Native High Density Lipoproteins (HDL) Interfere with Platelet Activation Induced by Oxidized Low Density Lipoproteins (OxLDL)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
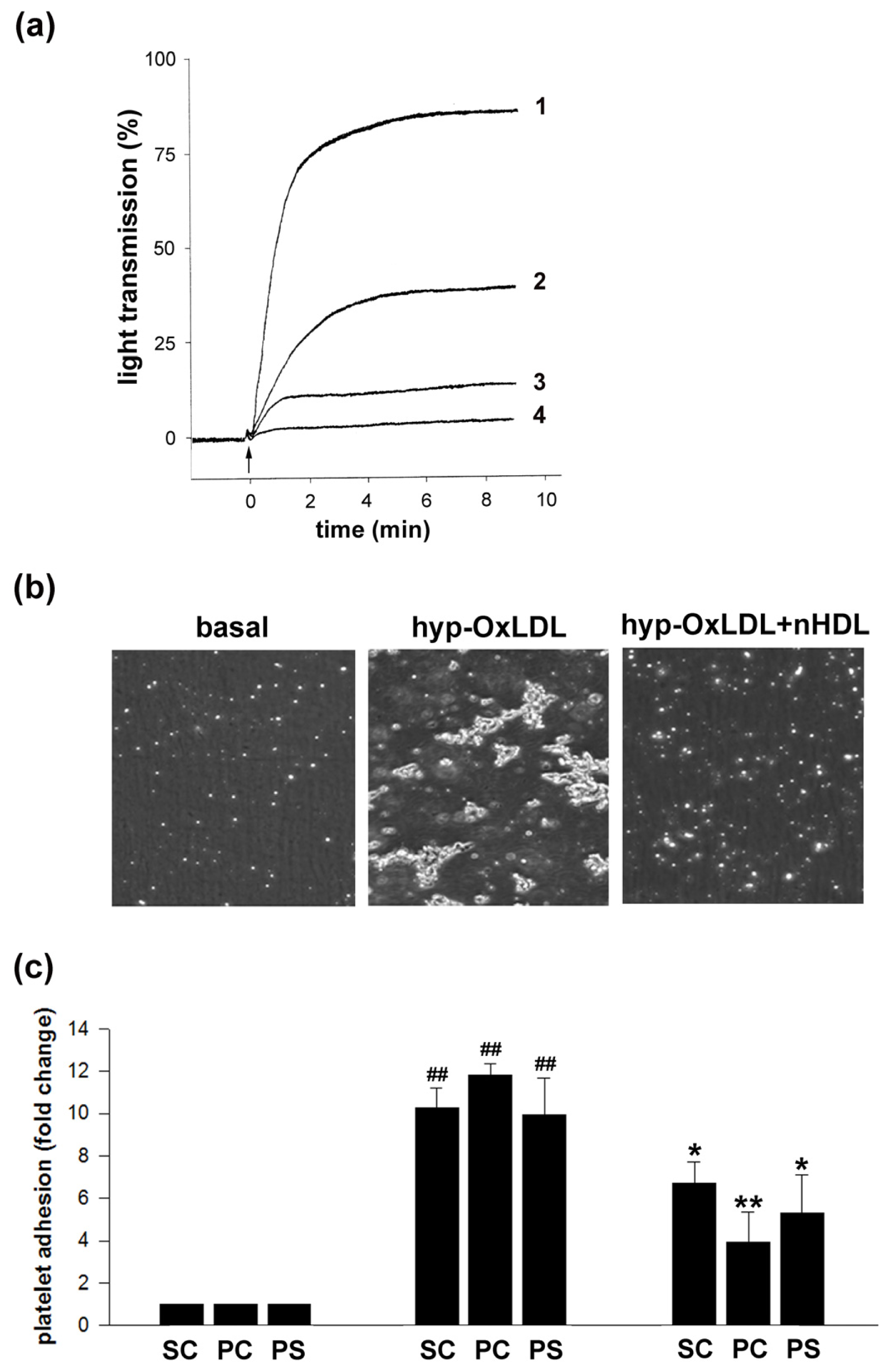
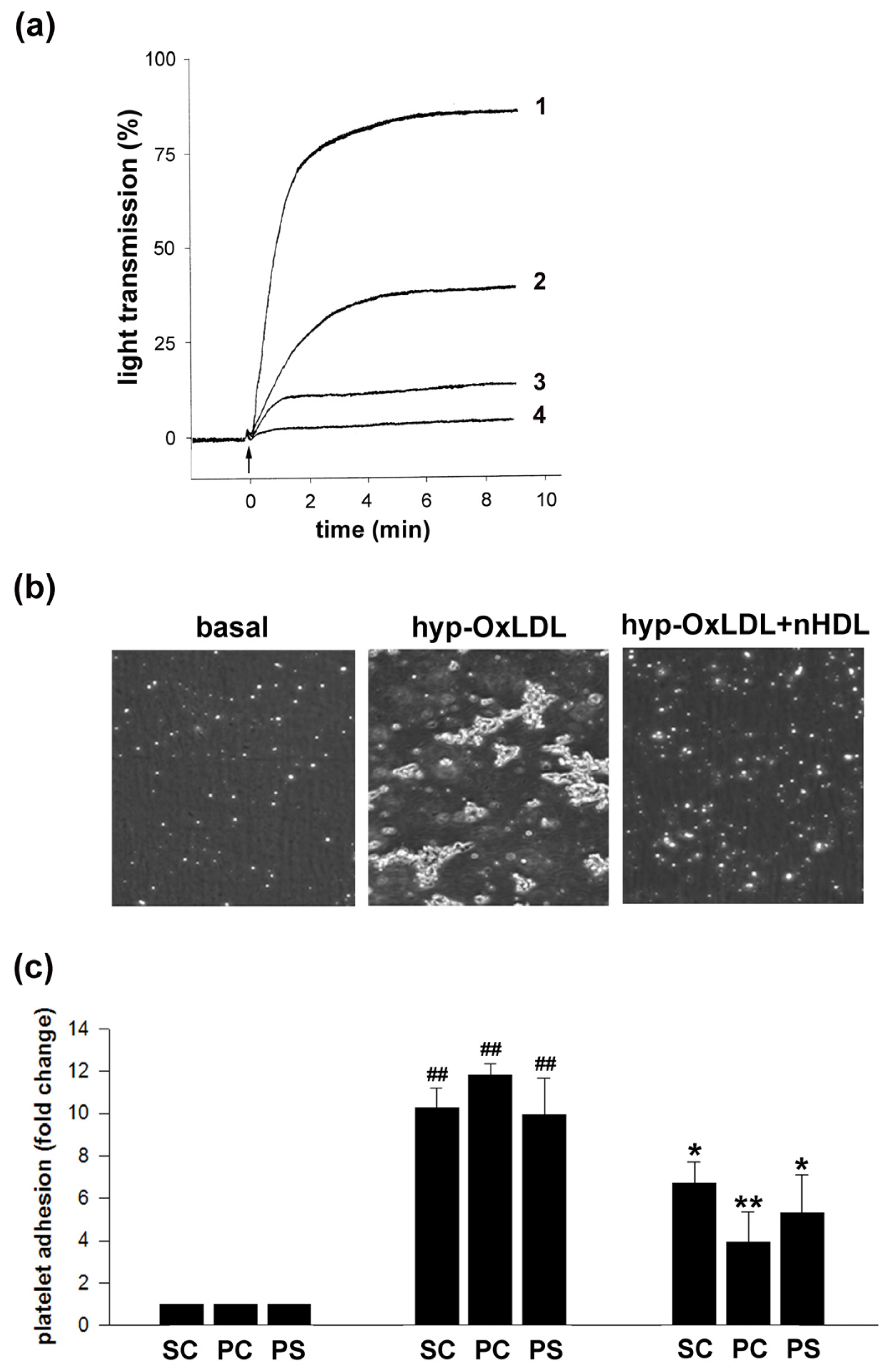
2.1. Native HDL Counteract Hyp-OxLDL-Induced Platelet Aggregation and Adhesion
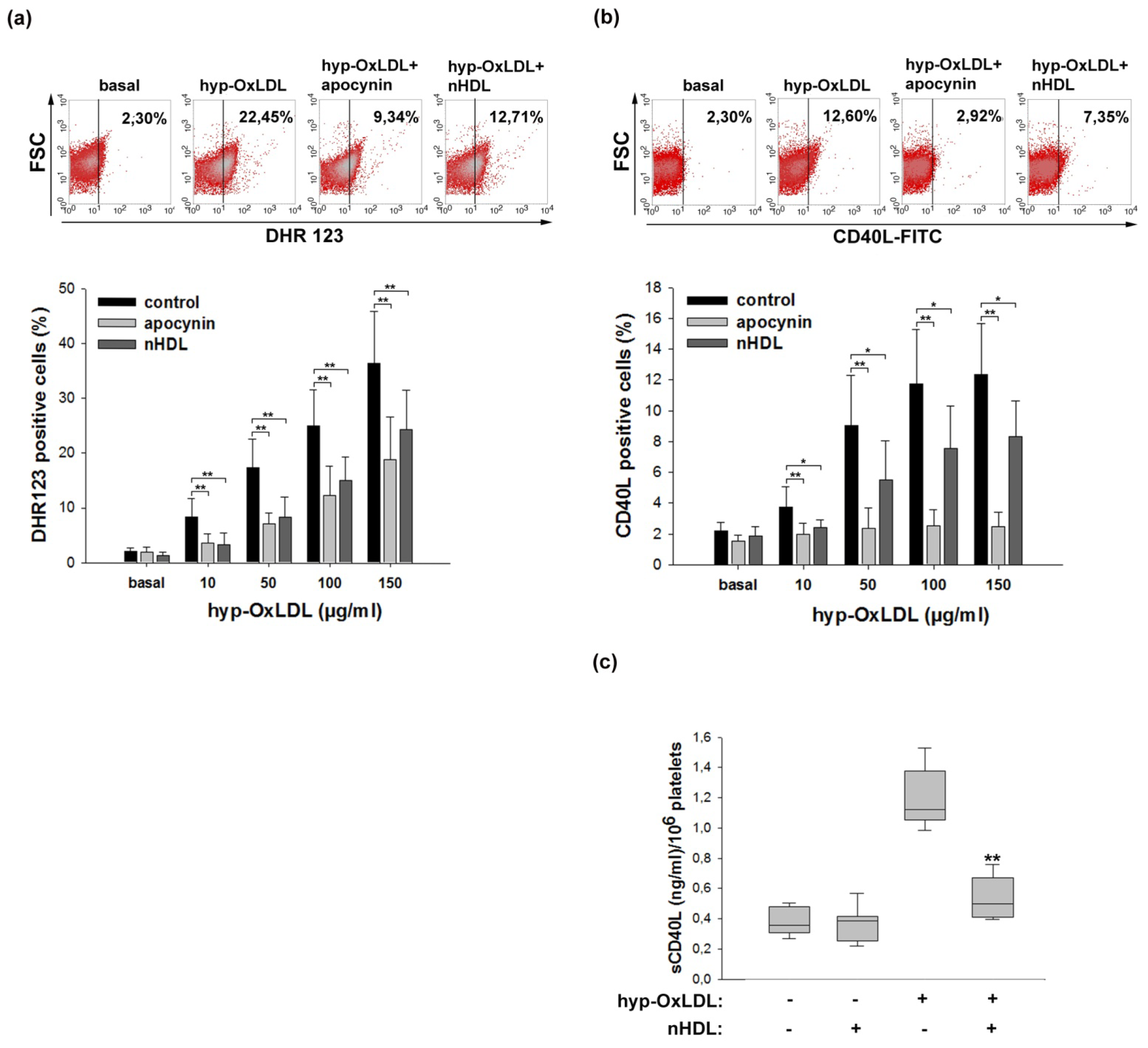
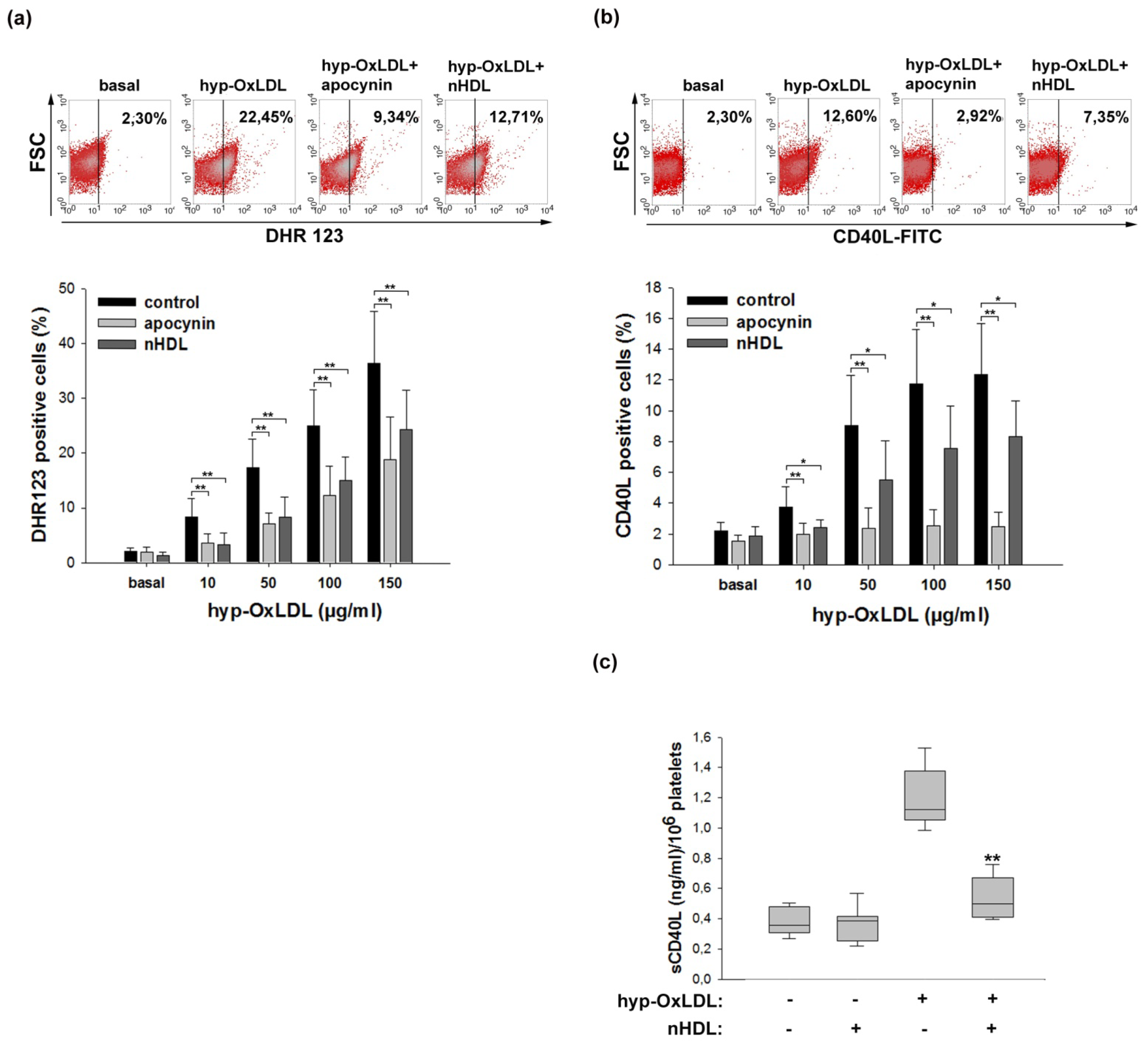
2.2. Native HDL Attenuate Intraplatelet ROS Formation and CD40L Expression Induced by Hyp-OxLDL
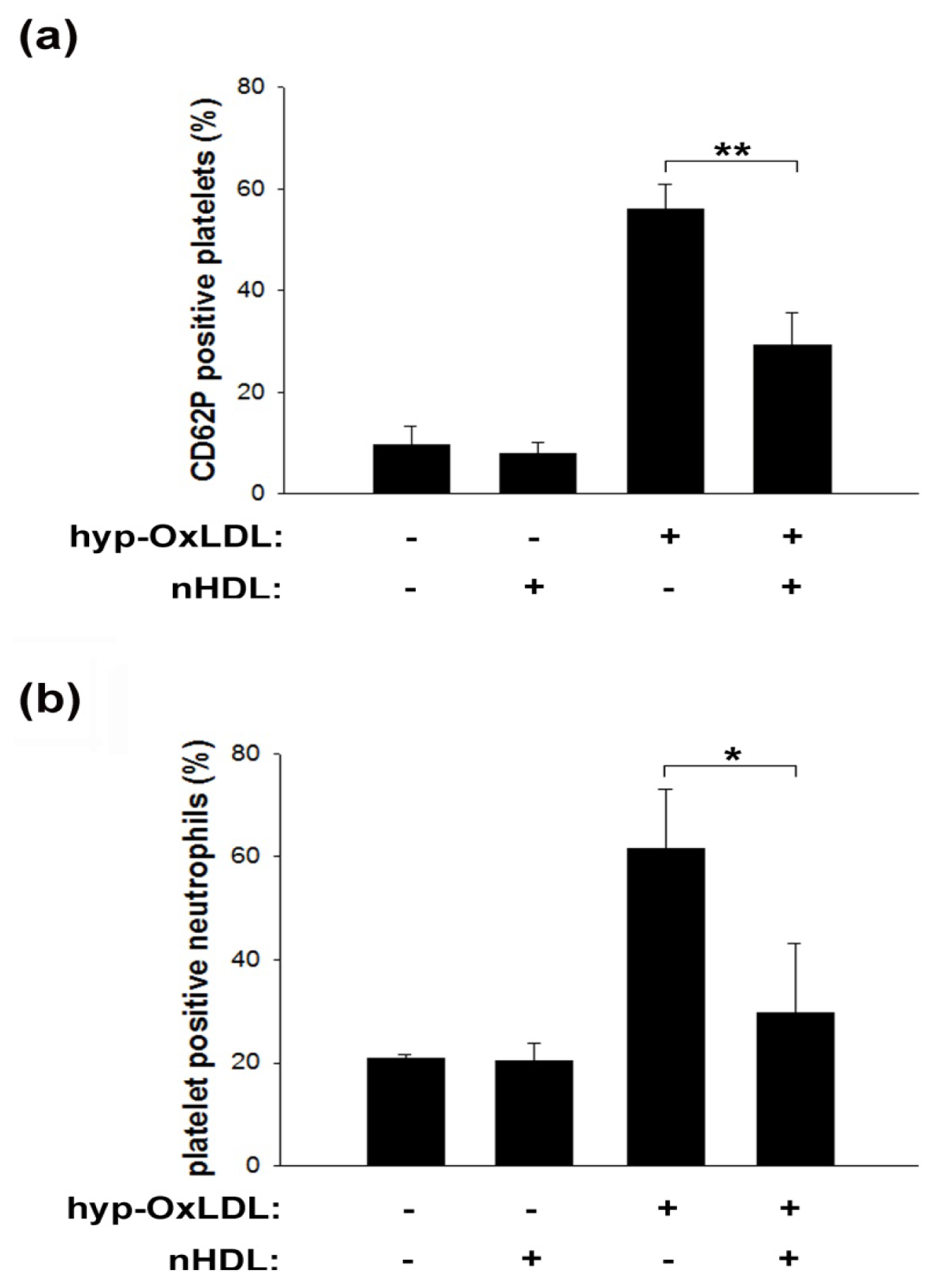
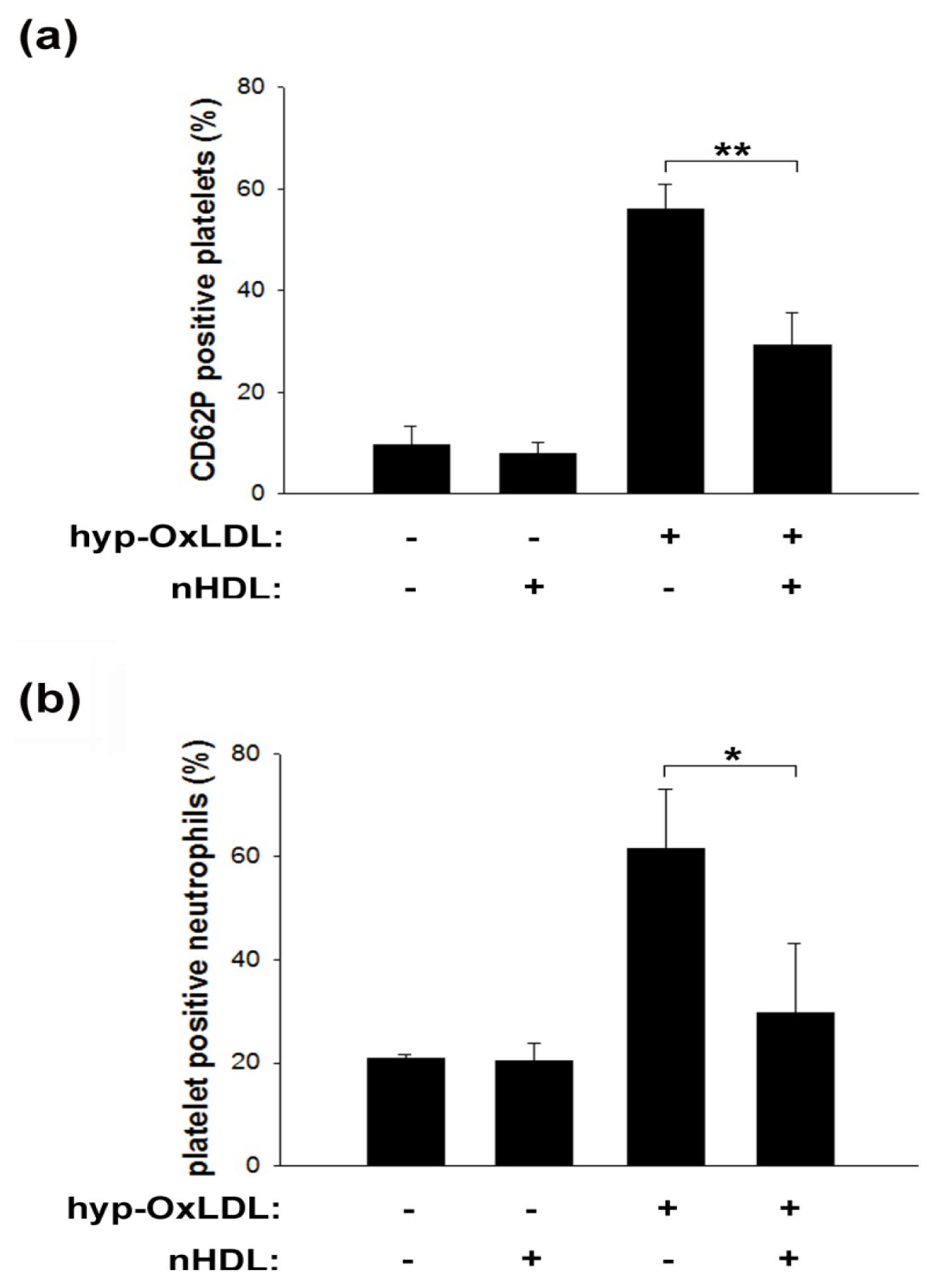
2.3. Native HDL Modulate Expression of CD62P and Formation of Platelet-Neutrophil Aggregates
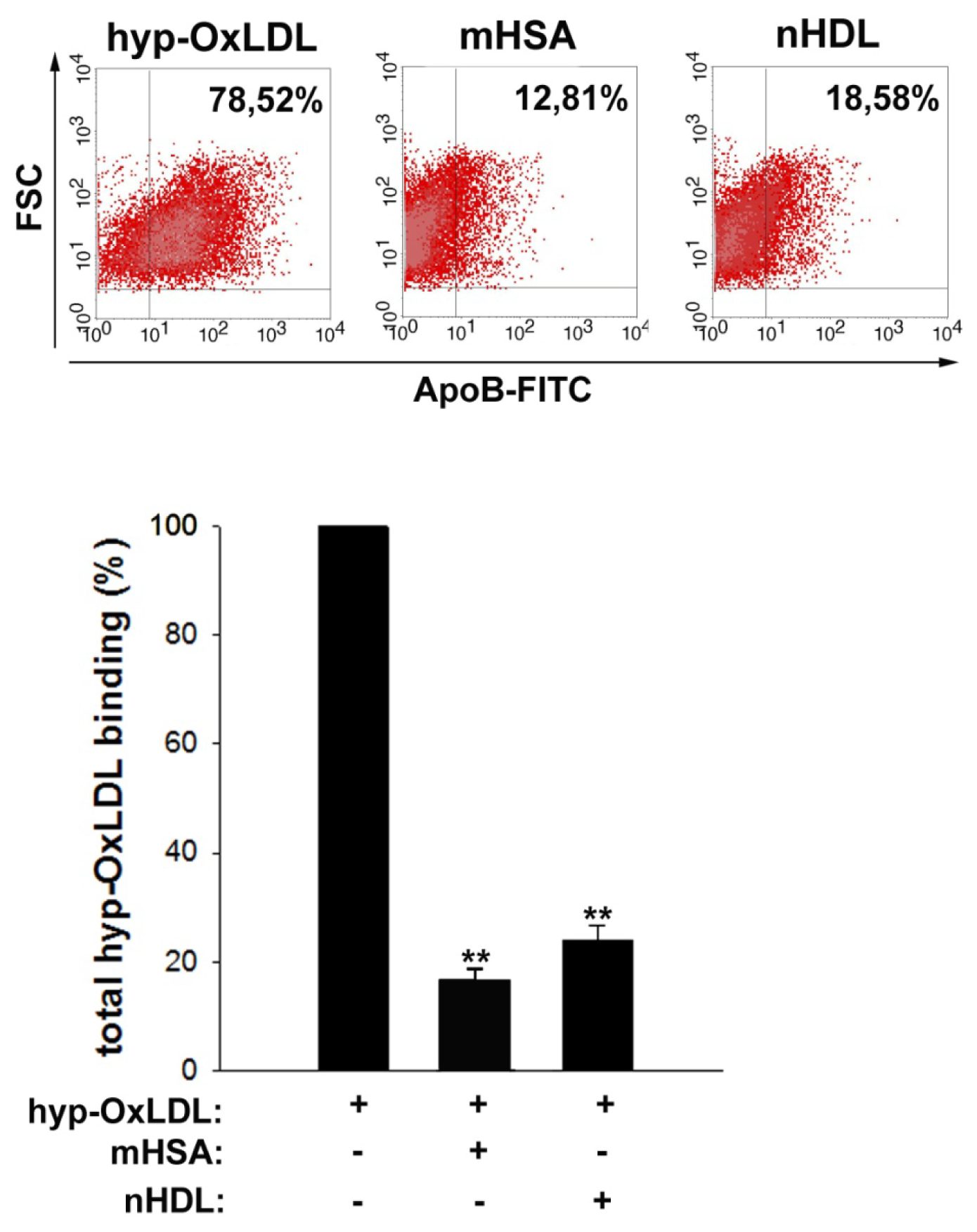
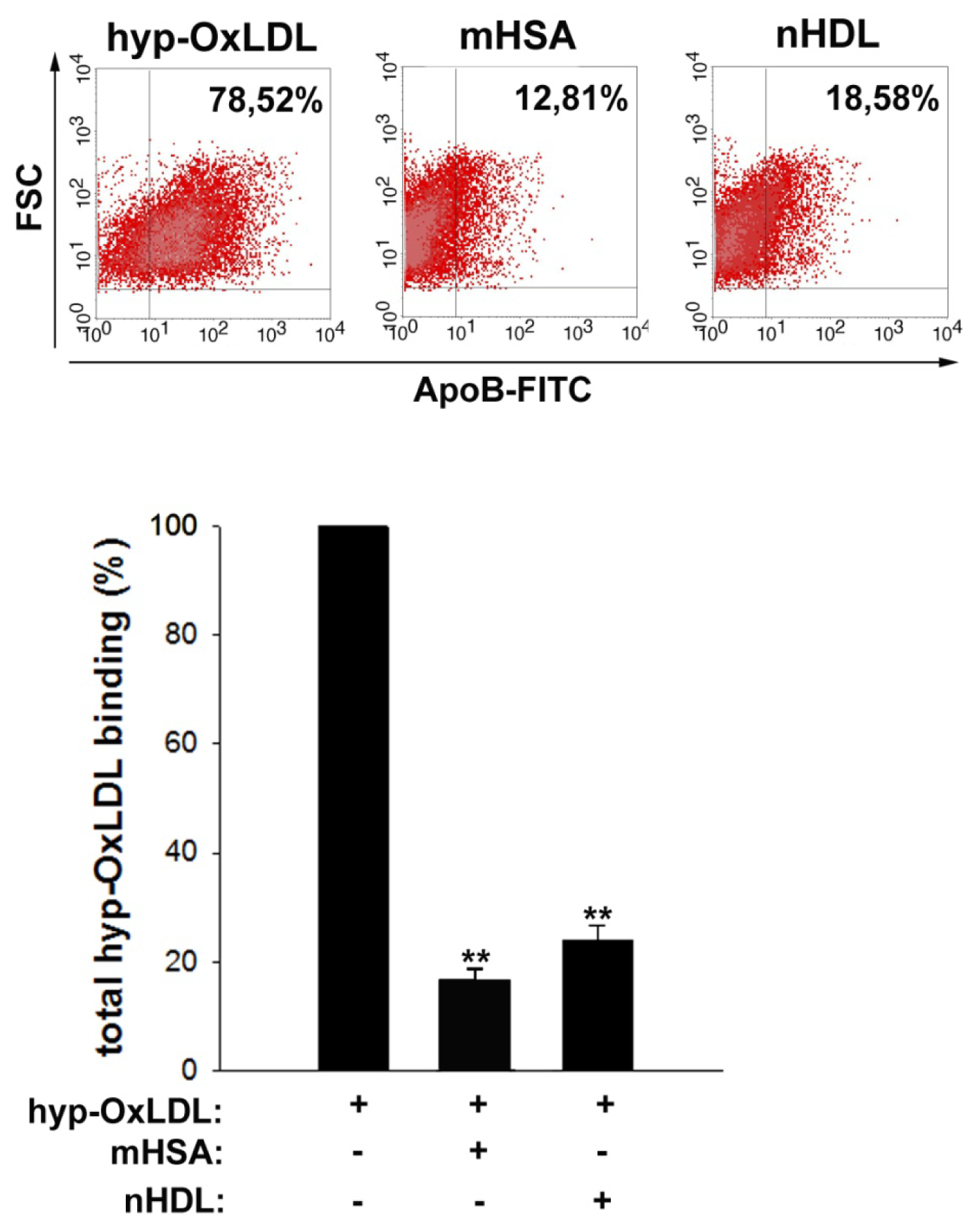
2.4. Native HDL Compete with Binding of Hyp-OxLDL to Human Platelets
3. Experimental Section
3.1. Blood Collection and Isolation of Human Platelets
3.2. Isolation of Lipoproteins and Oxidative Modification of LDL
3.3. Platelet Adhesion under Shear Stress
3.4. Aggregation
3.5. Quantification of CD62P
3.6. Quantification of CD40L Surface Expression and Reactive Oxygen Species (ROS)
3.7. Quantification of Soluble CD40L (sCD40L)
3.8. Determination of Platelet-Neutrophil Aggregates
3.9. Binding Studies
3.10. Statistical Analysis
4. Conclusions
Supplementary Information
ijms-14-10107-s001.pdfConflict of Interest
References
- Stocker, R.; Keaney, J.F. Role of oxidative modifications in atherosclerosis. Physiol. Rev 2004, 84, 1381–1478. [Google Scholar]
- Yla Herttuala, S. Is oxidized low-density lipoprotein present in vivo? Curr. Opin. Lipidol 1998, 9, 337–344. [Google Scholar]
- Lievens, D.; von Hundelshausen, P. Platelets in atherosclerosis. Thromb. Haemost 2011, 106, 827–838. [Google Scholar]
- Gordon, T.; Castelli, W.P.; Hjortland, M.C.; Kannel, W.B.; Dawber, T.R. High density lipoprotein as a protective factor against coronary heart disease. The Framingham Study. Am. J. Med 1977, 62, 707–714. [Google Scholar]
- Castelli, W.P.; Garrison, R.J.; Wilson, P.W.; Abbott, R.D.; Kalousdian, S.; Kannel, W.B. Incidence of coronary heart disease and lipoprotein cholesterol levels. The Framingham Study. J. Am. Med. Assoc 1986, 256, 2835–2838. [Google Scholar]
- Toikka, J.O.; Ahotupa, M.; Viikari, J.S.; Niinikoski, H.; Taskinen, M.; Irjala, K.; Hartiala, J.J.; Raitakari, O.T. Constantly low HDL-cholesterol concentration relates to endothelial dysfunction and increased in vivo LDL-oxidation in healthy young men. Atherosclerosis 1999, 147, 133–138. [Google Scholar]
- Fielding, C.J.; Fielding, P.E. Molecular physiology of reverse cholesterol transport. J. Lipid Res 1995, 36, 211–228. [Google Scholar]
- Mineo, C.; Shaul, P.W. Modulation of endothelial NO production by high-density lipoprotein. Cold Spring Harbor Symp. Quant. Biol 2002, 67, 459–469. [Google Scholar]
- Nofer, J.R.; Kehrel, B.; Fobker, M.; Levkau, B.; Assmann, G.; von Eckardstein, A. HDL and arteriosclerosis: Beyond reverse cholesterol transport. Atherosclerosis 2002, 161, 1–16. [Google Scholar]
- Galle, J.; Ochslen, M.; Schollmeyer, P.; Wanner, C. Oxidized lipoproteins inhibit endothelium-dependent vasodilation. Effects of pressure and high-density lipoprotein. Hypertension 1994, 23, 556–564. [Google Scholar]
- Kopprasch, S.; Pietzsch, J.; Graessler, J. The protective effects of HDL and its constituents against neutrophil respiratory burst activation by hypochlorite-oxidized LDL. Mol. Cell. Biochem 2004, 258, 121–127. [Google Scholar]
- Navab, M.; Imes, S.S.; Hama, S.Y.; Hough, G.P.; Ross, L.A.; Bork, R.W.; Valente, A.J.; Berliner, J.A.; Drinkwater, D.C.; Laks, H. Monocyte transmigration induced by modification of low density lipoprotein in cocultures of human aortic wall cells is due to induction of monocyte chemotactic protein 1 synthesis and is abolished by high density lipoprotein. J. Clin. Invest 1991, 88, 2039–2046. [Google Scholar]
- Mertens, A.; Holvoet, P. Oxidized LDL and HDL: Antagonists in atherothrombosis. FASEB J 2001, 15, 2073–2084. [Google Scholar]
- Takahashi, Y.; Chiba, H.; Matsuno, K.; Akita, H.; Hui, S.P.; Nagasaka, H.; Nakamura, H.; Kobayashi, K.; Tandon, N.N.; Jamieson, G.A. Native lipoproteins inhibit platelet activation induced by oxidized lipoproteins. Biochem. Biophys. Res. Commun 1996, 222, 453–459. [Google Scholar]
- Mehta, J.L.; Chen, L.Y. Reversal by high-density lipoprotein of the effect of oxidized low-density lipoprotein on nitric oxide synthase protein expression in human platelets. J. Lab. Clin. Med 1996, 127, 287–295. [Google Scholar]
- Volf, I.; Bielek, E.; Moeslinger, T.; Koller, F.; Koller, E. Modification of protein moiety of human low density lipoprotein by hypochlorite generates strong platelet agonist. Arterioscler. Thromb. Vasc. Biol 2000, 20, 2011–2018. [Google Scholar]
- Daugherty, A.; Dunn, J.L.; Rateri, D.L.; Heinecke, J.W. Myeloperoxidase, a catalyst for lipoprotein oxidation, is expressed in human atherosclerotic lesions. J. Clin. Invest 1994, 94, 437–444. [Google Scholar]
- Volf, I.; Roth, A.; Cooper, J.; Moeslinger, T.; Koller, E. Hypochlorite modified LDL are a stronger agonist for platelets than copper oxidized LDL. FEBS Lett 2000, 483, 155–159. [Google Scholar]
- Nergiz-Unal, R.; Lamers, M.M.; van Kruchten, R.; Luiken, J.J.; Cosemans, J.M.; Glatz, J.F.; Kuijpers, M.J.; Heemskerk, J.W. Signaling role of CD36 in platelet activation and thrombus formation on immobilized thrombospondin or oxidized low-density lipoprotein. J. Thromb. Haemost 2011, 9, 1835–1846. [Google Scholar]
- Krotz, F.; Sohn, H.Y.; Pohl, U. Reactive oxygen species: Players in the platelet game. Arterioscler. Thromb. Vasc. Biol 2004, 24, 1988–1996. [Google Scholar]
- Nofer, J.R.; Brodde, M.F.; Kehrel, B.E. High-density lipoproteins, platelets and the pathogenesis of atherosclerosis. Clin. Exp. Pharmacol. Physiol 2010, 37, 726–735. [Google Scholar]
- Cettour-Rose, P.; Nguyen, T.X.; Serrander, L.; Kaufmann, M.T.; Dayer, J.M.; Burger, D.; Roux-Lombard, P. T cell contact-mediated activation of respiratory burst in human polymorphonuclear leukocytes is inhibited by high-density lipoproteins and involves CD18. J. Leukoc. Biol 2005, 77, 52–58. [Google Scholar]
- Pignatelli, P.; Sanguigni, V.; Lenti, L.; Ferro, D.; Finocchi, A.; Rossi, P.; Violi, F. gp91phox-dependent expression of platelet CD40 ligand. Circulation 2004, 110, 1326–1329. [Google Scholar]
- Rizvi, M.; Pathak, D.; Freedman, J.E.; Chakrabarti, S. CD40-CD40 ligand interactions in oxidative stress, inflammation and vascular disease. Trends Mol. Med 2008, 14, 530–538. [Google Scholar]
- Andre, P.; Nannizzi-Alaimo, L.; Prasad, S.K.; Phillips, D.R. Platelet-derived CD40L: The switch-hitting player of cardiovascular disease. Circulation 2002, 106, 896–899. [Google Scholar]
- Totani, L.; Evangelista, V. Platelet-leukocyte interactions in cardiovascular disease and beyond. Arterioscler. Thromb. Vasc. Biol 2010, 30, 2357–2361. [Google Scholar]
- De Groot, P.G.; Urbanus, R.T.; Roest, M. Platelet interaction with the vessel wall. Handb. Exp. Pharmacol 2012, 210, 87–110. [Google Scholar]
- Michelson, A.D.; Barnard, M.R.; Krueger, L.A.; Valeri, C.R.; Furman, M.I. Circulating monocyte-platelet aggregates are a more sensitive marker of in vivo platelet activation than platelet surface P-selectin: Studies in baboons, human coronary intervention, and human acute myocardial infarction. Circulation 2001, 104, 1533–1537. [Google Scholar]
- Badrnya, S.; Butler, L.M.; Soderberg-Naucler, C.; Volf, I.; Assinger, A. Platelets directly enhance neutrophil transmigration in response to oxidised low-density lipoprotein. Thromb. Haemost 2012, 108, 719–729. [Google Scholar]
- Soehnlein, O. Multiple roles for neutrophils in atherosclerosis. Circ. Res 2012, 110, 875–888. [Google Scholar]
- Naruko, T.; Ueda, M.; Haze, K.; van der Wal, A.C.; van der Loos, C.M.; Itoh, A.; Komatsu, R.; Ikura, Y.; Ogami, M.; Shimada, Y.; et al. Neutrophil infiltration of culprit lesions in acute coronary syndromes. Circulation 2002, 106, 2894–2900. [Google Scholar]
- Maugeri, N.; Rovere-Querini, P.; Evangelista, V.; Godino, C.; Demetrio, M.; Baldini, M.; Figini, F.; Coppi, G.; Slavich, M.; Camera, M.; et al. An intense and short-lasting burst of neutrophil activation differentiates early acute myocardial infarction from systemic inflammatory syndromes. PLoS One 2012, 7, e39484. [Google Scholar]
- Murphy, A.J.; Woollard, K.J.; Suhartoyo, A.; Stirzaker, R.A.; Shaw, J.; Sviridov, D.; Chin-Dusting, J.P. Neutrophil activation is attenuated by high-density lipoprotein and apolipoprotein A–I in in vitro and in vivo models of inflammation. Arterioscler. Thromb. Vasc. Biol 2011, 31, 1333–1341. [Google Scholar]
- Faraday, N.; Scharpf, R.B.; Dodd-o, J.M.; Martinez, E.A.; Rosenfeld, B.A.; Dorman, T. Leukocytes can enhance platelet-mediated aggregation and thromboxane release via interaction of P-selectin glycoprotein ligand 1 with P-selectin. Anesthesiology 2001, 94, 145–151. [Google Scholar]
- Weyrich, A.S.; Elstad, M.R.; McEver, R.P.; McIntyre, T.M.; Moore, K.L.; Morrissey, J.H.; Prescott, S.M.; Zimmerman, G.A. Activated platelets signal chemokine synthesis by human monocytes. J. Clin. Invest 1996, 97, 1525–1534. [Google Scholar]
- Nagata, K.; Tsuji, T.; Todoroki, N.; Katagiri, Y.; Tanoue, K.; Yamazaki, H.; Hanai, N.; Irimura, T. Activated platelets induce superoxide anion release by monocytes and neutrophils through P-selectin (CD62). J. Immunol 1993, 151, 3267–3273. [Google Scholar]
- Korporaal, S.J.; van Eck, M.; Adelmeijer, J.; Ijsseldijk, M.; Out, R.; Lisman, T.; Lenting, P.J.; van Berkel, T.J.; Akkerman, J.W. Platelet activation by oxidized low density lipoprotein is mediated by CD36 and scavenger receptor-A. Arterioscler. Thromb. Vasc. Biol 2007, 27, 2476–2483. [Google Scholar]
- Chen, R.; Chen, X.; Salomon, R.G.; McIntyre, T.M. Platelet activation by low concentrations of intact oxidized LDL particles involves the PAF receptor. Arterioscler. Thromb. Vasc. Biol 2008, 29, 363–371. [Google Scholar]
- Assinger, A.; Koller, F.; Schmid, W.; Zellner, M.; Koller, E.; Volf, I. Hypochlorite-oxidized LDL induces intraplatelet ROS formation and surface exposure of CD40L—A prominent role of CD36. Atherosclerosis 2010, 213, 129–134. [Google Scholar]
- Calvo, D.; Gomez Coronado, D.; Suarez, Y.; Lasuncion, M.A.; Vega, M.A. Human CD36 is a high affinity receptor for the native lipoproteins HDL, LDL, and VLDL. J. Lipid Res 1998, 39, 777–788. [Google Scholar]
- Volf, I.; Moeslinger, T.; Cooper, J.; Schmid, W.; Koller, E. Human platelets exclusively bind oxidized LDL showing no specificity for acetylated LDL. FEBS Lett 1999, 449, 141–145. [Google Scholar]
- Chen, L.Y.; Mehta, J.L. Inhibitory effect of high-density lipoprotein on platelet function is mediated by increase in nitric oxide synthase activity in platelets. Life Sci 1994, 55, 1815–1821. [Google Scholar]
- Nofer, J.R.; Walter, M.; Kehrel, B.; Wierwille, S.; Tepel, M.; Seedorf, U.; Assmann, G. HDL3-mediated inhibition of thrombin-induced platelet aggregation and fibrinogen binding occurs via decreased production of phosphoinositide-derived second messengers 1,2-diacylglycerol and inositol 1,4,5-tris-phosphate. Arterioscler. Thromb. Vasc. Biol 1998, 18, 861–869. [Google Scholar]
- Brodde, M.F.; Korporaal, S.J.; Herminghaus, G.; Fobker, M.; van Berkel, T.J.; Tietge, U.J.; Robenek, H.; van Eck, M.; Kehrel, B.E.; Nofer, J.R. Native high-density lipoproteins inhibit platelet activation via scavenger receptor BI: Role of negatively charged phospholipids. Atherosclerosis 2011, 215, 374–382. [Google Scholar]
- Assinger, A.; Schmid, W.; Eder, S.; Schmid, D.; Koller, E.; Volf, I. Oxidation by hypochlorite converts protective HDL into a potent platelet agonist. FEBS Lett 2008, 582, 778–784. [Google Scholar]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem 1951, 193, 265–275. [Google Scholar]
- Assinger, A.; Laky, M.; Schabbauer, G.; Hirschl, A.; Buchberger, E.; Binder, B.R.; Volf, I. Efficient phagocytosis of periodontopathogens by neutrophils requires plasma factors, platelets and TLR2. J. Thromb. Haemost 2011, 9, 799–809. [Google Scholar]
- Shaw, J.A.; Bobik, A.; Murphy, A.; Kanellakis, P.; Blombery, P.; Mukhamedova, N.; Woollard, K.; Lyon, S.; Sviridov, D.; Dart, A.M. Infusion of reconstituted high-density lipoprotein leads to acute changes in human atherosclerotic plaque. Circ. Res 2008, 103, 1084–1091. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Badrnya, S.; Assinger, A.; Volf, I. Native High Density Lipoproteins (HDL) Interfere with Platelet Activation Induced by Oxidized Low Density Lipoproteins (OxLDL). Int. J. Mol. Sci. 2013, 14, 10107-10121. https://doi.org/10.3390/ijms140510107
Badrnya S, Assinger A, Volf I. Native High Density Lipoproteins (HDL) Interfere with Platelet Activation Induced by Oxidized Low Density Lipoproteins (OxLDL). International Journal of Molecular Sciences. 2013; 14(5):10107-10121. https://doi.org/10.3390/ijms140510107
Chicago/Turabian StyleBadrnya, Sigrun, Alice Assinger, and Ivo Volf. 2013. "Native High Density Lipoproteins (HDL) Interfere with Platelet Activation Induced by Oxidized Low Density Lipoproteins (OxLDL)" International Journal of Molecular Sciences 14, no. 5: 10107-10121. https://doi.org/10.3390/ijms140510107