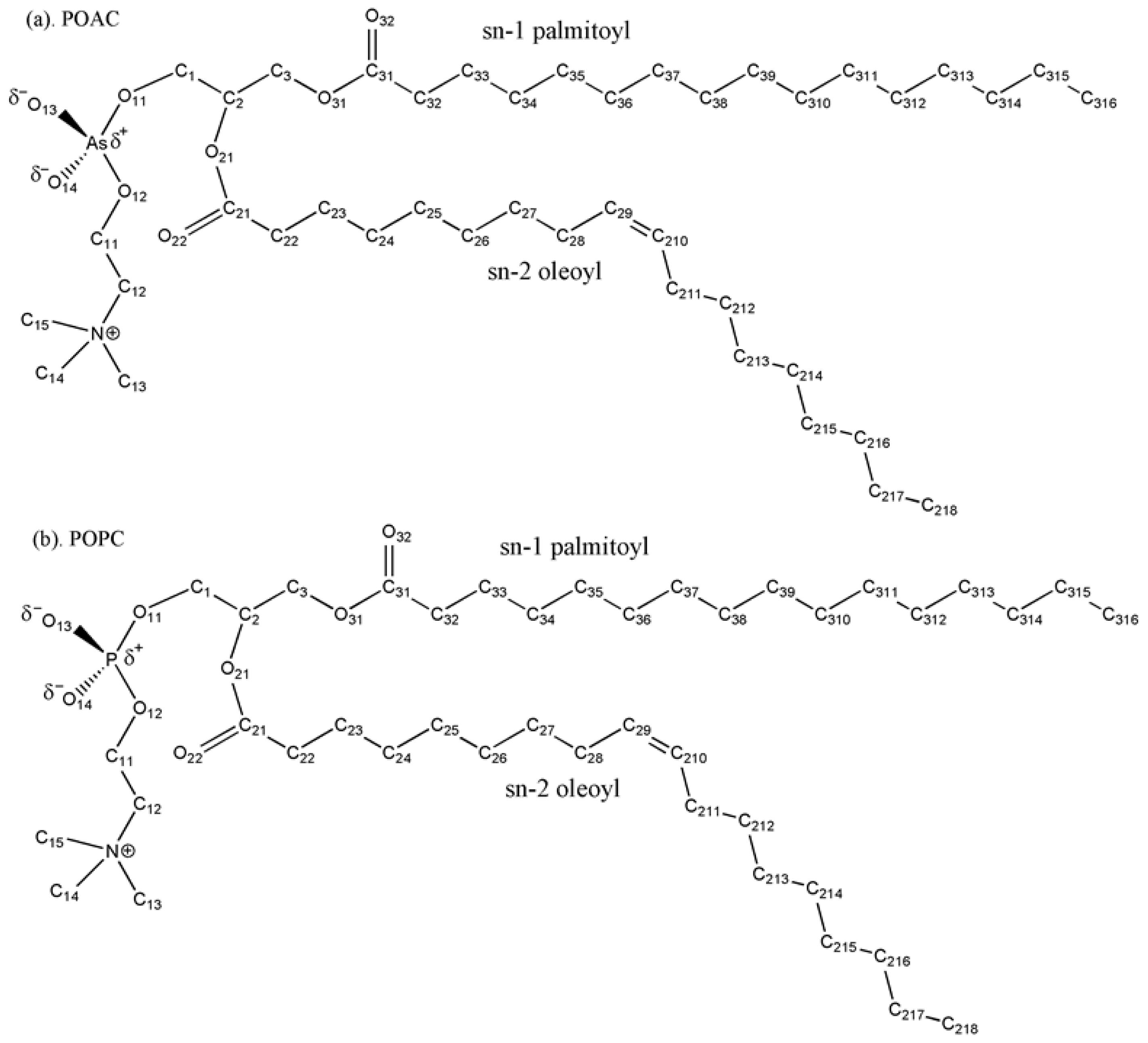
A Molecular Dynamics Study of the Structural and Dynamical Properties of Putative Arsenic Substituted Lipid Bilayers

Abstract
:
1. Introduction
2. Results and Discussion
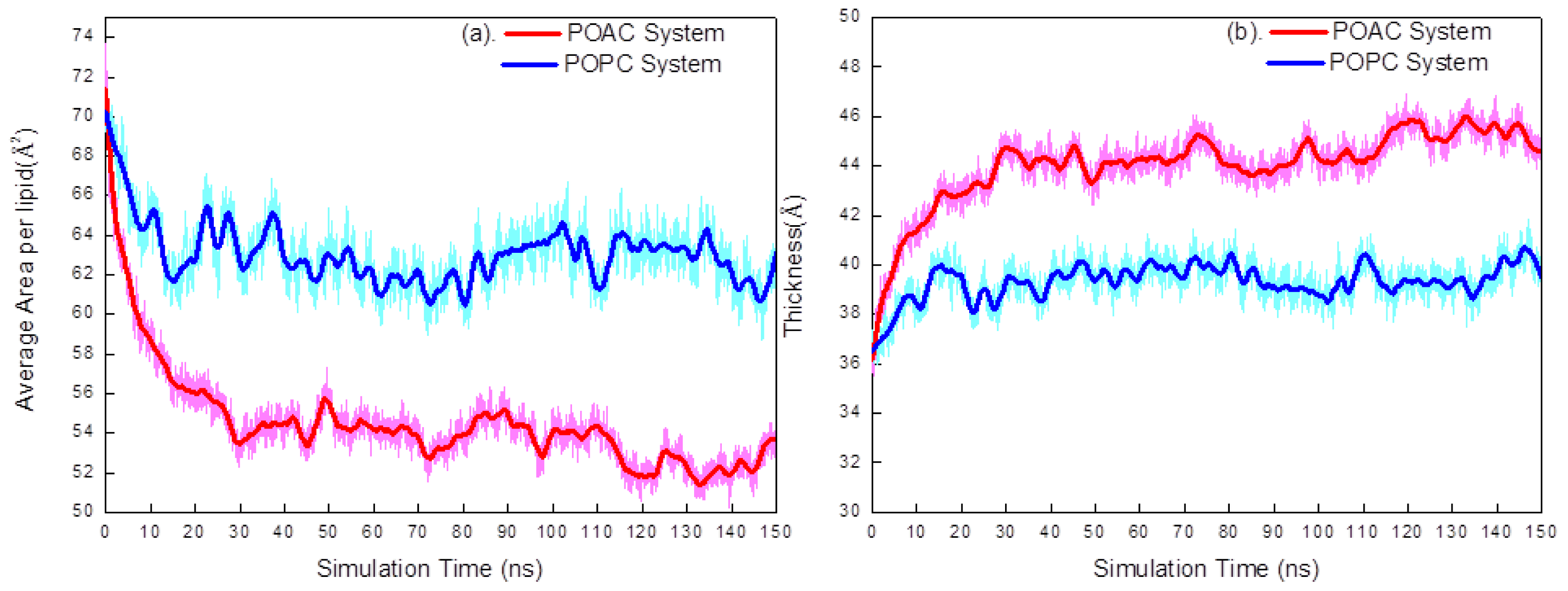
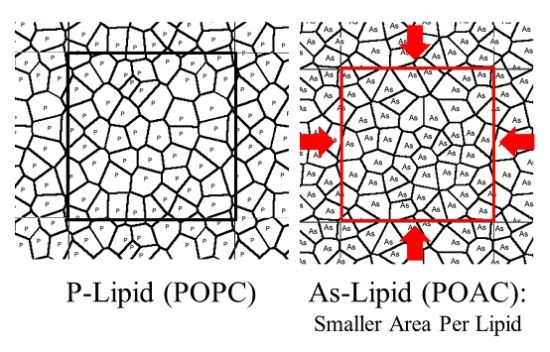
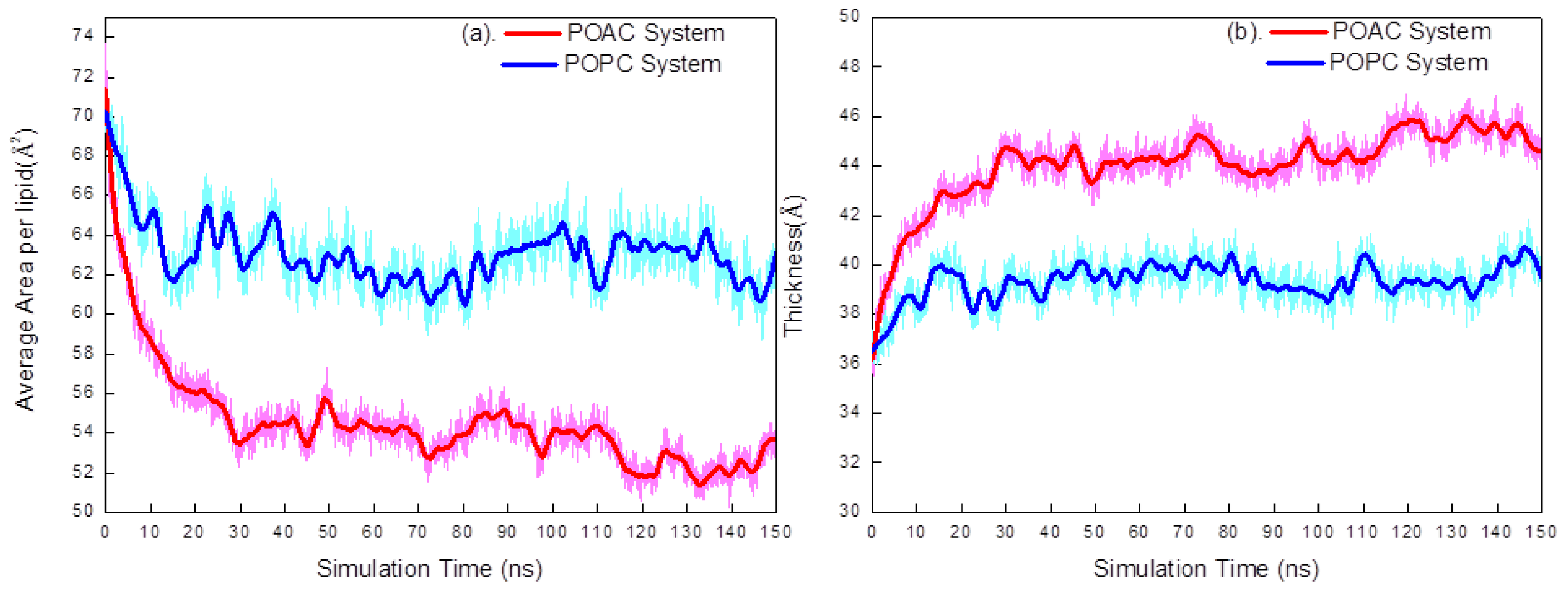
2.1. Area per Lipid and Membrane Thickness
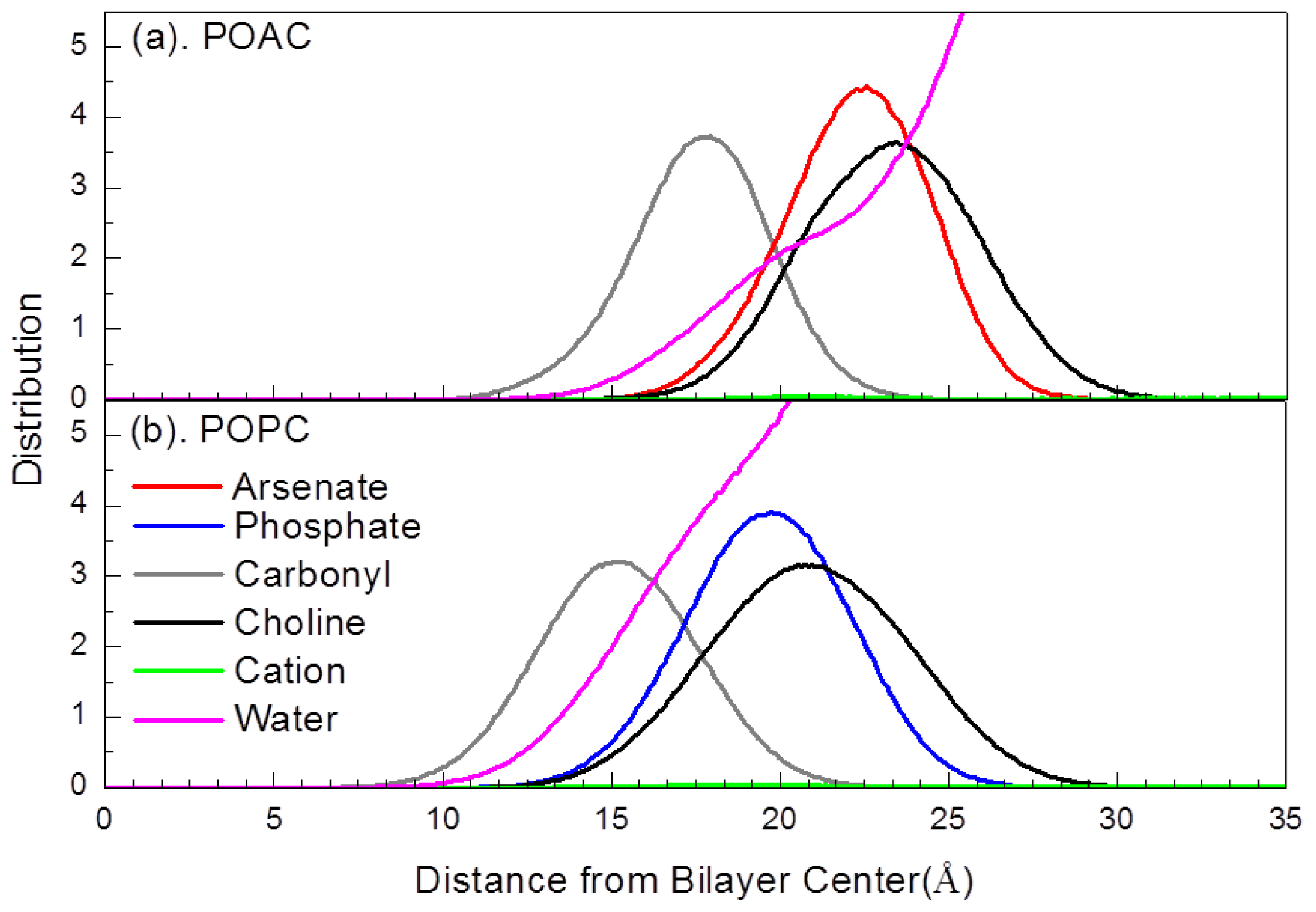
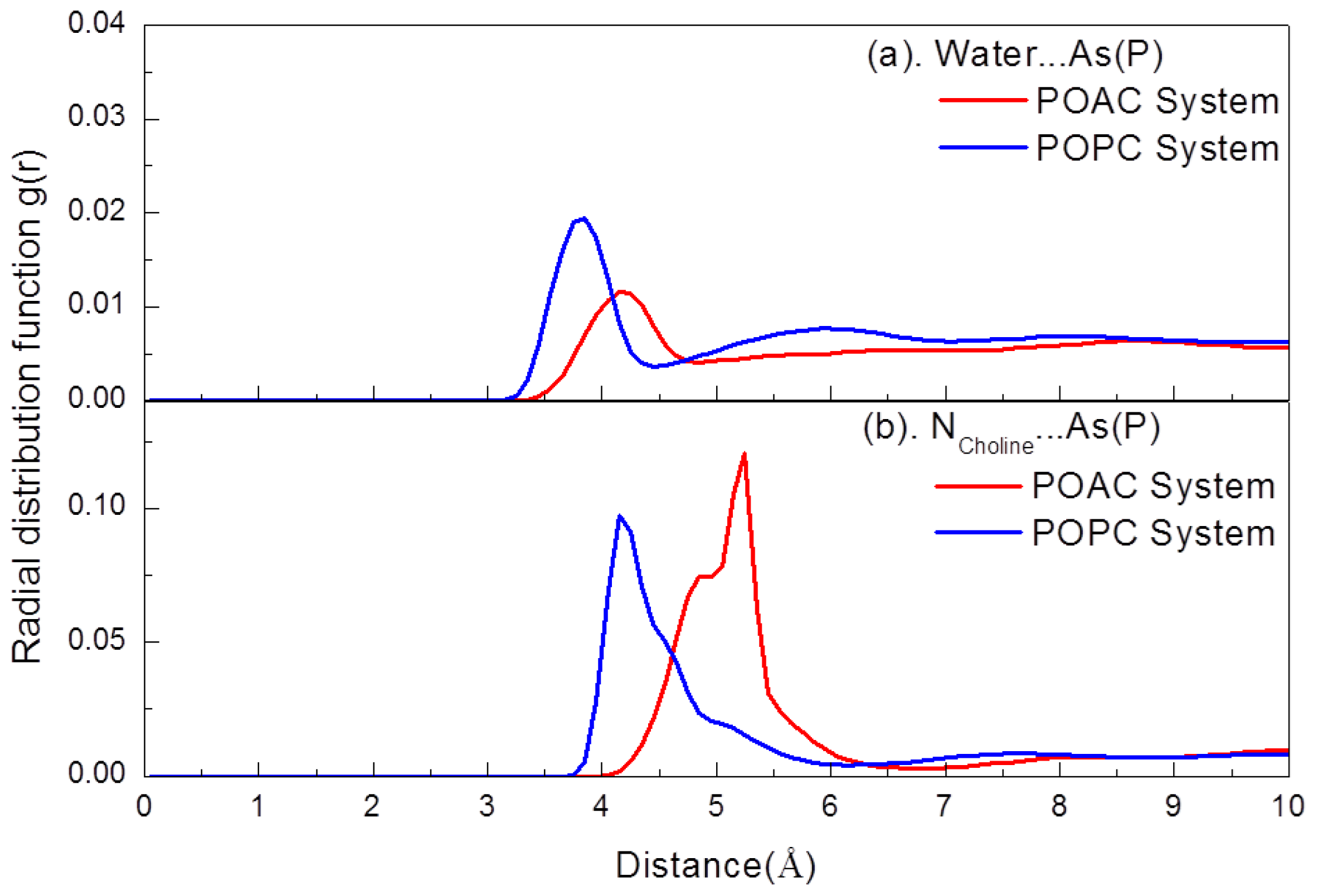
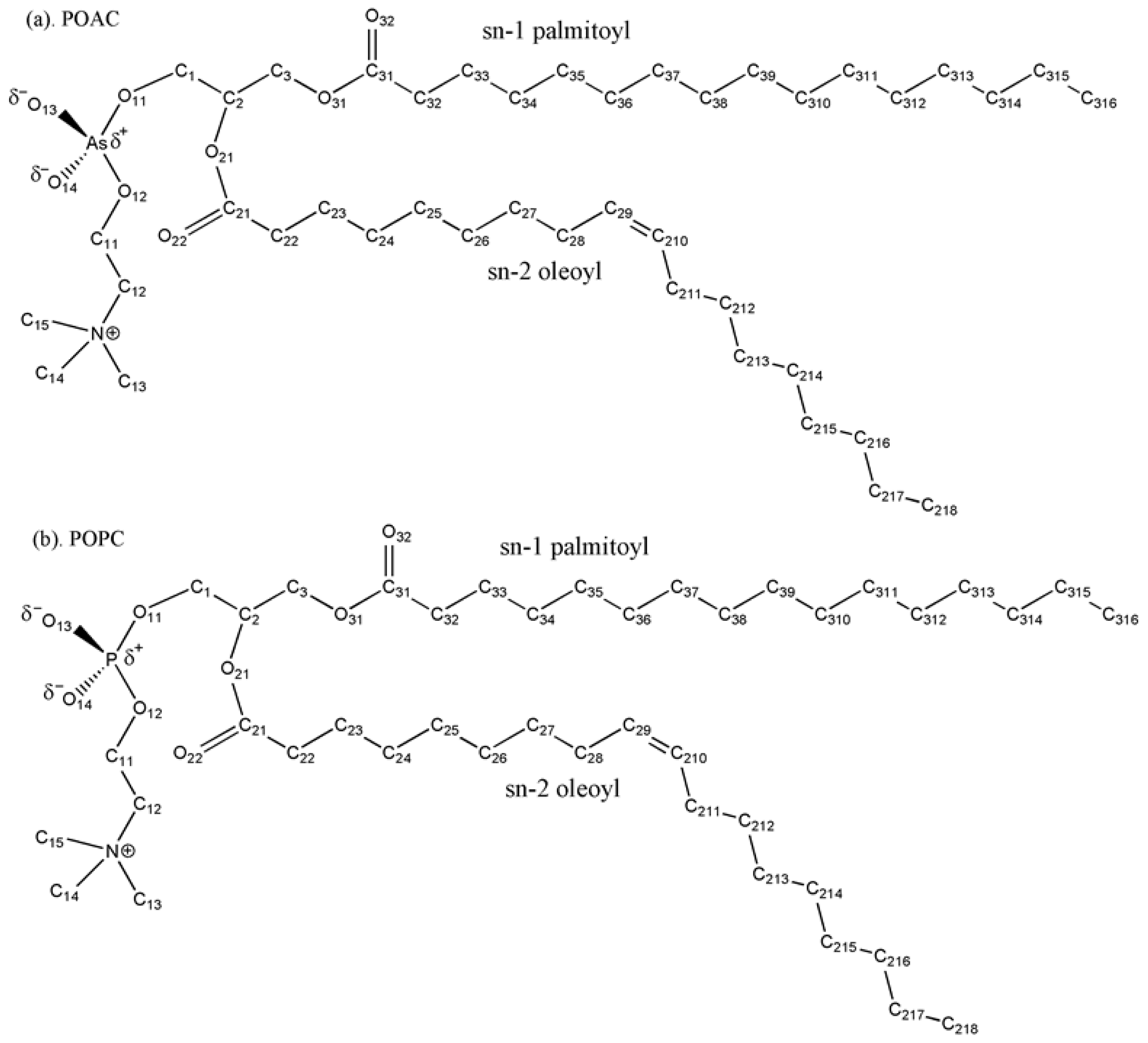
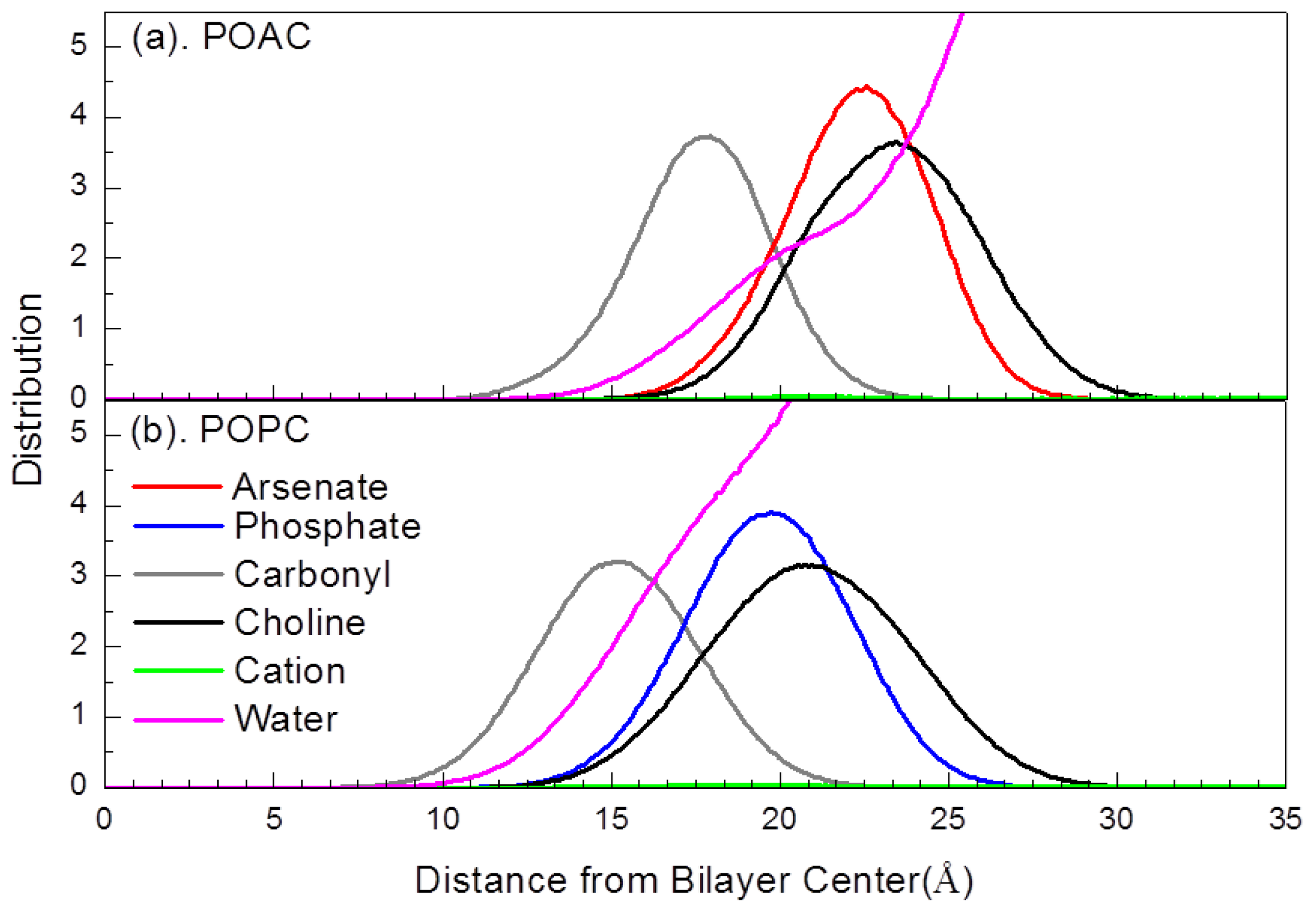
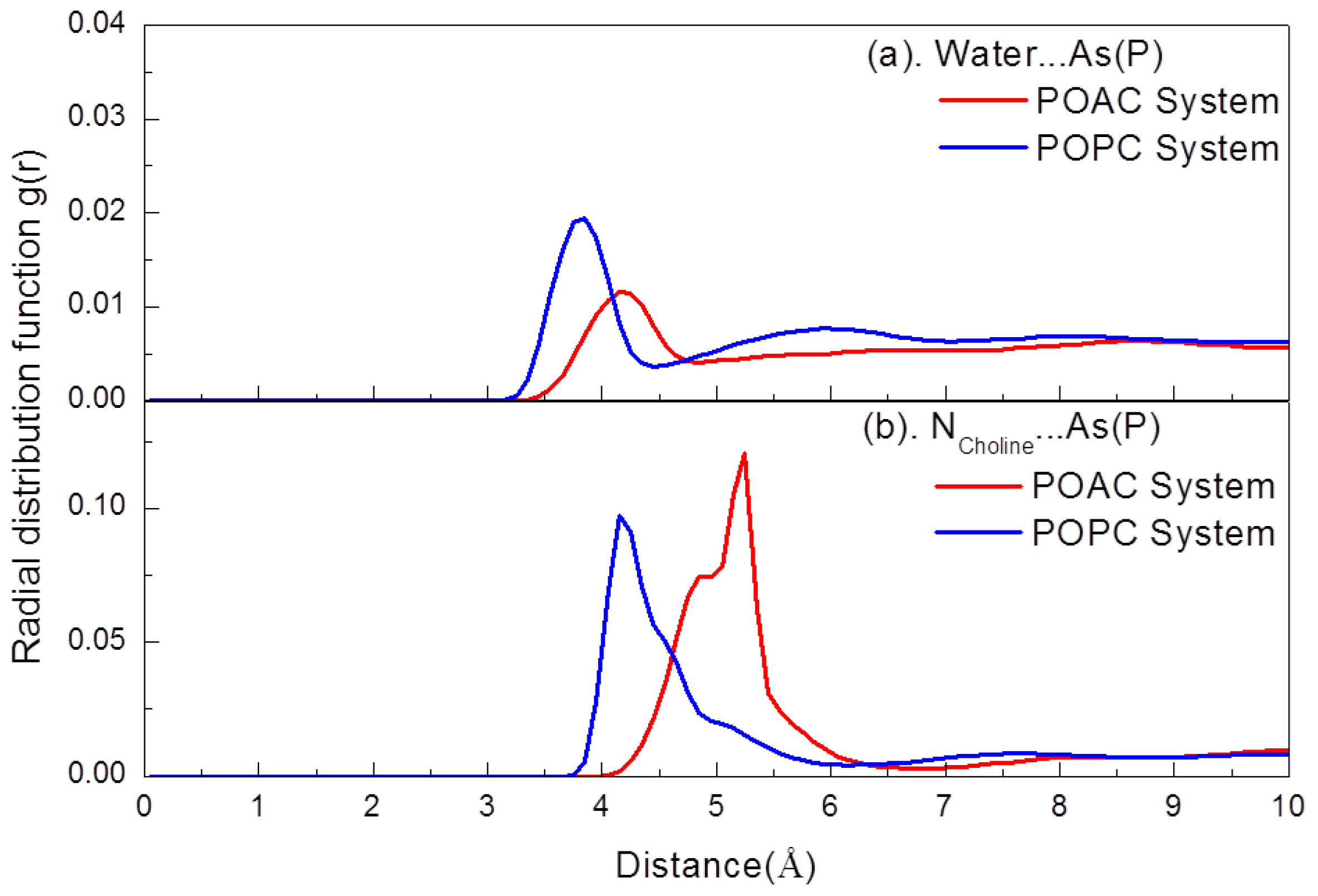
2.2. Atom Distribution
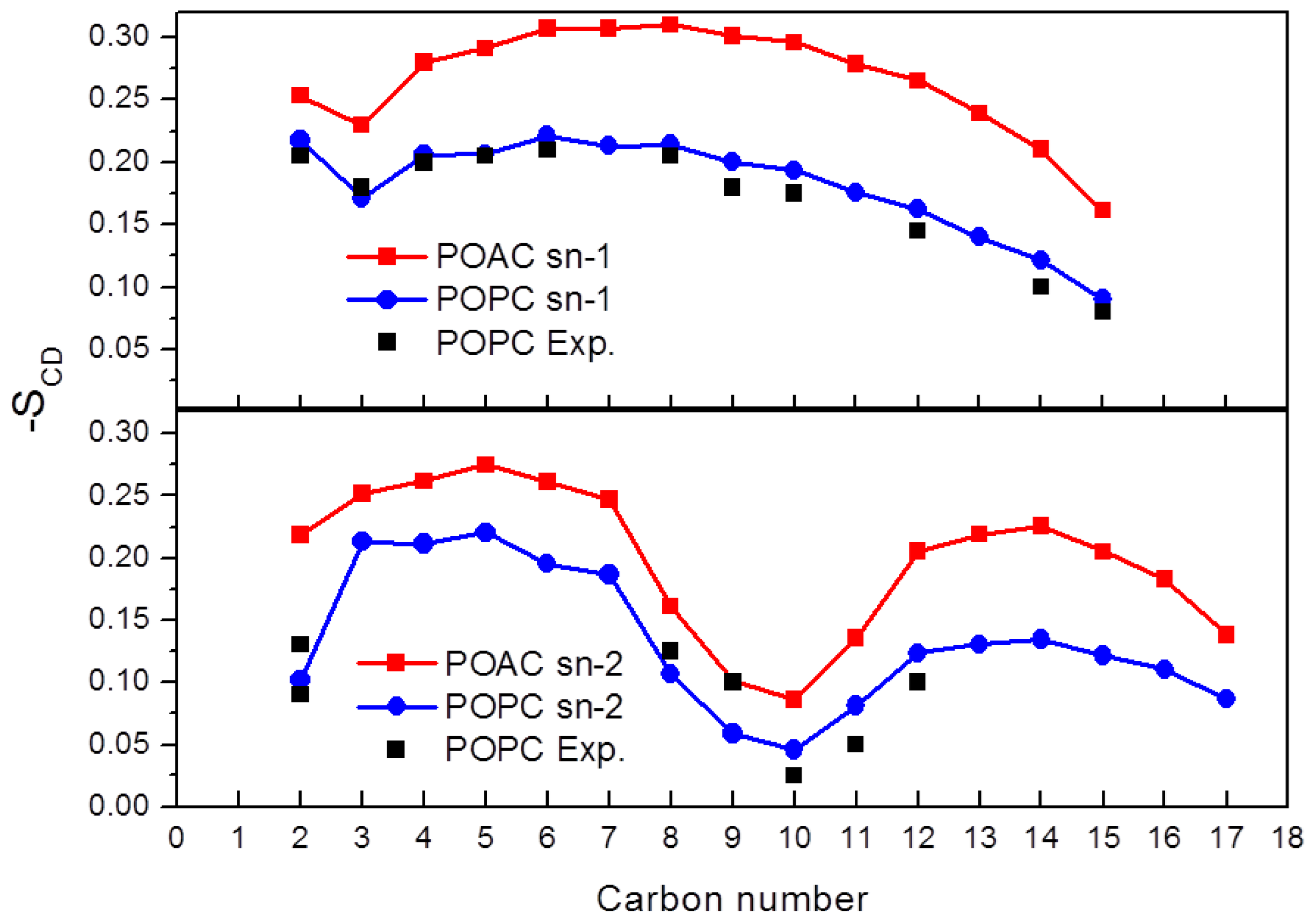
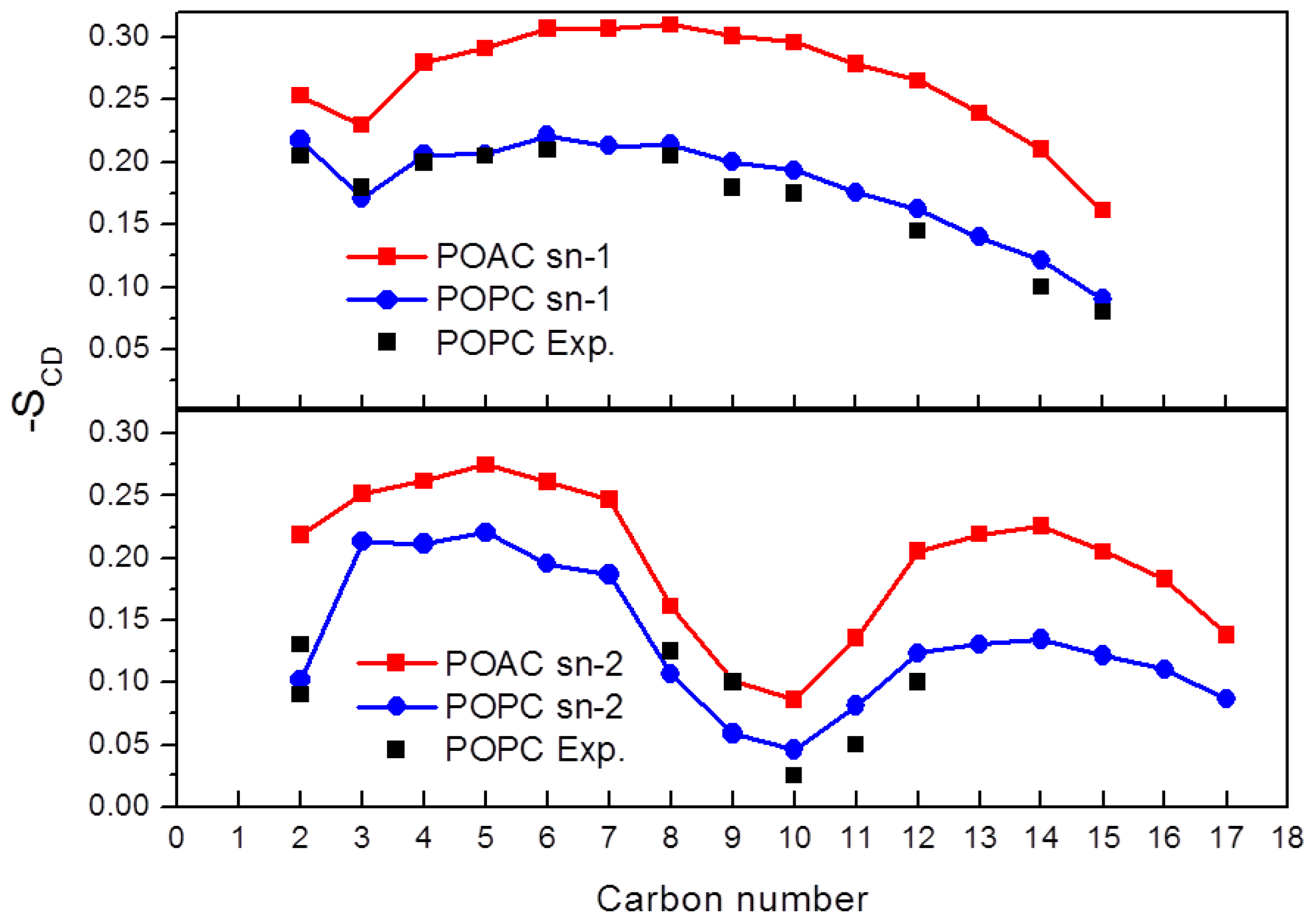
2.3. Order Parameters
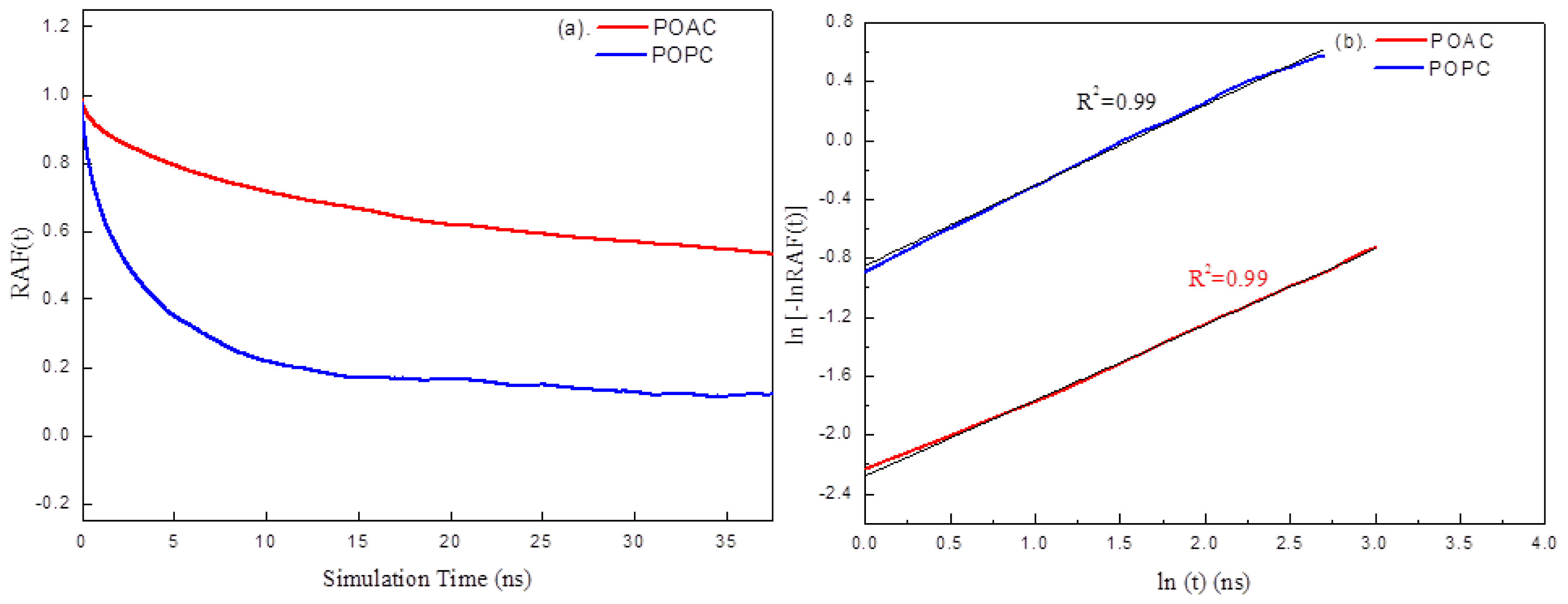
2.4. Dynamics of Lipids
3. Computational Methods
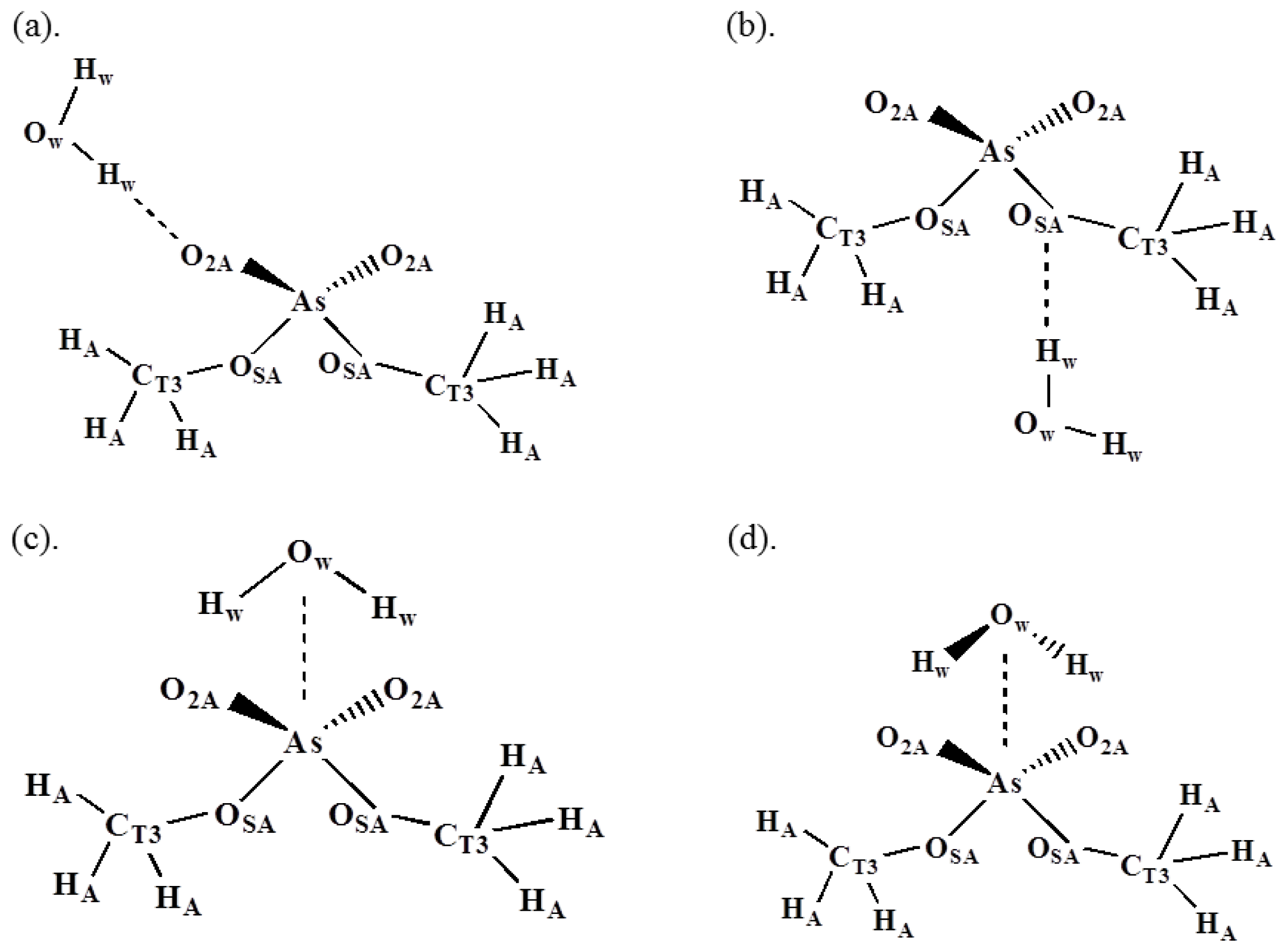
3.1. Arsenate Force Field Parameterization
3.2. Molecular Dynamics Simulations
4. Conclusions and Summary
Supplementary Information
ijms-14-07702-s001.pdfAcknowledgments
References
- Wolfe-Simon, F.; Blum, S.J.; Kulp, T.R.; Gordon, G.W.; Hoeft, S.E.; Pett-Ridge, J.; Stolz, J.F.; Webb, S.M.; Weber, P.K.; Davies, P.C.; et al. A bacterium that can grow by using arsenic instead of phosphorus. Science 2011, 332, 1163–1166. [Google Scholar]
- Fekry, M.I.; Tipton, P.A.; Gates, K.S. Kinetic consequences of replacing the internucleotide phosphorus atoms in DNA with arsenic. ACS Chem. Biol 2011, 6, 127–130. [Google Scholar]
- Wang, J.; Gu, J.; Leszczynski, J. Could hydrolysis of arsenic substituted DNA be prevented? Protection arises from stacking interactions. Chem. Commun. (Camb) 2012, 48, 3626–3628. [Google Scholar]
- Reaves, M.L.; Sinha, S.; Rabinowitz, J.D.; Kruglyak, L.; Redfield, R.J. Absence of detectable arsenate in DNA from arsenate-grown GFAJ-1 cells. Science 2012, 337, 470–473. [Google Scholar]
- Erb, T.J.; Kiefer, P.; Hattendorf, B.; Günther, D.; Vorholt, J.A. GFAJ-1 Is an Arsenate-Resistant, Phosphate-Dependent Organism. Science 2012, 337, 467–470. [Google Scholar]
- Elias, M.; Wellner, A.; Goldin-Azulay, K.; Chabriere, E.; Vorholt, J.A.; Erb, T.J.; Tawfik, D.S. The molecular basis of phosphate discrimination in arsenate-rich environments. Nature 2012, 491, 134–137. [Google Scholar]
- Denning, E.J.; Mackerell, A.D., Jr. Impact of arsenic/phosphorus substitution on the intrinsic conformational properties of the phosphodiester backbone of DNA investigated using ab initio quantum mechanical calculations. J. Am. Chem. Soc 2011, 133, 5770–5772. [Google Scholar]
- Xu, Y.; Ma, B.; Nussinov, R. Structural and functional consequences of phosphate-arsenate substitutions in selected nucleotides: DNA, RNA, and ATP. J. Phys. Chem. B 2012, 116, 4801–4811. [Google Scholar]
- Rosen, B.P.; Ajees, A.A.; McDermott, T.R. Life and death with arsenic. Arsenic life: An analysis of the recent report “A bacterium that can grow by using arsenic instead of phosphorus”. Bioessays 2011, 33, 350–357. [Google Scholar]
- Stryer, L. Biochemistry, 3rd ed; W.H. Freeman and Co: New York, NY, USA, 1988. [Google Scholar]
- Pabst, G.; Rappolt, M.; Amenitsch, H.; Laggner, P. Structural information from multilamellar liposomes at full hydration: Full q-range fitting with high quality X-ray data. Phys. Rev. E 2000, 62, 4000–4009. [Google Scholar]
- Smaby, J.M.; Momsen, M.M.; Brockman, H.L.; Brown, R.E. Phosphatidylcholine acyl unsaturation modulates the decrease in interfacial elasticity induced by cholesterol. Biophys. J 1997, 73, 1492–1505. [Google Scholar]
- Kučerka, N.; Tristram-Nagle, S.; Nagle, J. Structure of Fully Hydrated Fluid Phase Lipid Bilayers with Monounsaturated Chains. J. Membr. Biol 2006, 208, 193–202. [Google Scholar]
- Hyslop, P.A.; Morel, B.; Sauerheber, R.D. Organization and interaction of cholesterol and phosphatidylcholine in model bilayer membranes. Biochemistry 1990, 29, 1025–1038. [Google Scholar]
- Poger, D.; Mark, A.E. On the validation of molecular dynamics simulations of saturated and cis-monounsaturated phosphatidylcholine lipid bilayers: A comparison with experiment. J. Chem. Theory Comput 2009, 6, 325–336. [Google Scholar]
- Poger, D.; Mark, A.E. Lipid bilayers: The effect of force field on ordering and dynamics. J. Chem. Theory Comput 2012, 8, 4807–4817. [Google Scholar]
- Seelig, J.; Waespe-Sarcevic, N. Molecular order in cis and trans unsaturated phospholipid bilayers. Biochemistry 1978, 17, 3310–3315. [Google Scholar]
- Williams, G.; Watts, D.C. Non-Symmetrical dielectric relaxation behaviour arising from a simple empirical decay function. Trans. Faraday Soc 1970, 66, 80–85. [Google Scholar]
- Williams, G.; Watts, D.C.; Dev, S.B.; North, A.M. Further considerations of non symmetrical dielectric relaxation behaviour arising from a simple empirical decay function. Trans. Faraday Soc 1971, 67, 1323–1335. [Google Scholar]
- Mackerell, A.D.; Wiorkiewiczkuczera, J.; Karplus, M. An all-atom empirical energy function for the simulation of nucleic-acids. J. Am. Chem. Soc 1995, 117, 11946–11975. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.1; Gaussian, Inc: Wallingford, CT, USA, 2009. [Google Scholar]
- Brooks, B.R.; Brooks, C.L.; Mackerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem 2009, 30, 1545–1614. [Google Scholar]
- Simon, S.L.; Duran, M.; Dannenberg, J.J. How does basis set superposition error change the potential surfaces for hydrogen-bonded dimers? J. Chem. Phys 1996, 105, 11024. [Google Scholar]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem 2008, 29, 1859–1865. [Google Scholar]
- Klauda, J.B.; Venable, R.M.; Freites, J.A.; O’Connor, J.W.; Tobias, D.J.; Mondragon-Ramirez, C.; Vorobyov, I.; MacKerell, A.D.; Pastor, R.W. Update of the CHARMM All-Atom additive force field for lipids: Validation on six lipid types. J. Phys. Chem. B 2010, 114, 7830–7843. [Google Scholar]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys 1983, 79, 926–935. [Google Scholar]
- Kale, L.; Skeel, R.; Bhandarkar, M.; Brunner, R.; Gursoy, A.; Krawetz, N.; Phillips, J.; Shinozaki, A.; Varadarajan, K.; Schulten, K. NAMD2: Greater scalability for parallel molecular dynamics. J. Comp. Phys 1999, 151, 283–312. [Google Scholar]
- Feller, S.E.; Zhang, Y.; Pastor, R.W.; Brooks, B.R. Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys 1995, 103, 4613–4621. [Google Scholar]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald—An N.Log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys 1993, 98, 10089–10092. [Google Scholar]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comp. Phys 1977, 23, 327–341. [Google Scholar]
- Tsai, H.H.; Lai, W.X.; Lin, H.D.; Lee, J.B.; Juang, W.F.; Tseng, W.H. Molecular dynamics simulation of cation-phospholipid clustering in phospholipid bilayers: Possible role in stalk formation during membrane fusion. Biochim. Biophys. Acta 2012, 1818, 2742–2755. [Google Scholar]
- Tsai, H.-H.G.; Lee, J.-B.; Tseng, S.-S.; Pan, X.-A.; Shih, Y.-C. Folding and membrane insertion of amyloid-beta (25–35) peptide and its mutants: Implications for aggregation and neurotoxicity. Proteins Struct. Funct. Bioinform 2010, 78, 1909–1925. [Google Scholar]
- Tsai, H.H.; Reches, M.; Tsai, C.J.; Gunasekaran, K.; Gazit, E.; Nussinov, R. Energy landscape of amyloidogenic peptide oligomerization by parallel-tempering molecular dynamics simulation: Significant role of Asn ladder. Proc. Natl. Acad. Sci. USA 2005, 102, 8174–8179. [Google Scholar]
- Tsai, C.-W.; Hsu, N.-Y.; Wang, C.-H.; Lu, C.-Y.; Chang, Y.; Tsai, H.-H.G.; Ruaan, R.-C. Coupling molecular dynamics simulations with experiments for the rational design of indolicidin-analogous antimicrobial peptides. J. Mol. Biol 2009, 392, 837–854. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
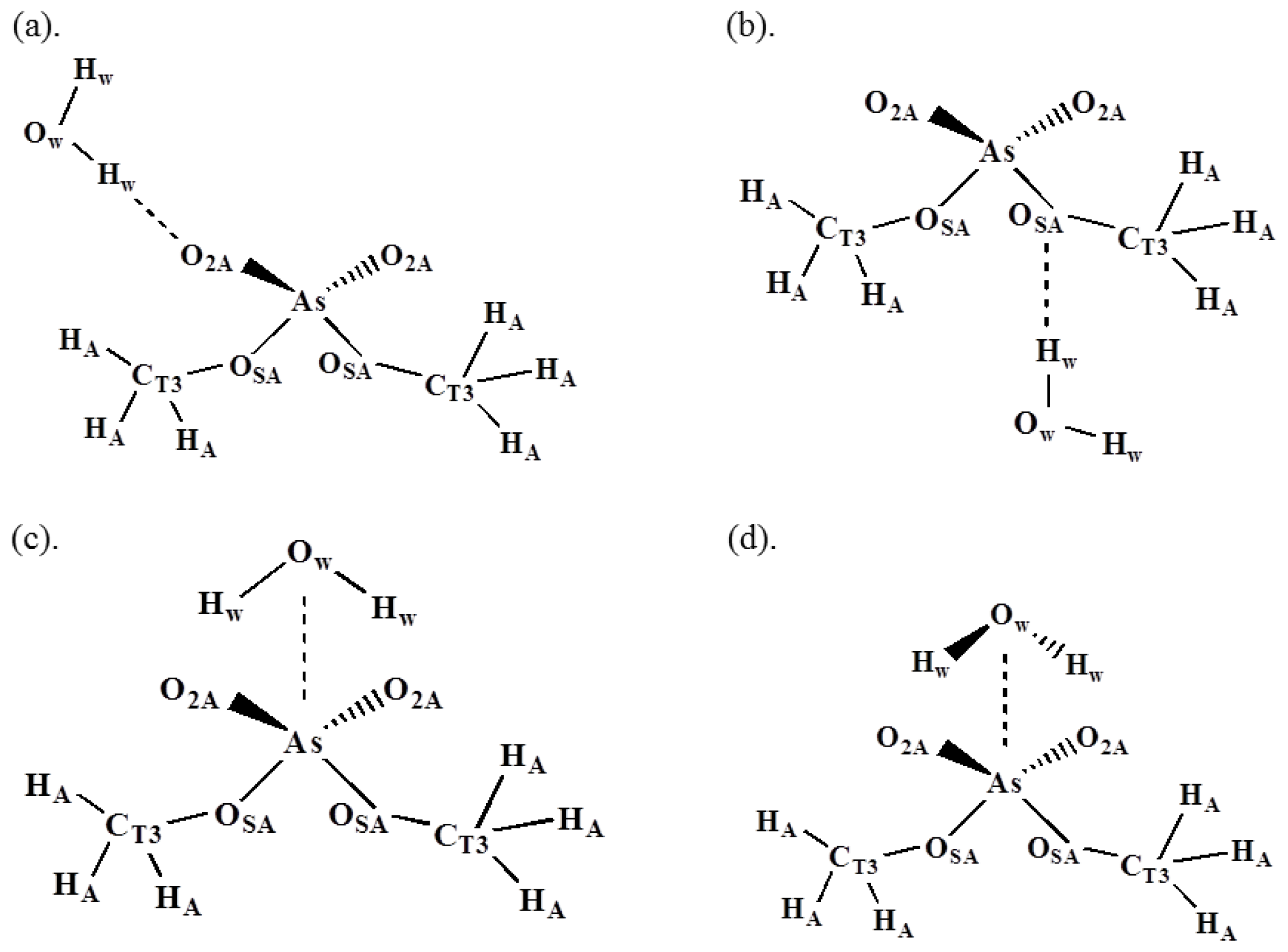
| HF/6-31G* | Empirical b | |||
|---|---|---|---|---|
| Interaction | Emin | Rmin | Emin | Rmin |
| (a) O2A–Hw | −11.4 | 1.87 | −11.64 | 1.88 |
| (b) OSA–Hw | −4.77 | 2.35 | −5.03 | 2.34 |
| (c) As–Owc | −7.76 | 4.05 | 8.24 | 3.86 |
| (d) As–Owd | −13.29 | 3.48 | −12.83 | 3.50 |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tsai, H.-H.G.; Lee, J.-B.; Huang, J.-M.; Juwita, R. A Molecular Dynamics Study of the Structural and Dynamical Properties of Putative Arsenic Substituted Lipid Bilayers. Int. J. Mol. Sci. 2013, 14, 7702-7715. https://doi.org/10.3390/ijms14047702
Tsai H-HG, Lee J-B, Huang J-M, Juwita R. A Molecular Dynamics Study of the Structural and Dynamical Properties of Putative Arsenic Substituted Lipid Bilayers. International Journal of Molecular Sciences. 2013; 14(4):7702-7715. https://doi.org/10.3390/ijms14047702
Chicago/Turabian StyleTsai, Hui-Hsu Gavin, Jian-Bin Lee, Jian-Ming Huang, and Ratna Juwita. 2013. "A Molecular Dynamics Study of the Structural and Dynamical Properties of Putative Arsenic Substituted Lipid Bilayers" International Journal of Molecular Sciences 14, no. 4: 7702-7715. https://doi.org/10.3390/ijms14047702