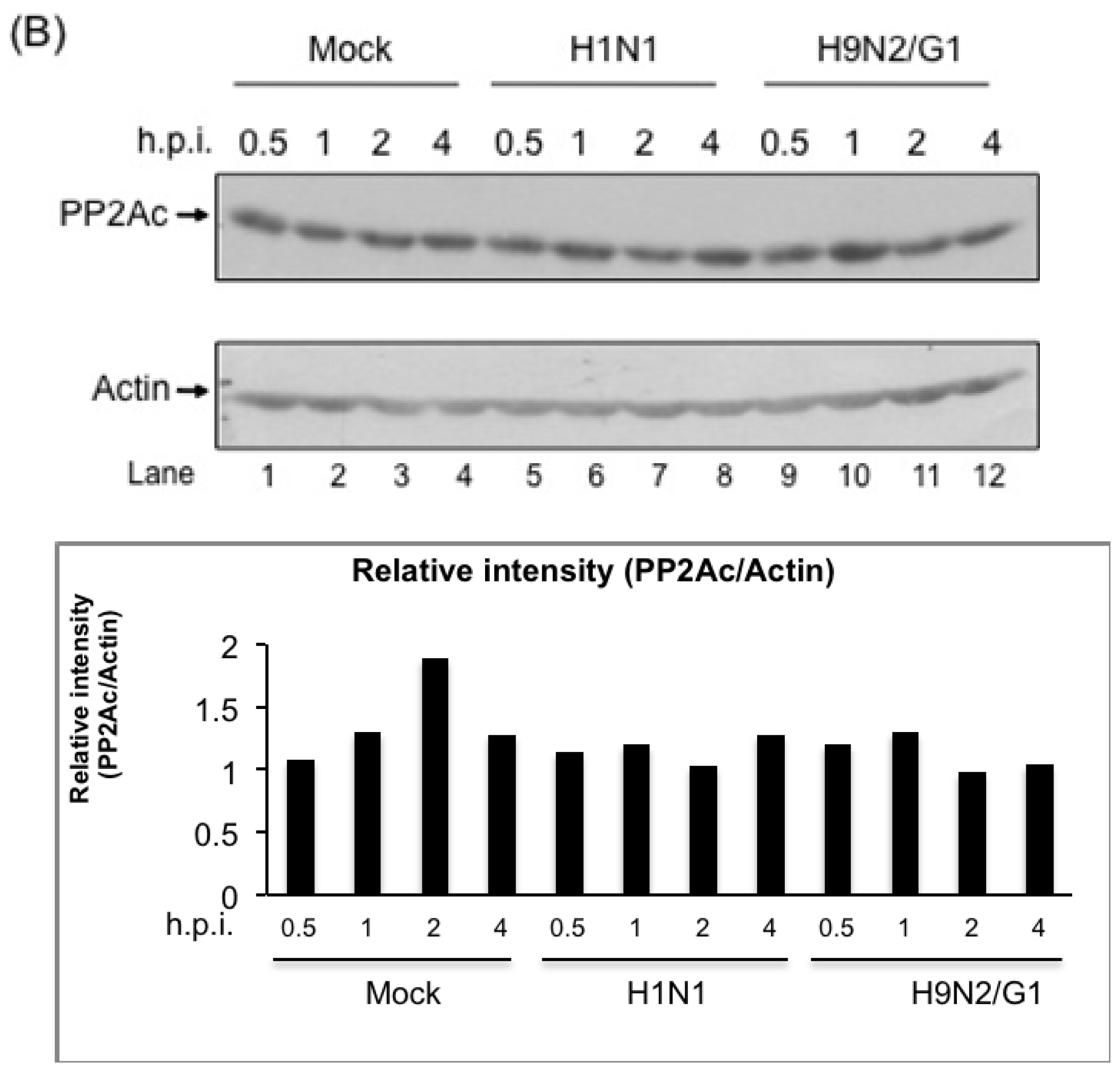
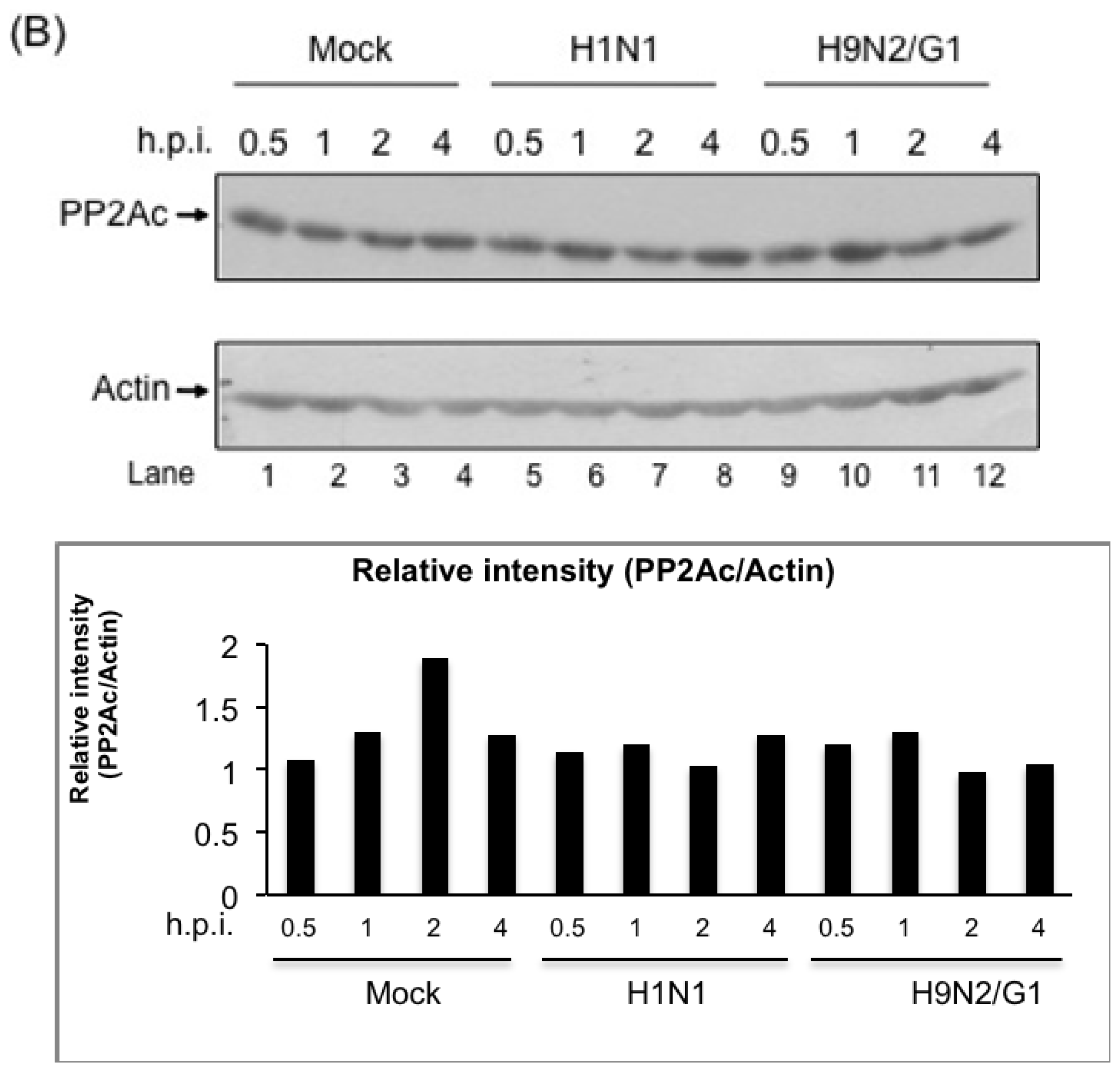
2.2. H9N2/G1 Virus Did Not Alter the Cellular Protein Levels of MKP-1 and PP2A Catalytic Subunit
2.3. H9N2/G1 Induced PP2A Activity in Primary Human Blood Macrophages
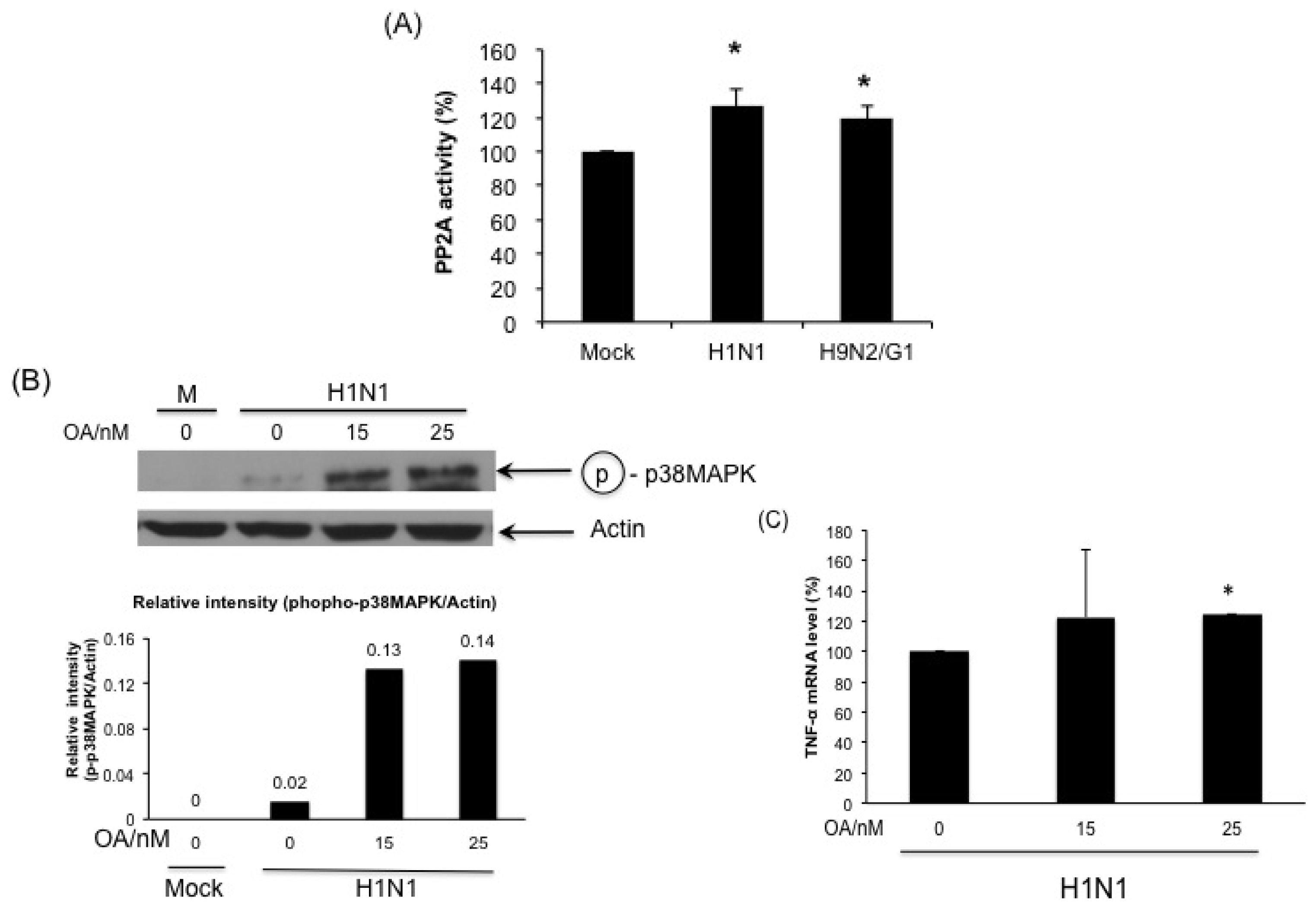
2.4. PP2A Is a Negative Regulator in H9N2/G1-Induced p38MAPK Activation and TNF-Alpha Expression
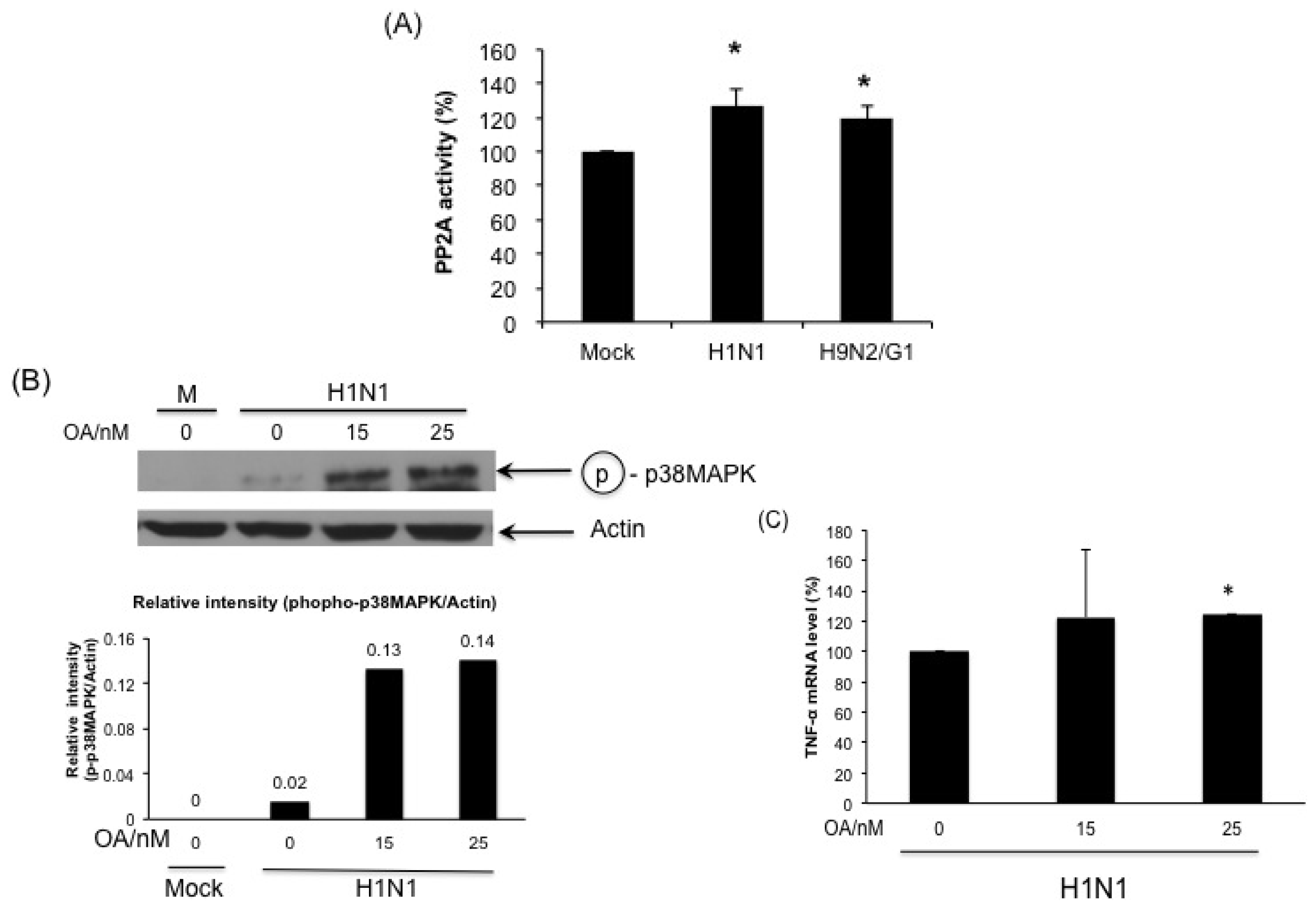
2.5. H1N1 also Induces PP2A Activation in Primary Human Blood Macrophages
3. Experimental Section
3.1. Reagents and Antibodies
3.2. Cells and Viruses
3.3. Virus Infection
3.4. Protein Extraction
3.5. Quantitative Reverse Transcription-PCR Analyses
3.6. Quantitative Analyses of TNF-Alpha by Enzyme-Linked Immunosorbent Assays (ELISA)
3.7. Protein Phosphatase 2A Activity Assay
3.8. Statistical Analysis
4. Conclusions
Supplementary Information
ijms-14-07327-s001.docxAcknowledgements
References
- World Health Organization (WHO). Cumulative number of confirmed human cases for avian influenza A (H5N1) reported to WHO, 2003–2013. Available online: http://www.who.int/influenza/human_animal_interface/EN_GIP_20130312CumulativeNumberH5N1cases.pdf (accessed on 12 March 2013).
- De Jong, M.D.; Simmons, C.P.; Thanh, T.T.; Hien, V.M.; Smith, G.J.; Chau, T.N.; Hoang, D.M.; Chau, N.V.; Khanh, T.H.; Dong, V.C.; et al. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat. Med 2006, 12, 1203–1207. [Google Scholar]
- Guan, Y.; Shortridge, K.F.; Krauss, S.; Webster, R.G. Molecular characterization of H9N2 influenza viruses: Were they the donors of the “internal” genes of H5N1 viruses in Hong Kong? Proc. Natl. Acad. Sci. USA 1999, 96, 9363–9367. [Google Scholar]
- Xu, K.M.; Li, K.S.; Smith, G.J.; Li, J.W.; Tai, H.; Zhang, J.X.; Webster, R.G.; Peiris, J.S.; Chen, H.; Guan, Y. Evolution and molecular epidemiology of H9N2 influenza A viruses from quail in southern China, 2000 to 2005. J. Virol 2007, 81, 2635–2645. [Google Scholar]
- Butt, K.M.; Smith, G.J.; Chen, H.; Zhang, L.J.; Leung, Y.H.; Xu, K.M.; Lim, W.; Webster, R.G.; Yuen, K.Y.; Peiris, J.S.; Guan, Y. Human infection with an avian H9N2 influenza A virus in Hong Kong in 2003. J. Clin. Microbiol 2005, 43, 5760–5767. [Google Scholar]
- Peiris, M.; Yuen, K.Y.; Leung, C.W.; Chan, K.H.; Ip, P.L.; Lai, R.W.; Orr, W.K.; Shortridge, K.F. Human infection with influenza H9N2. Lancet 1999, 354, 916–917. [Google Scholar]
- Lin, Y.P.; Shaw, M.; Gregory, V.; Cameron, K.; Lim, W.; Klimov, A.; Subbarao, K.; Guan, Y.; Krauss, S.; Shortridge, K.; et al. Avian-to-human transmission of H9N2 subtype influenza A viruses: Relationship between H9N2 and H5N1 human isolates. Proc. Natl. Acad. Sci. USA 2000, 97, 9654–9658. [Google Scholar]
- Cheng, V.C.; Chan, J.F.; Wen, X.; Wu, W.L.; Que, T.L.; Chen, H.; Chan, K.H.; Yuen, K.Y. Infection of immunocompromised patients by avian H9N2 influenza A virus. J. Infect 2011, 62, 394–399. [Google Scholar]
- Wan, H.; Sorrell, E.M.; Song, H.; Hossain, M.J.; Ramirez-Nieto, G.; Monne, I.; Stevens, J.; Cattoli, G.; Capua, I.; Chen, L.M.; et al. Replication and transmission of H9N2 influenza viruses in ferrets: Evaluation of pandemic potential. PLoS One 2008, 3, e2923. [Google Scholar]
- Lee, D.C.; Cheung, C.Y.; Law, A.H.; Mok, C.K.; Peiris, M.; Lau, A.S. p38 mitogen-activated protein kinase-dependent hyperinduction of tumor necrosis factor alpha expression in response to avian influenza virus H5N1. J. Virol 2005, 79, 10147–10154. [Google Scholar]
- Cheung, C.Y.; Poon, L.L.; Lau, A.S.; Luk, W.; Lau, Y.L.; Shortridge, K.F.; Gordon, S.; Guan, Y.; Peiris, J.S. Induction of proinflammatory cytokines in human macrophages by influenza A (H5N1) viruses: A mechanism for the unusual severity of human disease? Lancet 2002, 360, 1831–1837. [Google Scholar]
- Law, A.H.; Lee, D.C.; Yuen, K.Y.; Peiris, M.; Lau, A.S. Cellular response to influenza virus infection: A potential role for autophagy in CXCL10 and interferon-alpha induction. Cell. Mol. Immunol 2010, 7, 263–270. [Google Scholar]
- Zhao, Q.; Wang, X.; Nelin, L.D.; Yao, Y.; Matta, R.; Manson, M.E.; Baliga, R.S.; Meng, X.; Smith, C.V.; Bauer, J.A.; et al. MAP kinase phosphatase 1 controls innate immune responses and suppresses endotoxic shock. J. Exp. Med 2006, 203, 131–140. [Google Scholar]
- Hammer, M.; Mages, J.; Dietrich, H.; Servatius, A.; Howells, N.; Cato, A.C.; Lang, R. Dual specificity phosphatase 1 (DUSP1) regulates a subset of LPS-induced genes and protects mice from lethal endotoxin shock. J. Exp. Med 2006, 203, 15–20. [Google Scholar]
- Chi, H.; Barry, S.P.; Roth, R.J.; Wu, J.J.; Jones, E.A.; Bennett, A.M.; Flavell, R.A. Dynamic regulation of pro- and anti-inflammatory cytokines by MAPK phosphatase 1 (MKP-1) in innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 2274–2279. [Google Scholar]
- Sontag, E. Protein phosphatase 2A: The Trojan Horse of cellular signaling. Cell. Signal 2001, 13, 7–16. [Google Scholar]
- Alvarado-Kristensson, M.; Andersson, T. Protein phosphatase 2A regulates apoptosis in neutrophils by dephosphorylating both p38 MAPK and its substrate caspase 3. J. Biol. Chem 2005, 280, 6238–6244. [Google Scholar]
- Bae, D.; Ceryak, S. Raf-independent, PP2A-dependent MEK activation in response to ERK silencing. Biochem. Biophys. Res. Commun 2009, 385, 523–527. [Google Scholar]
- Zhao, B.; Sun, L.; Haas, M.; Denenberg, A.G.; Wong, H.R.; Shanley, T.P. PP2A regulates upstream members of the c-jun N-terminal kinase mitogen-activated protein kinase signaling pathway. Shock 2008, 29, 181–188. [Google Scholar]
- Guergnon, J.; Godet, A.N.; Galioot, A.; Falanga, P.B.; Colle, J.H.; Cayla, X.; Garcia, A. PP2A targeting by viral proteins: A widespread biological strategy from DNA/RNA tumor viruses to HIV-1. Biochim. Biophys. Acta 2011, 1812, 1498–1507. [Google Scholar]
- Liao, Y.; Wang, X.; Huang, M.; Tam, J.P.; Liu, D.X. Regulation of the p38 mitogen-activated protein kinase and dual-specificity phosphatase 1 feedback loop modulates the induction of interleukin 6 and 8 in cells infected with coronavirus infectious bronchitis virus. Virology 2011, 420, 106–116. [Google Scholar]
- Tachado, S.D.; Zhang, J.; Zhu, J.; Patel, N.; Koziel, H. HIV impairs TNF-alpha release in response to Toll-like receptor 4 stimulation in human macrophages in vitro. Am. J. Respir. Cell Mol. Biol 2005, 33, 610–621. [Google Scholar]
- Bialojan, C.; Takai, A. Inhibitory effect of a marine-sponge toxin, okadaic acid, on protein phosphatases. Specificity and kinetics. Biochem. J 1988, 256, 283–290. [Google Scholar]
- Cohen, P.; Klumpp, S.; Schelling, D.L. An improved procedure for identifying and quantitating protein phosphatases in mammalian tissues. FEBS Lett 1989, 250, 596–600. [Google Scholar]
- Marchant, D.; Singhera, G.K.; Utokaparch, S.; Hackett, T.L.; Boyd, J.H.; Luo, Z.; Si, X.; Dorscheid, D.R.; McManus, B.M.; Hegele, R.G. Toll-like receptor 4-mediated activation of p38 mitogen-activated protein kinase is a determinant of respiratory virus entry and tropism. J. Virol 2010, 84, 11359–11373. [Google Scholar]
- Shi, Y. Serine/threonine phosphatases: Mechanism through structure. Cell 2009, 139, 468–484. [Google Scholar]


© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Law, A.H.Y.; Tam, A.H.M.; Lee, D.C.W.; Lau, A.S.Y. A Role for Protein Phosphatase 2A in Regulating p38 Mitogen Activated Protein Kinase Activation and Tumor Necrosis Factor-Alpha Expression during Influenza Virus Infection. Int. J. Mol. Sci. 2013, 14, 7327-7340. https://doi.org/10.3390/ijms14047327
Law AHY, Tam AHM, Lee DCW, Lau ASY. A Role for Protein Phosphatase 2A in Regulating p38 Mitogen Activated Protein Kinase Activation and Tumor Necrosis Factor-Alpha Expression during Influenza Virus Infection. International Journal of Molecular Sciences. 2013; 14(4):7327-7340. https://doi.org/10.3390/ijms14047327
Chicago/Turabian StyleLaw, Anna H. Y., Alex H. M. Tam, Davy C. W. Lee, and Allan S. Y. Lau. 2013. "A Role for Protein Phosphatase 2A in Regulating p38 Mitogen Activated Protein Kinase Activation and Tumor Necrosis Factor-Alpha Expression during Influenza Virus Infection" International Journal of Molecular Sciences 14, no. 4: 7327-7340. https://doi.org/10.3390/ijms14047327