Resistin Promotes the Expression of Vascular Endothelial Growth Factor in Ovary Carcinoma Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
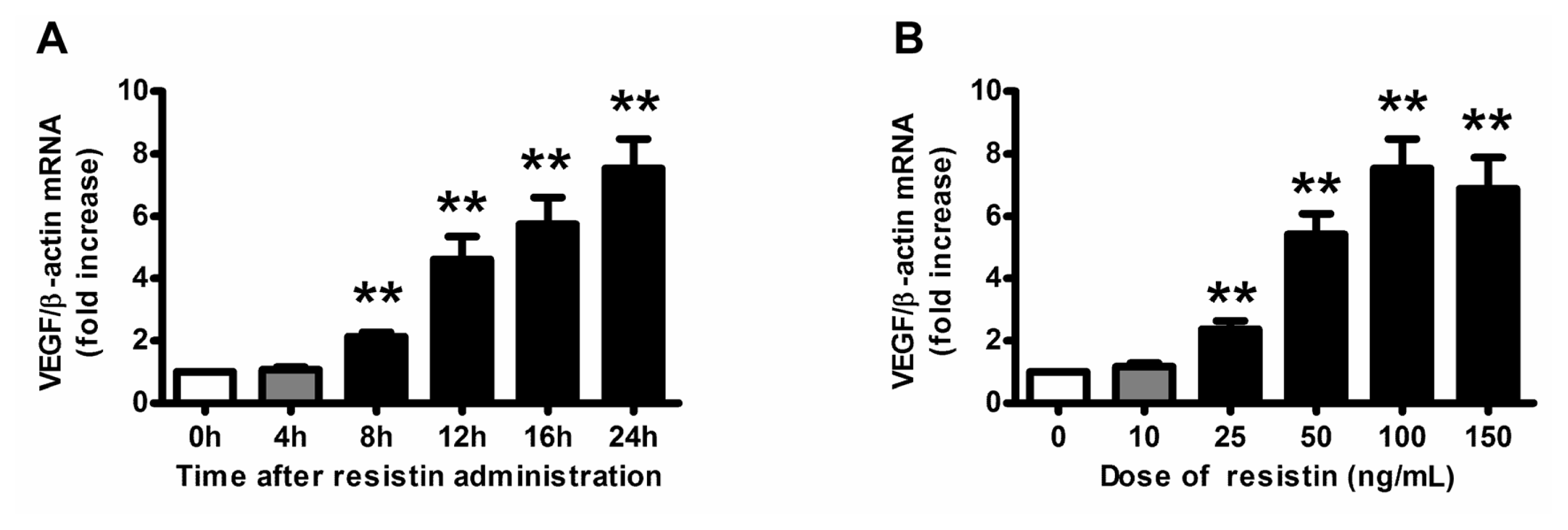
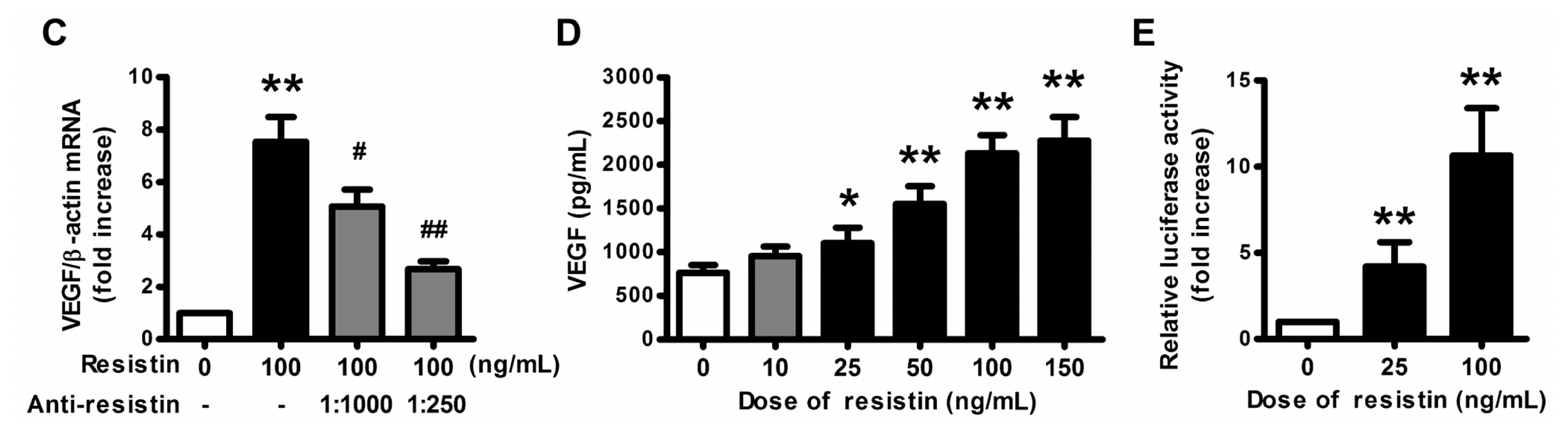
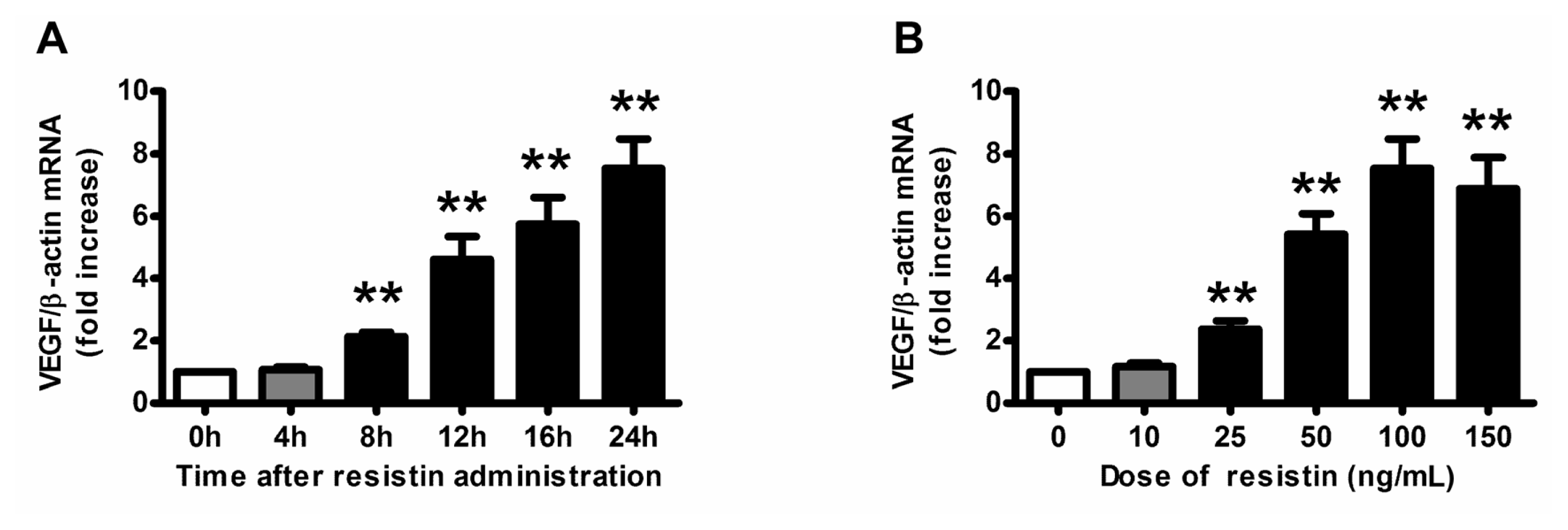
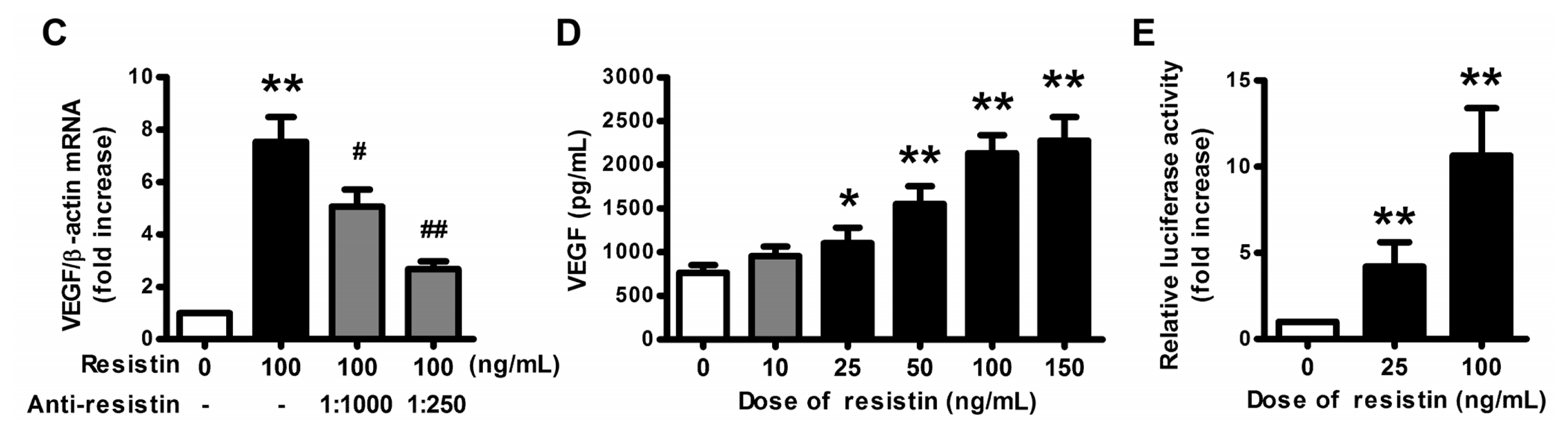
2.1. VEGF Expression and Production in Ovarian Epithelial Carcinoma Cells Was Induced by Resistin
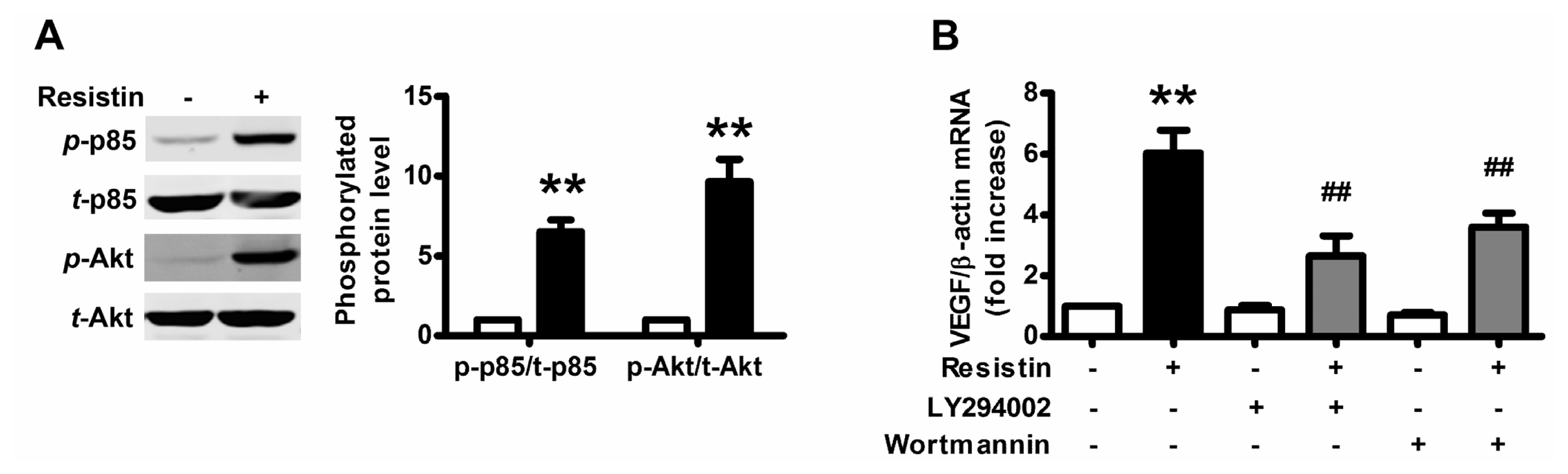
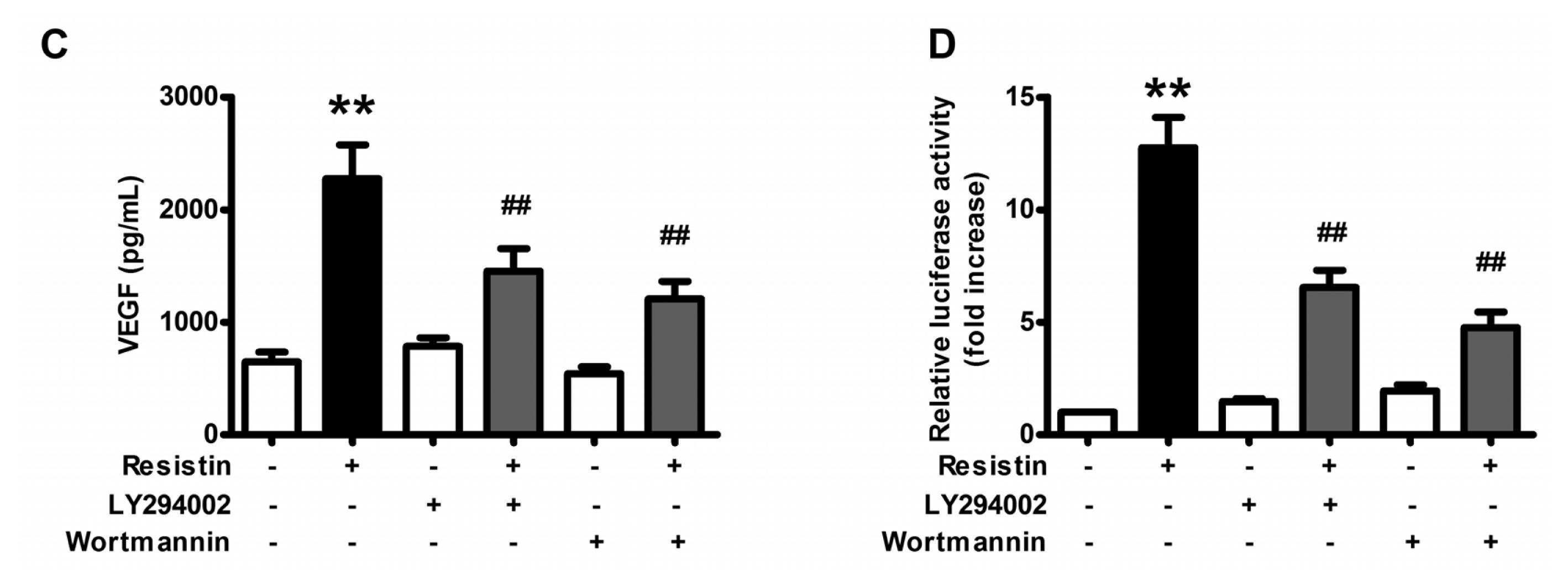
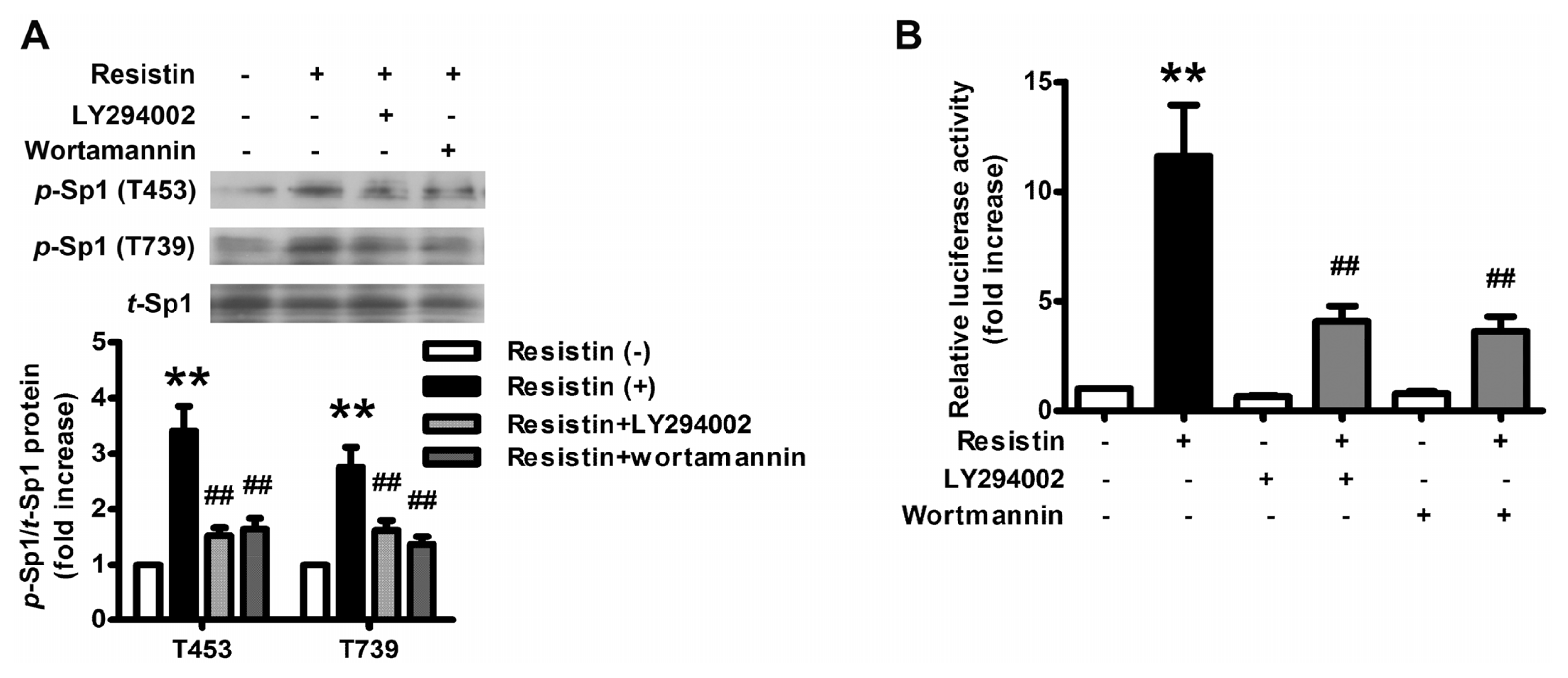
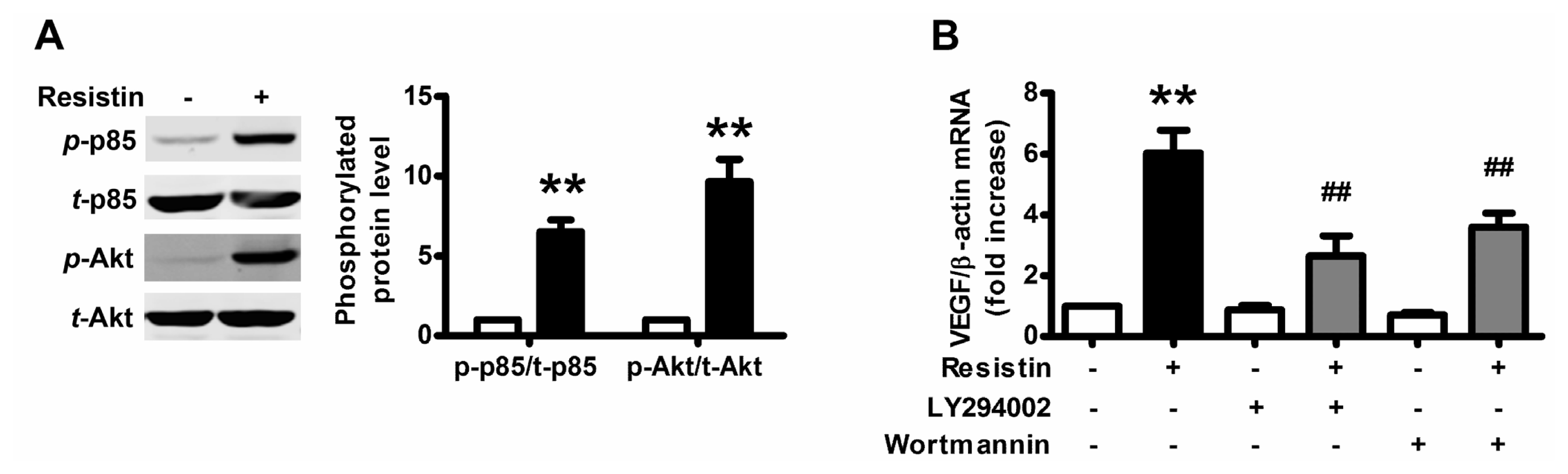
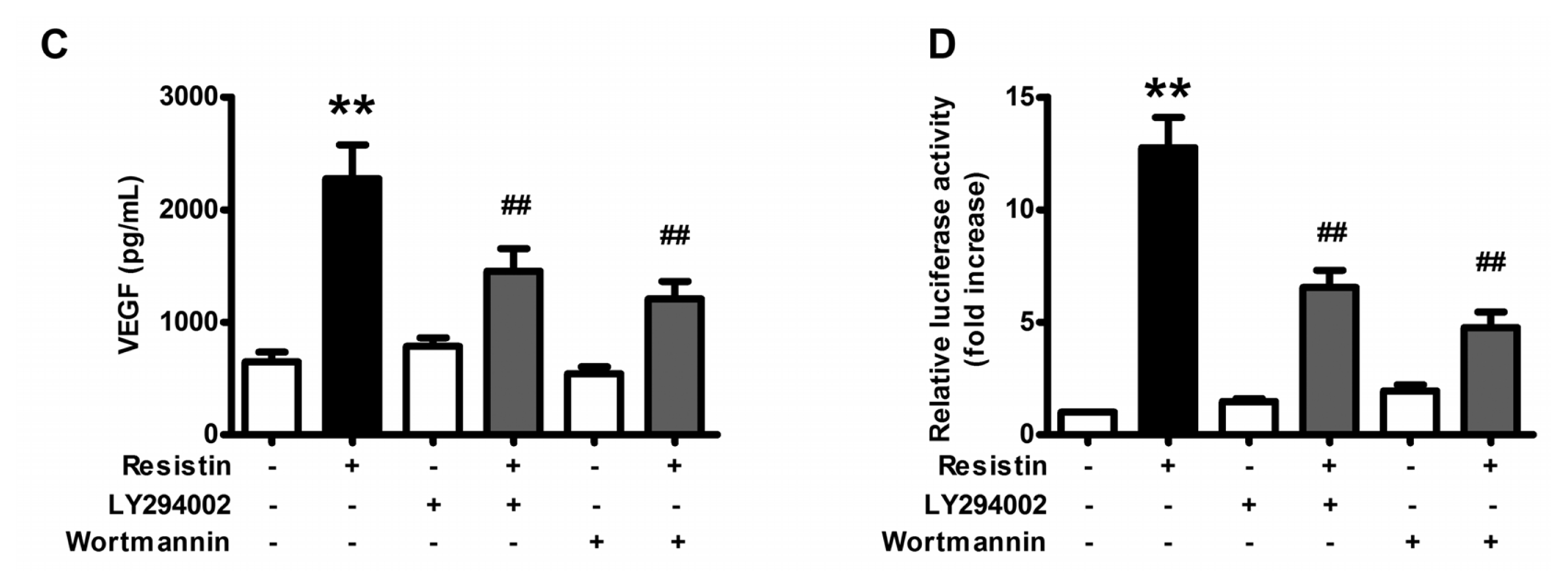
2.2. PI3K/Akt Pathway Is Activated upon Resistin Stimulation
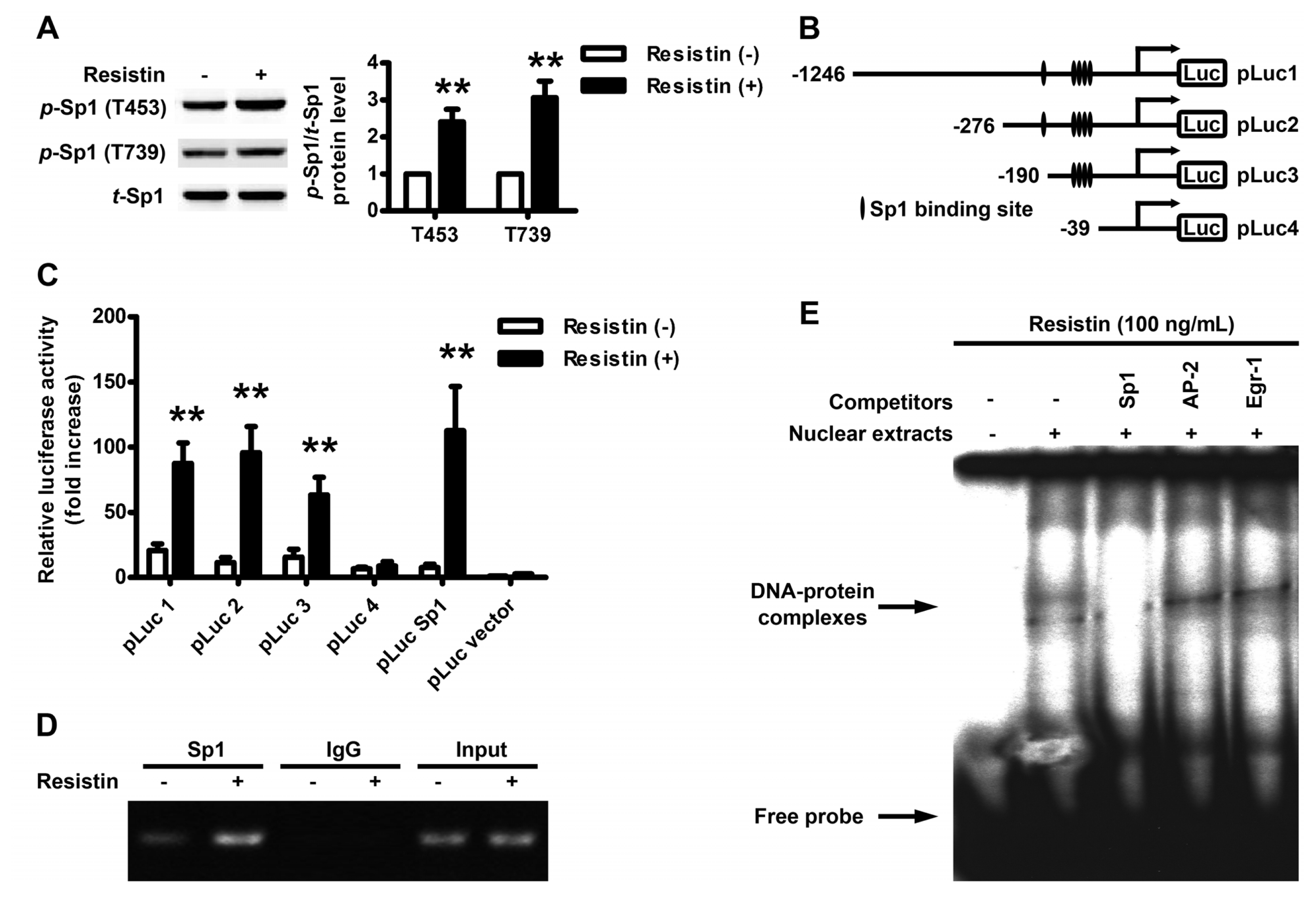
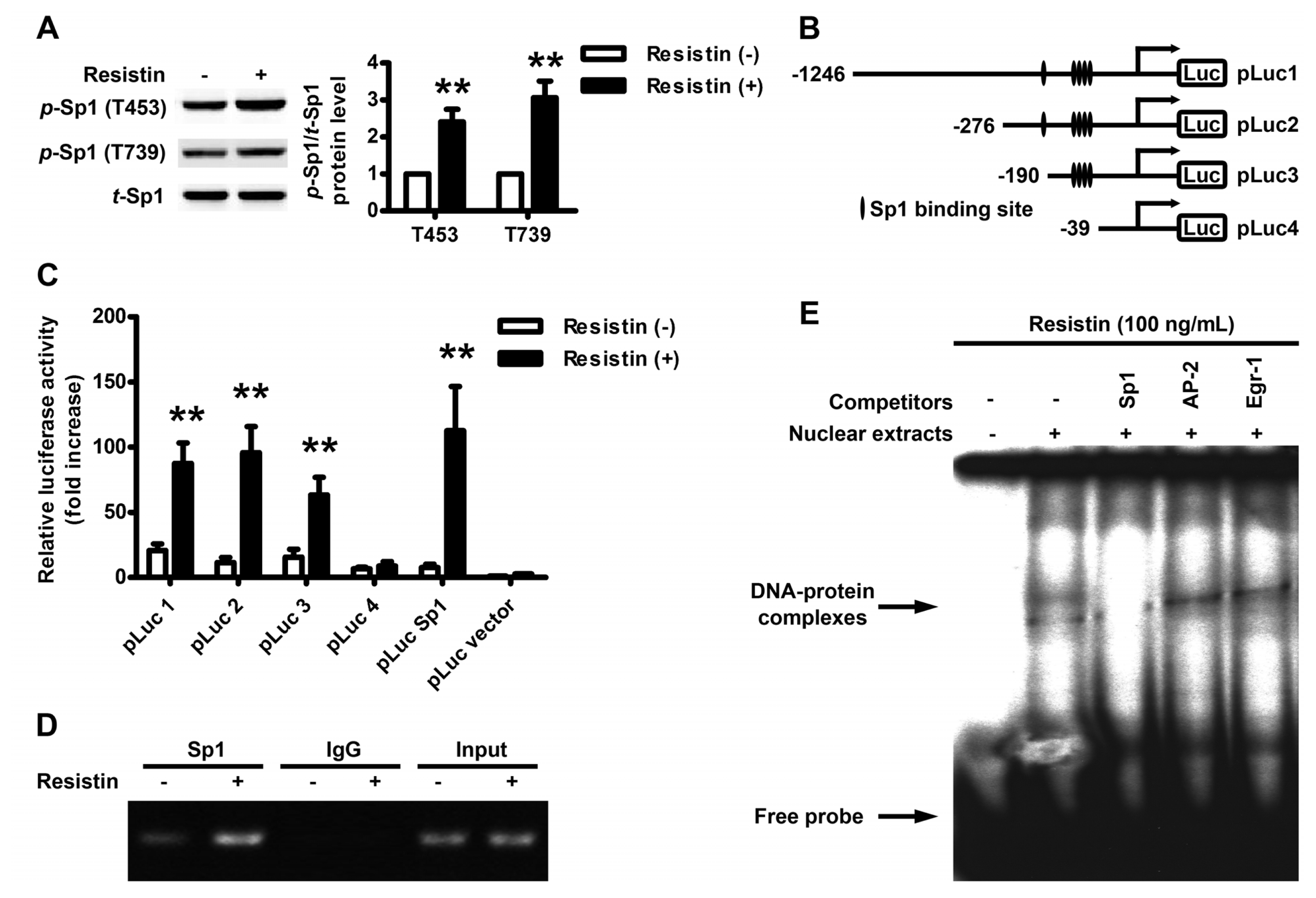
2.3. Localization of the Resistin Regulatory Element in the VEGF Gene Promoter
2.4. Resistin Induces PI3K/Akt-dependent Phosphorylation of Sp1
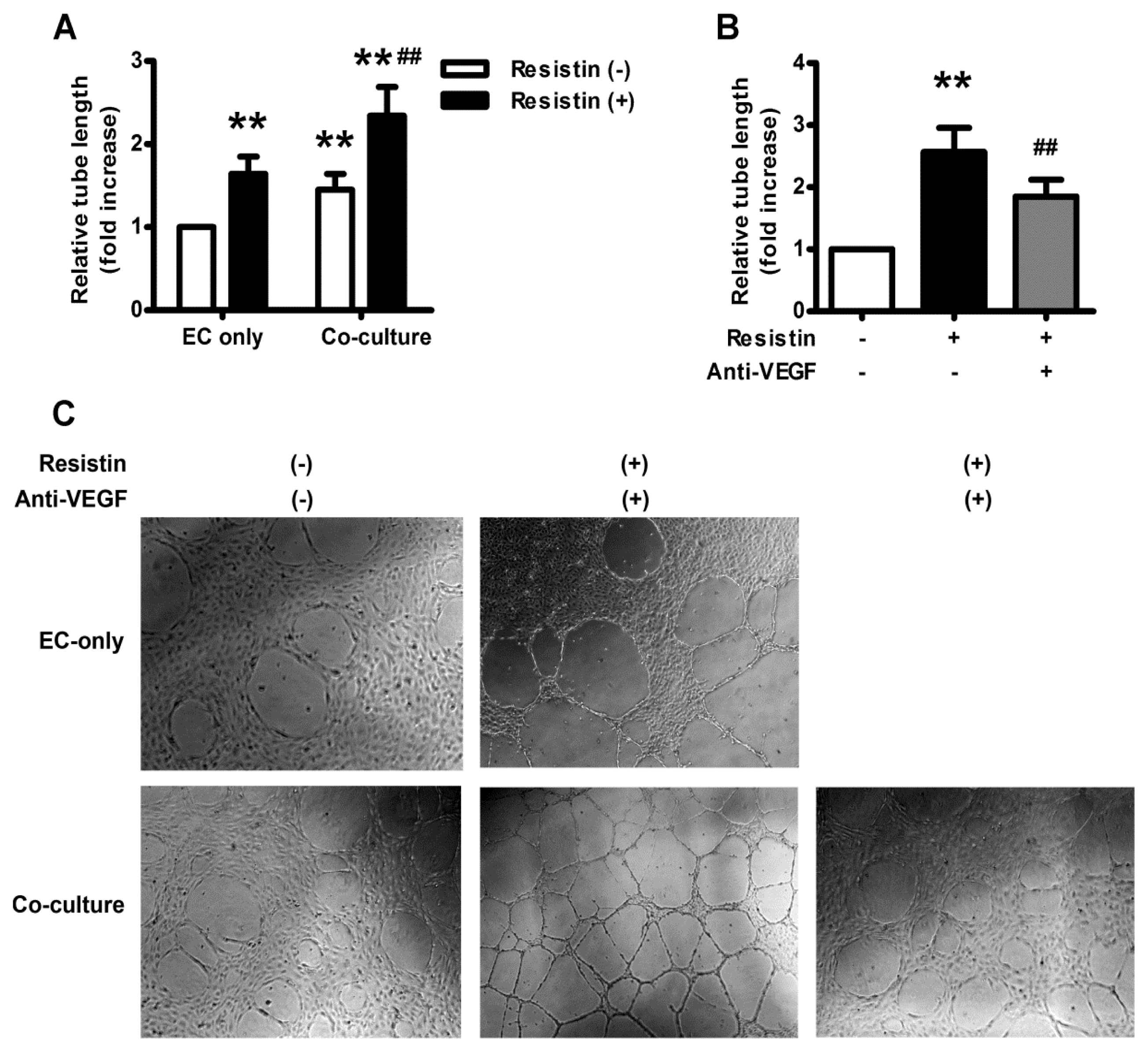
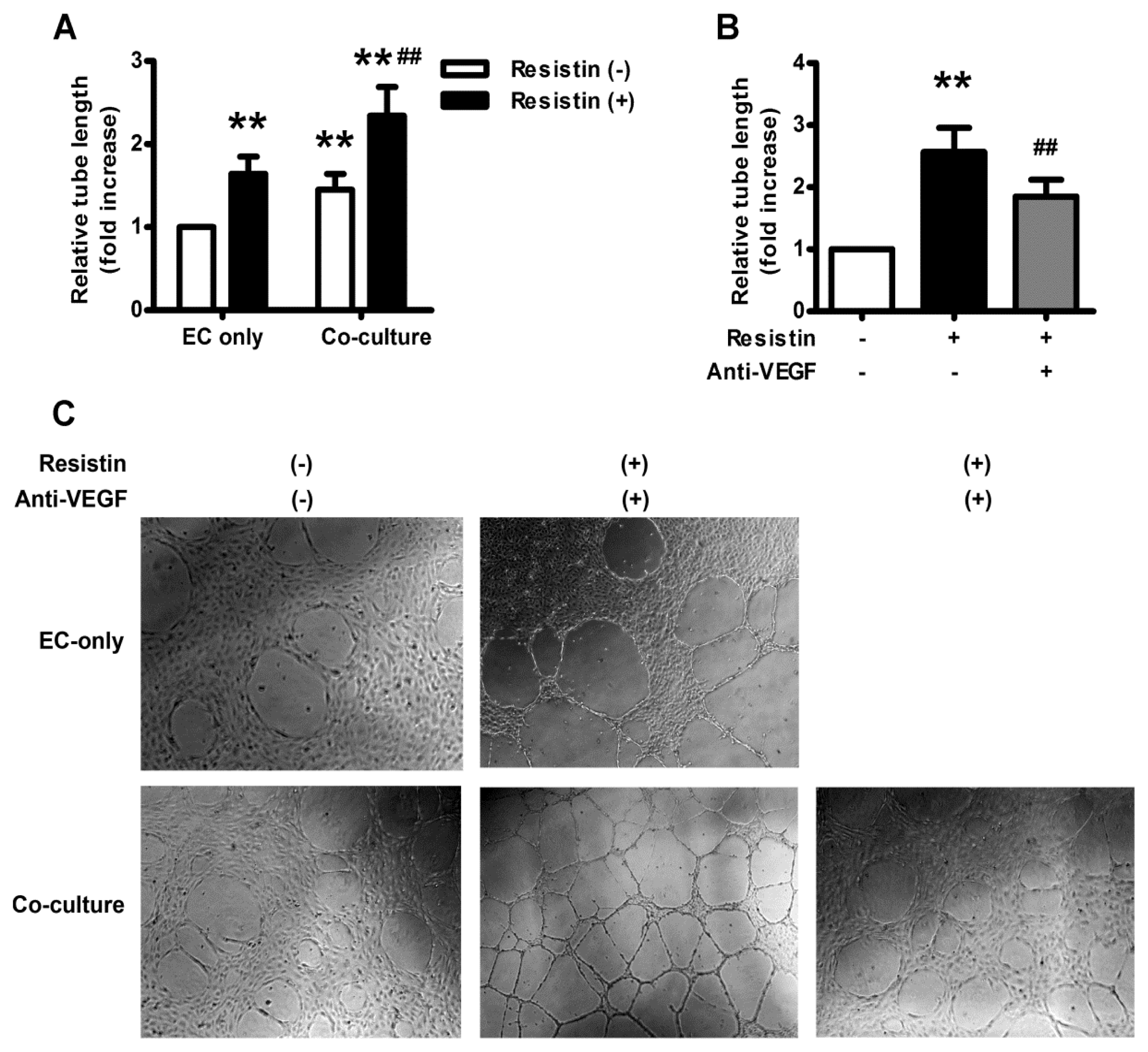
2.5. VEGF Mediates Resistin-Induced Angiogenesis of Ovary Carcinoma in Vitro
2.6. Discussion
3. Material and Methods
3.1. Chemicals and Reagents
3.2. Cell Culture
3.3. RNA Extraction and Quantitative Real Time PCR Analysis
3.4. Quantitation of VEGF by ELISA
3.5. Preparation of Cytosolic Proteins and Western Blot Analysis
3.6. Plasmid Construction and Transient Transfection Assays
3.7. Preparation of Nuclear Proteins and Electrophoretic Mobility Shift Assay (EMSA)
3.8. Chromatin Immunoprecipitation (ChIP) and PCR Analysis
3.9. Capillary-Like Tube Formation Assay
3.10. Statistical Analysis
4. Conclusions
Conflict of Interest
References
- Billottet, C.; Janji, B.; Thiery, J.P.; Jouanneau, J. Rapid tumor development and potent vascularization are independent events in carcinoma producing FGF-1 or FGF-2. Oncogene 2002, 21, 8128–8139. [Google Scholar]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar]
- Asahara, T.; Kalka, C.; Isner, J.M. Stem cell therapy and gene transfer for regeneration. Gene Ther 2000, 7, 451–457. [Google Scholar]
- Rafii, S. Circulating endothelial precursors: Mystery, reality, and promise. J. Clin. Invest 2000, 105, 17–19. [Google Scholar]
- Carmeliet, P. Mechanisms of angiogenesis and arteriogenesis. Nat. Med 2000, 6, 389–395. [Google Scholar]
- Ferrara, N.; Alitalo, K. Clinical applications of angiogenic growth factors and their inhibitors. Nat. Med 1999, 5, 1359–1364. [Google Scholar]
- Bernatchez, P.N.; Rollin, S.; Soker, S.; Sirois, M.G. Relative effects of VEGF-A and VEGF-C on endothelial cell proliferation, migration and PAF synthesis: Role of neuropilin-1. J. Cell. Biochem 2002, 85, 629–639. [Google Scholar]
- Fukumura, D.; Xavier, R.; Sugiura, T.; Chen, Y.; Park, E.C.; Lu, N.; Selig, M.; Nielsen, G.; Taksir, T.; Jain, R.K.; et al. Tumor induction of VEGF promoter activity in stromal cells. Cell 1998, 94, 715–725. [Google Scholar]
- Trayhurn, P.; Beattie, J.H. Physiological role of adipose tissue: White adipose tissue as an endocrine and secretory organ. Proc. Nutr. Soc 2001, 60, 329–339. [Google Scholar]
- Yang, H.S.; Yoon, C.; Myung, S.K.; Park, S.M. Effect of obesity on survival of women with epithelial ovarian cancer: A systematic review and meta-analysis of observational studies. Int. J. Gynecol. Cancer 2011, 21, 1525–1532. [Google Scholar]
- Steppan, C.M.; Bailey, S.T.; Bhat, S.; Brown, E.J.; Banerjee, R.R.; Wright, C.M.; Patel, H.R.; Ahima, R.S.; Lazar, M.A. The hormone resistin links obesity to diabetes. Nature 2001, 409, 307–312. [Google Scholar]
- Patel, L.; Buckels, A.C.; Kinghorn, I.J.; Murdock, P.R.; Holbrook, J.D.; Plumpton, C.; Macphee, C.H.; Smith, S.A. Resistin is expressed in human macrophages and directly regulated by PPAR gamma activators. Biochem. Biophys. Res. Commun 2003, 300, 472–476. [Google Scholar]
- Ohmori, R.; Momiyama, Y.; Kato, R.; Taniguchi, H.; Ogura, M.; Ayaori, M.; Nakamura, H.; Ohsuzu, F. Associations between serum resistin levels and insulin resistance, inflammation, and coronary artery disease. J. Am. Coll. Cardiol 2005, 46, 379–380. [Google Scholar]
- Jiang, C.Y.; Wang, W.; Yin, Y.L.; Yuan, Z.R.; Wang, L.B. Expression of the adipocytokine resistin and its association with the clinicopathological features and prognosis of pancreatic ductal adenocarcinoma. Oncol. Lett 2012, 4, 960–964. [Google Scholar]
- Mu, H.; Ohashi, R.; Yan, S.; Chai, H.; Yang, H.; Lin, P.; Yao, Q.; Chen, C. Adipokine resistin promotes in vitro angiogenesis of human endothelial cells. Cardiovasc. Res 2006, 70, 146–157. [Google Scholar]
- Di Simone, N.; di Nicuolo, F.; Sanguinetti, M.; Castellani, R.; D’Asta, M.; Caforio, L.; Caruso, A. Resistin regulates human choriocarcinoma cell invasive behaviour and endothelial cell angiogenic processes. J. Endocrinol 2006, 189, 691–699. [Google Scholar]
- Jiang, C.; Zhang, H.; Zhang, W.; Kong, W.; Zhu, Y.; Zhang, H.; Xu, Q.; Li, Y.; Wang, X. Homocysteine promotes vascular smooth muscle cell migration by induction of the adipokine resistin. Am. J. Physiol. Cell Physiol 2009, 297, C1466–C1476. [Google Scholar]
- Pavelka, J.C.; Brown, R.S.; Karlan, B.Y.; Cass, I.; Leuchter, R.S.; Lagasse, L.D.; Li, A.J. Effect of obesity on survival in epithelial ovarian cancer. Cancer 2006, 107, 1520–1524. [Google Scholar]
- Conde, J.; Scotece, M.; Gomez, R.; Lopez, V.; Gomez-Reino, J.J.; Lago, F.; Gualillo, O. Adipokines: Biofactors from white adipose tissue. A complex hub among inflammation, metabolism, and immunity. Biofactors 2011, 37, 413–420. [Google Scholar]
- Dalamaga, M.; Diakopoulos, K.N.; Mantzoros, C.S. The role of adiponectin in cancer: A review of current evidence. Endocr. Rev 2012, 33, 547–594. [Google Scholar]
- Li, Y.; Jiang, C.; Xu, G.; Wang, N.; Zhu, Y.; Tang, C.; Wang, X. Homocysteine upregulates resistin production from adipocytes in vivo and in vitro. Diabetes 2008, 57, 817–827. [Google Scholar]
- Filkova, M.; Haluzik, M.; Gay, S.; Senolt, L. The role of resistin as a regulator of inflammation: Implications for various human pathologies. Clin. Immunol 2009, 133, 157–170. [Google Scholar] [Green Version]
- Dalamaga, M.; Sotiropoulos, G.; Karmaniolas, K.; Pelekanos, N.; Papadavid, E.; Lekka, A. Serum resistin: A biomarker of breast cancer in postmenopausal women? Association with clinicopathological characteristics, tumor markers, inflammatory and metabolic parameters. Clin. Biochem 2013, 46, 584–590. [Google Scholar]
- Nakajima, T.E.; Yamada, Y.; Hamano, T.; Furuta, K.; Gotoda, T.; Katai, H.; Kato, K.; Hamaguchi, T.; Shimada, Y. Adipocytokine levels in gastric cancer patients: Resistin and visfatin as biomarkers of gastric cancer. J. Gastroenterol 2009, 44, 685–690. [Google Scholar]
- Danese, E.; Montagnana, M.; Minicozzi, A.M.; Bonafini, S.; Ruzzenente, O.; Gelati, M.; de Manzoni, G.; Lippi, G.; Guidi, G.C. The role of resistin in colorectal cancer. Clin. Chim. Acta 2012, 413, 760–764. [Google Scholar]
- Hlavna, M.; Kohut, L.; Lipkova, J.; Bienertova-Vasku, J.; Dostalova, Z.; Chovanec, J.; Vasku, A. Relationship of resistin levels with endometrial cancer risk. Neoplasma 2011, 58, 124–128. [Google Scholar]
- Lee, Y.C.; Chen, Y.J.; Wu, C.C.; Lo, S.; Hou, M.F.; Yuan, S.S. Resistin expression in breast cancer tissue as a marker of prognosis and hormone therapy stratification. Gynecol. Oncol 2012, 125, 742–750. [Google Scholar]
- Zheng, L.D.; Yang, C.L.; Qi, T.; Qi, M.; Tong, L.; Tong, Q.S. Effects of resistin-like molecule beta over-expression on gastric cancer cells in vitro. World J. Gastroenterol 2012, 18, 754–766. [Google Scholar]
- Kim, H.J.; Lee, Y.S.; Won, E.H.; Chang, I.H.; Kim, T.H.; Park, E.S.; Kim, M.K.; Kim, W.; Myung, S.C. Expression of resistin in the prostate and its stimulatory effect on prostate cancer cell proliferation. BJU Int 2011, 108, E77–E83. [Google Scholar]
- Pan, B.; Zhao, M.H.; Chen, Z.; Lu, L.; Wang, Y.; Shi, D.W.; Han, P.Z. Inhibitory effects of resistin-13-peptide on the proliferation, adhesion, and invasion of MDA-MB-231 in human breast carcinoma cells. Endocr. Relat. Cancer 2007, 14, 1063–1071. [Google Scholar]
- Claffey, K.P.; Robinson, G.S. Regulation of VEGF/VPF expression in tumor cells: Consequences for tumor growth and metastasis. Cancer Metastasis Rev 1996, 15, 165–176. [Google Scholar]
- Claffey, K.P.; Brown, L.F.; del Aguila, L.F.; Tognazzi, K.; Yeo, K.T.; Manseau, E.J.; Dvorak, H.F. Expression of vascular permeability factor/vascular endothelial growth factor by melanoma cells increases tumor growth, angiogenesis, and experimental metastasis. Cancer Res 1996, 56, 172–181. [Google Scholar]
- Schweizer, M.T.; Carducci, M.A. From bevacizumab to tasquinimod: Angiogenesis as a therapeutic target in prostate cancer. Cancer J 2013, 19, 99–106. [Google Scholar]
- Pages, G.; Pouyssegur, J. Transcriptional regulation of the Vascular Endothelial Growth Factor gene—A concert of activating factors. Cardiovasc. Res 2005, 65, 564–573. [Google Scholar]
- Gille, J.; Swerlick, R.A.; Caughman, S.W. Transforming growth factor-alpha-induced transcriptional activation of the vascular permeability factor (VPF/VEGF) gene requires AP-2-dependent DNA binding and transactivation. EMBO J 1997, 16, 750–759. [Google Scholar]
- Hanson, J.; Gorman, J.; Reese, J.; Fraizer, G. Regulation of vascular endothelial growth factor, VEGF, gene promoter by the tumor suppressor, WT1. Front. Biosci 2007, 12, 2279–2290. [Google Scholar]
- Shima, D.T.; Gougos, A.; Miller, J.W.; Tolentino, M.; Robinson, G.; Adamis, A.P.; D’Amore, P.A. Cloning and mRNA expression of vascular endothelial growth factor in ischemic retinas of Macaca fascicularis. Invest. Ophthalmol. Vis. Sci 1996, 37, 1334–1340. [Google Scholar]
- Li, D.Q.; Pakala, S.B.; Reddy, S.D.; Ohshiro, K.; Zhang, J.X.; Wang, L.; Zhang, Y.; Moreno de Alboran, I.; Pillai, M.R.; Eswaran, J.; et al. Bidirectional autoregulatory mechanism of metastasis-associated protein 1-alternative reading frame pathway in oncogenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 8791–8796. [Google Scholar]
- Gong, M.; Yu, W.; Pei, F.; You, J.; Cui, X.; McNutt, M.A.; Li, G.; Zheng, J. KLF6/Sp1 initiates transcription of the tmsg-1 gene in human prostate carcinoma cells: An exon involved mechanism. J. Cell. Biochem 2012, 113, 329–339. [Google Scholar]
- Santiago, F.S.; Ishii, H.; Shafi, S.; Khurana, R.; Kanellakis, P.; Bhindi, R.; Ramirez, M.J.; Bobik, A.; Martin, J.F.; Chesterman, C.N.; et al. Yin Yang-1 inhibits vascular smooth muscle cell growth and intimal thickening by repressing p21WAF1/Cip1 transcription and p21WAF1/Cip1-Cdk4-cyclin D1 assembly. Circ. Res 2007, 101, 146–155. [Google Scholar]
- Grinstein, E.; Jundt, F.; Weinert, I.; Wernet, P.; Royer, H.D. Sp1 as G1 cell cycle phase specific transcription factor in epithelial cells. Oncogene 2002, 21, 1485–1492. [Google Scholar]
- Feng, J.; Zhang, Y.; Xing, D. Low-power laser irradiation (LPLI) promotes VEGF expression and vascular endothelial cell proliferation through the activation of ERK/Sp1 pathway. Cell Signal 2012, 24, 1116–1125. [Google Scholar]
- Schafer, G.; Cramer, T.; Suske, G.; Kemmner, W.; Wiedenmann, B.; Hocker, M. Oxidative stress regulates vascular endothelial growth factor-A gene transcription through Sp1- and Sp3-dependent activation of two proximal GC-rich promoter elements. J. Biol. Chem 2003, 278, 8190–8198. [Google Scholar]
- Gonzalez-Perez, R.R.; Xu, Y.; Guo, S.; Watters, A.; Zhou, W.; Leibovich, S.J. Leptin upregulates VEGF in breast cancer via canonic and non-canonical signalling pathways and NFkappaB/HIF-1alpha activation. Cell Signal 2010, 22, 1350–1362. [Google Scholar]
- Lin, H.H.; Lai, S.C.; Chau, L.Y. Heme oxygenase-1/carbon monoxide induces vascular endothelial growth factor expression via p38 kinase-dependent activation of Sp1. J. Biol. Chem 2011, 286, 3829–3838. [Google Scholar]
- Kim, H.A.; Seo, K.H.; Kang, Y.R.; Ko, H.M.; Kim, K.J.; Back, H.K.; Lee, H.K.; Im, S.Y. Mechanisms of platelet-activating factor-induced enhancement of VEGF expression. Cell Physiol. Biochem 2011, 27, 55–62. [Google Scholar]
- Chuang, J.Y.; Wang, Y.T.; Yeh, S.H.; Liu, Y.W.; Chang, W.C.; Hung, J.J. Phosphorylation by c-Jun NH2-terminal kinase 1 regulates the stability of transcription factor Sp1 during mitosis. Mol. Biol. Cell 2008, 19, 1139–1151. [Google Scholar]
- Dwivedi, P.P.; Gao, X.H.; Tan, J.C.; Evdokiou, A.; Ferrante, A.; Morris, H.A.; May, B.K.; Hii, C.S. A role for the phosphatidylinositol 3-kinase—Protein kinase C zeta—Sp1 pathway in the 1,25-dihydroxyvitamin D3 induction of the 25-hydroxyvitamin D3 24-hydroxylase gene in human kidney cells. Cell Signal 2010, 22, 543–552. [Google Scholar]
- Bae, I.H.; Park, M.J.; Yoon, S.H.; Kang, S.W.; Lee, S.S.; Choi, K.M.; Um, H.D. Bcl-w promotes gastric cancer cell invasion by inducing matrix metalloproteinase-2 expression via phosphoinositide 3-kinase, Akt, and Sp1. Cancer Res 2006, 66, 4991–4995. [Google Scholar]
- Su, Y.; Zheng, L.; Wang, Q.; Bao, J.; Cai, Z.; Liu, A. The PI3K/Akt pathway upregulates Id1 and integrin alpha4 to enhance recruitment of human ovarian cancer endothelial progenitor cells. BMC Cancer 2010, 10, 459. [Google Scholar]
- Zhang, L.; Yang, N.; Katsaros, D.; Huang, W.; Park, J.W.; Fracchioli, S.; Vezzani, C.; Rigault de la Longrais, I.A.; Yao, W.; Rubin, S.C.; et al. The oncogene phosphatidylinositol 3′-kinase catalytic subunit alpha promotes angiogenesis via vascular endothelial growth factor in ovarian carcinoma. Cancer Res 2003, 63, 4225–4231. [Google Scholar]
- Tischer, E.; Mitchell, R.; Hartman, T.; Silva, M.; Gospodarowicz, D.; Fiddes, J.C.; Abraham, J.A. The human gene for vascular endothelial growth factor. Multiple protein forms are encoded through alternative exon splicing. J. Biol. Chem 1991, 266, 11947–11954. [Google Scholar]

© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pang, L.; Zhang, Y.; Yu, Y.; Zhang, S. Resistin Promotes the Expression of Vascular Endothelial Growth Factor in Ovary Carcinoma Cells. Int. J. Mol. Sci. 2013, 14, 9751-9766. https://doi.org/10.3390/ijms14059751
Pang L, Zhang Y, Yu Y, Zhang S. Resistin Promotes the Expression of Vascular Endothelial Growth Factor in Ovary Carcinoma Cells. International Journal of Molecular Sciences. 2013; 14(5):9751-9766. https://doi.org/10.3390/ijms14059751
Chicago/Turabian StylePang, Li, Yi Zhang, Yu Yu, and Shulan Zhang. 2013. "Resistin Promotes the Expression of Vascular Endothelial Growth Factor in Ovary Carcinoma Cells" International Journal of Molecular Sciences 14, no. 5: 9751-9766. https://doi.org/10.3390/ijms14059751