Posttranslational Modifications of GLUT4 Affect Its Subcellular Localization and Translocation

{kind=link}
{kind=link}
Abstract
:1. Introduction
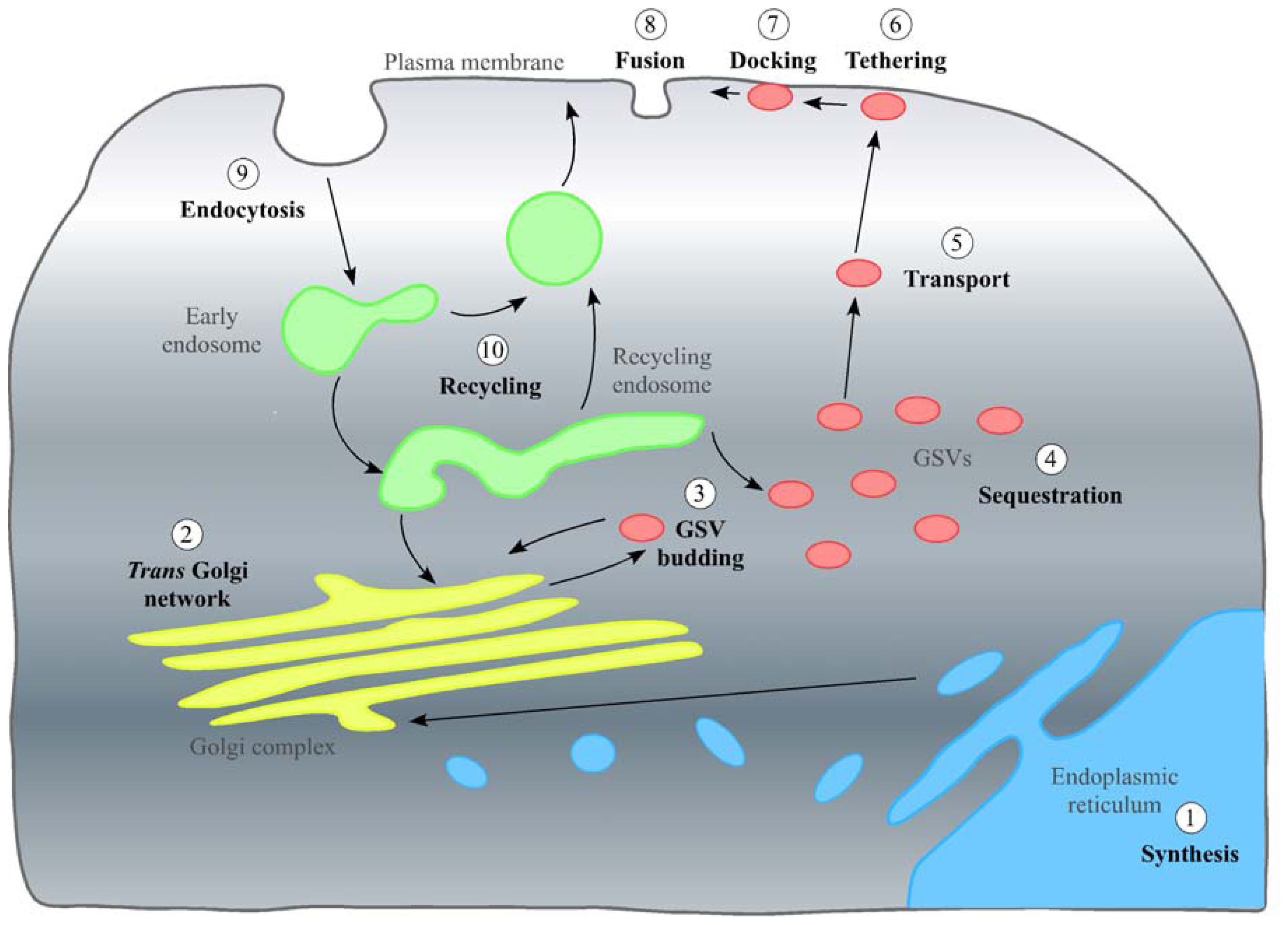
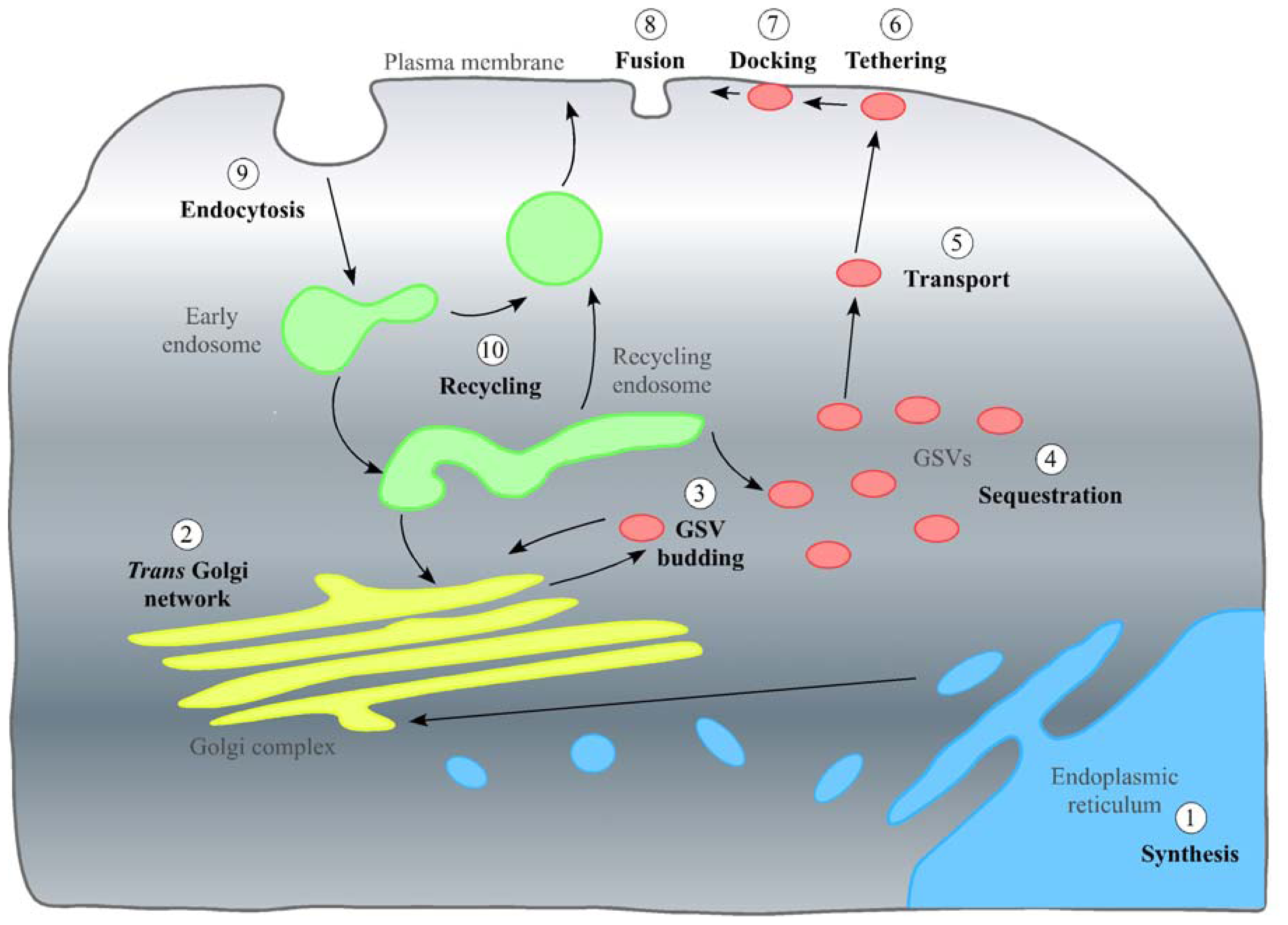
2. In the Beginning: GLUT4 Synthesis, the TGN and Inclusion into GSVs
2.1. Stage 1: Synthesis and the Endoplasmic Reticulum
2.2. Stage 2: The Trans Golgi Network
2.3. Stage 3: Inclusion in GSVs
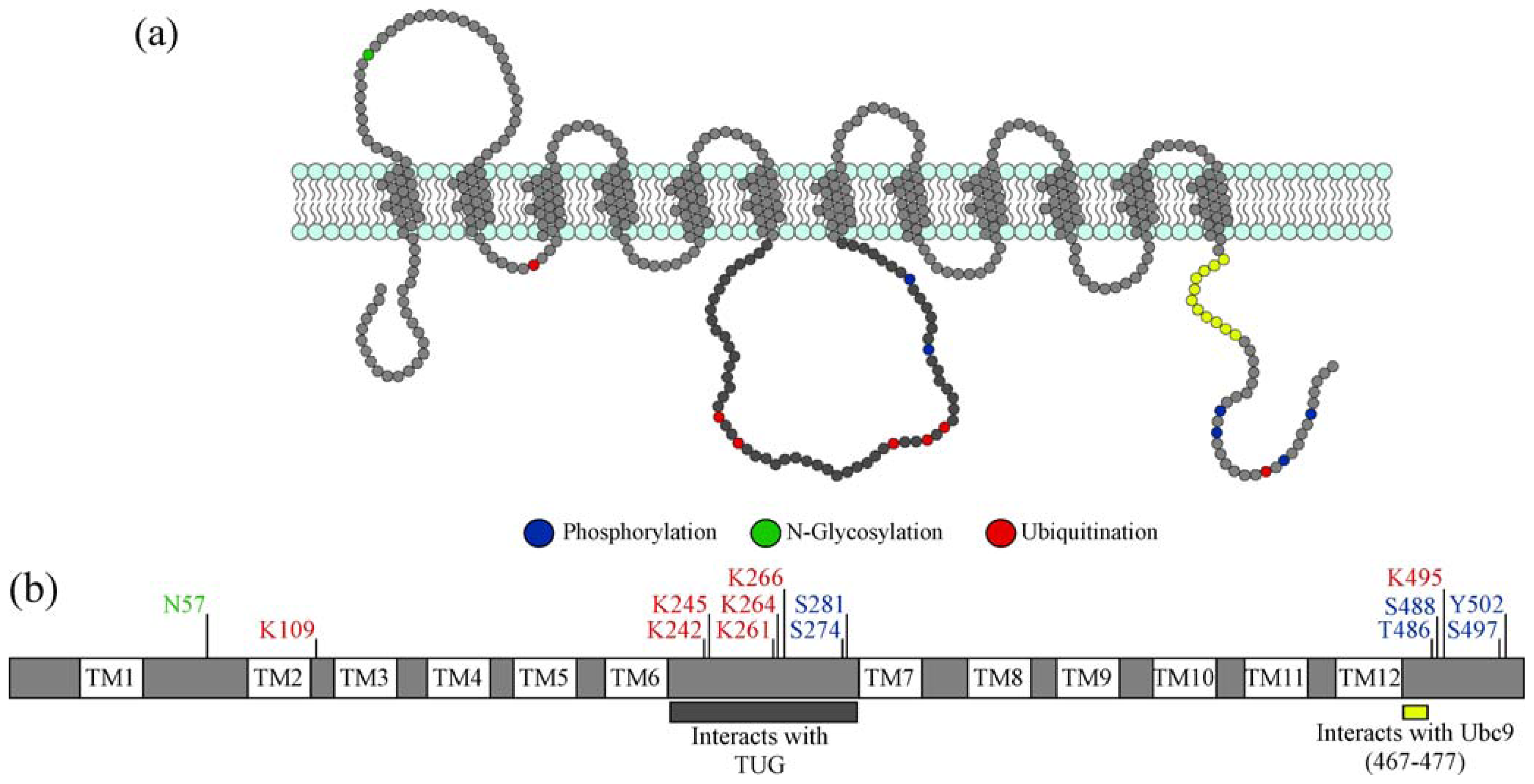
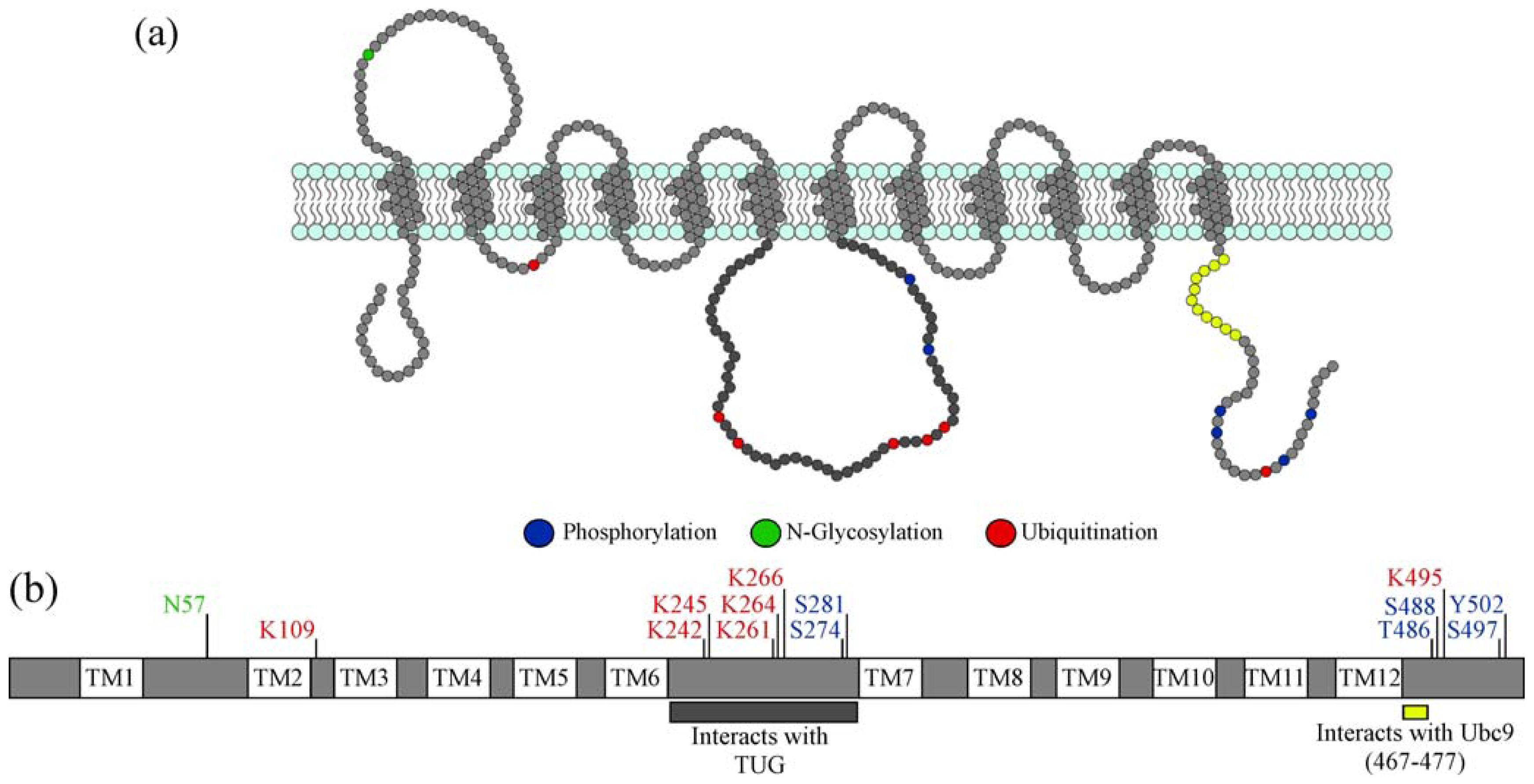
2.3.1. Effects of Post-Translational Modifications on GLUT4 Interactions with Proteins Required for Sorting into GSVs
2.3.2. GLUT4 Insulin Responsiveness: When Are Modifications Needed?
2.3.3. Role of Post-translational Modifications on GLUT4 Protein Stability
2.3.4. Effect on GSV Formation When Post-Translational Modifications Are Prevented
2.3.5. Effect of Increasing Levels of GLUT4 Post-translational Modification
3. The Pause and Release: Sequestering of GSVs and Their Release on Stimulation with Insulin
3.1. Stage 4: Sequestering of GSVs
3.2. Stage 5: Transport of GLUT4 to the Cell Surface on Stimulation with Insulin
4. Fulfilling Its Purpose: Stages 6–8, GLUT4 Insertion into the PM and Its Function as a Glucose Transporter
5. Re-Entry: Endocytosis and Transport of GLUT4 through the Endosome System
5.1. Stage 9: Endocytosis of GLUT4
5.2. Stage 10: The End
6. Summary
Acknowledgments
Conflict of Interest
References
- Bogan, J.S. Regulation of glucose transporter translocation in health and diabetes. Annu. Rev. Biochem 2012, 81, 507–532. [Google Scholar]
- Birnbaum, M.J. Identification of a novel gene encoding an insulin responsive glucose transporter protein. Cell 1989, 57, 305–315. [Google Scholar]
- James, D.E.; Strube, M.; Mueckler, M. Molecular cloning and characterization of an insulin-regulatable glucose transporter. Nature 1989, 338, 83–87. [Google Scholar]
- Pessin, J.E.; Bell, G.I. Mammalian facilitative glucose transporter family structure and molecular regulation. Annu. Rev. Physiol 1992, 54, 911–930. [Google Scholar]
- Huang, S.; Czech, M.P. The glut4 glucose transporter. Cell Metab 2007, 5, 237–252. [Google Scholar]
- Watson, R.T.; Khan, A.H.; Furukawa, M.; Hou, J.C.; Li, L.; Kanzaki, M.; Okada, S.; Kandror, K.V.; Pessin, J.E. Entry of newly synthesized glut4 into the insulin-responsive storage compartment is gga dependent. EMBO J 2004, 23, 2059–2070. [Google Scholar]
- Saltiel, A.R.; Kahn, C.R. Insulin signalling and the regulation of glucose and lipid metabolism. Nature 2001, 414, 799–806. [Google Scholar]
- Livingstone, C.; James, D.E.; Rice, J.E.; Hanpeter, D.; Gould, G. Compartment ablation analysis of the insulin-responsive glucose transporter (glut4) in 3t3-l1 adipocytes. Biochem. J 1996, 315, 487–495. [Google Scholar]
- Bryant, N.J.; Gould, G.W. Snare proteins underpin insulin-regulated glut4 traffic. Traffic 2011, 12, 657–664. [Google Scholar]
- Stöckli, J.; Fazakerley, D.J.; James, D.E. Glut4 exocytosis. J. Cell Sci 2011, 124, 4147–4159. [Google Scholar]
- Leto, D.; Saltiel, A.R. Regulation of glucose transport by insulin traffic control of glut4. Nat. Rev. Mol. Cell Biol 2012, 13, 383–396. [Google Scholar]
- Li, S.; Iakoucheva, L.M.; Mooney, S.D.; Radivojac, P. Loss of Post-translational Modification Sites in Disease. In Proceedings of the Pacific Symposium on Biocomputing; World Scientific Publishing Co: Kamuela, HI, USA, 2010; pp. 337–347. [Google Scholar]
- Garvey, W.T.; Huecksteadt, T.P.; Matthaei, S.; Olefsky, J.M. Role of glucose transporters in the cellular insulin resistance of type ii non-insulin-dependent diabetes mellitus. J. Clin. Investig 1988, 81, 1528–1536. [Google Scholar]
- Hardy, J. Amyloid, the presenilins and Alzheimer’s disease. Trends Neurol. Sci 1997, 20, 154–159. [Google Scholar]
- Toh, K.L.; Jones, C.R.; He, Y.; Eide, E.J.; Hinz, W.A.; Virshup, D.M.; Ptácek, L.J.; Fu, Y.H. An hper2 phosphorylation site mutation in familial advanced sleep phase syndrome. Science 2001, 291, 1040–1043. [Google Scholar]
- Grasbon-Frodl, E.; Lorenz, H.; Mann, U.; Nitsch, R.M.; Windl, O.; Kretzschmar, H.A. Loss of glycosylation associated with the t183a mutation in human prion disease. Acta Neuropathol 2004, 108, 476–484. [Google Scholar]
- Lau, K.S.; Partridge, E.A.; Grigorian, A.; Silvescu, C.I.; Reinhold, V.N.; Demetriou, M.; Dennis, J.W. Complex n-glycan number and degree of branching cooperate to regulate cell proliferation and differentiation. Cell 2007, 192, 123–134. [Google Scholar]
- Varki, A.; Cummings, R.D.; Esko, J.D.; Freeze, H.H.; Stanley, P.; Bertozzi, C.R.; Hart, G.W.; Etzler, M.E. Essentials of Glycobiology; Cold Spring Harbor Laboratories Press: Cold Spring Harbor, NY, USA, 2009. [Google Scholar]
- Haga, Y.; Ishii, K.; Suzuki, T. N-glycosylation is critical for the stability and intracellular trafficking of glucose transporter glut4. J. Biol. Chem 2011, 286, 31320–31327. [Google Scholar]
- Williams, D.; Hicks, S.W.; Machamer, C.E.; Pessin, J.E. Golgin-160 is required for the golgi membrane sorting of the insulin-responsive glucose transporter glut4 in adipocytes. Mol. Biol. Cell 2006, 17, 5346–5355. [Google Scholar]
- Ing, B.L.; Chen, H.; Robinson, K.A.; Buse, M.G.; Quon, M.J. Characterization of a mutant glut4 lacking the N-glycosylation site studies in transfected rat adipose cells. Biochem. Biophys. Res. Commun 1996, 218, 76–82. [Google Scholar]
- Zaarour, N.; Berenguer, M.; Le Marchand-Brustel, Y.; Govers, R. Deciphering the role of glut4 N-glycosylation in adipocyte and muscle cell models. Biochem. J 2012, 445, 265–273. [Google Scholar]
- McCormick, P.J.; Dumaresq-Doiron, K.; Pluviose, A.S.; Pichette, V.; Tosato, G.; Lefrancois, S. Palmitoylation controls recycling in lysosomal sorting and trafficking. Traffic 2008, 9, 1984–1997. [Google Scholar]
- Ren, W.; Jhala, U.; Du, K. Proteomic analysis of protein palmitoylation in adipocytes. Adipocyte 2013, 2, 17–27. [Google Scholar]
- Herrmann, J.; Lerman, L.O.; Lerman, A. Ubiquitin and ubiquitin-like proteins in protein regulation. Circ. Res 2007, 100, 1276–1291. [Google Scholar]
- Urbe, S. Ubiquitin and endocytic protein sorting. Essays Biochem 2005, 41, 81–98. [Google Scholar]
- Piper, R.C.; Luzio, J.P. Ubiquitin-dependent sorting of integralmembrane proteins for degradation in lysosomes. Curr. Opin. Cell Biol 2007, 19, 459–465. [Google Scholar]
- Hicke, L. Protein regulation by monoubiquitin. Nat. Rev. Mol. Cell Biol 2001, 2, 195–201. [Google Scholar]
- Lamb, C.A.; McCann, R.K.; Stockli, J.; James, D.E.; Bryant, N.J. Insulin-regulated trafficking of glut4 requires ubiquitination. Traffic 2010, 11, 1445–1454. [Google Scholar]
- Shi, J.; Kandror, K.V. Sortilin is essential and sufficient for the formation of glut4 storage vesicles in 3t3-l1 adipocytes. Dev. Cell 2005, 9, 99–108. [Google Scholar]
- Matunis, M.J.; Coutavas, E.; Blobel, G. A novel ubiquitin-like modification modulates the partitioning of the ran-gtpase-activating protein rangap1 between the cytosol and the nuclear pore complex. J. Cell Biol 1996, 135, 1457–1470. [Google Scholar]
- Mahajan, R.; Delphin, C.; Guan, T.; Gerace, L.; Melchior, F. A small ubiquitin-related polypeptide involved in targeting rangap1 to nuclear pore complex protein ranbp2. Cell 1997, 88, 97–107. [Google Scholar]
- Gill, G. Something about sumo inhibits transcription. Curr. Opin. Genet. Dev 2005, 15, 536–541. [Google Scholar]
- Bayer, P.; Arndt, A.; Metzger, S.; Mahajan, R.; Melchior, F.; Jaenicke, R.; Becker, J. Structure determination of the small ubiquitin-related modifier sumo-1. J. Mol. Biol 1998, 280, 275–286. [Google Scholar]
- Liu, L.; Omata, W.; Kojima, I.; Shibata, H. The sumo conjugating enzyme ubc9 is a regulator of glut4 turnover and targeting to the insulin-responsive storage compartment in 3t3-l1 adipocytes. Diabetes 2007, 56, 1977–1985. [Google Scholar]
- Pilch, P.F. The mass action hypothesis: Formation of glut4 storage vesicles, a tissue-specific, regulated exocytic compartment. Acta Physiol 2008, 192, 89–101. [Google Scholar]
- Shi, J.; Kandror, K.V. The luminal vps10p domain of sortilin plays the predominant role in targeting to insulin-responsive glut4-containing vesicles. J. Biol. Chem 2007, 282, 9008–9016. [Google Scholar]
- Giorgino, F.; de Robertis, O.; Laviola, L.; Montrone, C.; Perrini, S.; McCowen, K.C.; Smith, R.J. The sentrin-conjugating enzyme mubc9 interacts with glut4 and glut1 glucose transporters and regulates transporter levels in skeletal muscle cells. Proc. Natl. Acad. Sci. USA 2000, 97, 1125–1130. [Google Scholar]
- Perera, H.K.I.; Clarke, M.; Morris, N.J.; Hong, W.; Chamberlain, L.H.; Gould, G.W. Syntaxin 6 regulates glut4 trafficking in 3t3-l1 adipocytes. Mol. Biol. Cell 2003, 14, 2946–2958. [Google Scholar]
- Marshall, S.; Bacote, V.; Traxingerg, R.R. Discovery of a metabolic pathway mediating glucose-induced desensitization of the glucose transport system; role of hexosamine biosynthesis in the induction of insulin resistance. J. Biol. Chem 1991, 266, 4706–4712. [Google Scholar]
- Marshall, S.; Nadeau, O.; Yamasaki, K. Glucosamine-induced activation of glycogen biosynthesis in isolated adipocytes: Evidence for a rapid allosteric control mechanism within the hexosamine biosynthesis pathway. J. Biol. Chem 2005, 280, 11018–11024. [Google Scholar]
- Fujita, H.; Hatakeyama, H.; Watanabe, T.M.; Sato, M.; Higuchi, H.; Kanzaki, M. Identification of three distinct functional sites of insulin-mediated glut4 trafficking in adipocytes using quantitative single molecule imaging. Mol. Biol. Cell 2010, 21, 2721–2731. [Google Scholar]
- Govers, R.; Coster, A.C.F.; James, D.E. Insulin increases cell surface glut4 levels by dose dependent discharging glut4 into a cell surface recycling pathway. Mol. Cell. Biol 2004, 24, 6456–6466. [Google Scholar]
- Martin, O.J.; Lee, A.; McGraw, T.E. Glut4 distribution between the plasma membrane and the intracellular compartments is maintained by an insulinmodulated bipartite dynamic mechanism. J. Biol. Chem 2006, 281, 484–490. [Google Scholar]
- Karylowski, O.; Zeigerer, A.; Cohen, A.; McGraw, T.E. Glut4 is retained by an intracellular cycle of vesicle formation and fusion with endosomes. Mol. Biol. Cell 2004, 15, 870–882. [Google Scholar]
- Bogan, J.S.; Rubin, B.R.; Yu, C.; Löffler, M.G.; Orme, C.M.; Belman, J.P.; McNally, L.J.; Hao, M.; Cresswell, J.A. Endoproteolytic cleavage of tug regulates glut4 glucose transporter translocation. J. Biol. Chem 2012, 287, 23932–23947. [Google Scholar]
- Bogan, J.S.; Hendon, N.; McKee, A.E.; Tsao, T.S.; Lodish, H.F. Functional cloning of tug as a regulator of glut4 glucose transporter trafficking. Nature 2003, 425, 272–733. [Google Scholar]
- Yu, C.; Cresswell, J.; Loffler, M.G.; Bogan, J.S. The glucose transporter 4-regulating protein tug is essential for highly insulin-responsive glucose uptake in 3t3-l1 adipocytes. J. Biol. Chem 2007, 282, 7710–7722. [Google Scholar]
- Semiz, S.; Park, J.G.; Nicoloro, S.M.C.; Furcinitti, P.; Zhang, C.; Chawla, A.; Leszyk, J.; Czech, M.P. Conventional kinesin kif5b mediates insulinstimulated glut4 movements on microtubules. EMBO J 2003, 22, 2387–2399. [Google Scholar]
- Chang, L.; Chiang, S.H.; Saltiel, A.R. Tc10alpha is required for insulin-stimulated glucose uptake in adipocytes. Endocrinology 2007, 148, 27–33. [Google Scholar]
- Phosphosite. Available online: http://www.Phosphosite.Org/proteinaction.Do?Id=4961&showallsites=true (accessed on 1 February 2013).
- Jeyaraj, S.; Boehmer, C.; Lang, F.; Palmada, M. Role of sgk1 kinase in regulating glucose transport via glucose transporter glut4. Biochem. Biophys. Res. Commun 2007, 356, 629–635. [Google Scholar]
- Lawrence, J.C., Jr; Hiken, J.F.; James, D.E. Phosphorylation of the glucose transporter in rat adipocytes. Identification of the intracellular domain at the carboxyl terminus as a target for phosphorylation in intact-cells in vitro. J. Biol. Chem 1990, 265, 2324–2332. [Google Scholar]
- James, D.E.; Hiken, J.; Lawrence, J.C., Jr. Isoproterenol stimulates phosphorylation of the insulin-regulatable glucose transporter in rat adipocytes. Proc. Natl. Acad. Sci. USA 1989, 86, 8368–8372. [Google Scholar]
- Lawrence, J.C., Jr; Hiken, J.F.; James, D.E. Stimulation of glucose transport and glucose transporter phosphorylation by okadaic acid in rat adipocytes. J. Biol. Chem 1990, 265, 19768–19776. [Google Scholar]
- Muller, G.; Wied, S. The sulfonylurea drug, glimepiride, stimulates glucose transport, glucose transporter translocation, and dephosphorylation in insulin-resistant rat adipocytes in vitro. Diabetes 1993, 42, 1852–1867. [Google Scholar]
- Kupriyanova, T.A.; Kandror, K.V. Akt-2 binds to glut4-containing vesicles and phosphorylates their component proteins in response to insulin. J. Biol. Chem 1999, 274, 1458–1464. [Google Scholar]
- Vannucci, S.J.; Nishimura, H.; Satoh, S.; Cushman, S.W.; Holman, G.D.; Simpson, I.A. Cell surface accessibility of glut4 glucose transporters in insulin-stimulated rat adipose cells: Modulation by isoprenaline and adenosine. Biochem. J 1992, 288, 325–330. [Google Scholar]
- Yang, J.; Hodel, A.; Holman, G.D. Insulin and isoproterenol have opposing roles in the maintenance of cytosol ph and optimal fusion of glut4 vesicles with the plasma membrane. J. Biol. Chem 2002, 277, 6559–6566. [Google Scholar]
- Begum, N.; Draznin, B. Effect of streptozotocin-induced diabetes on glut-4 phosphorylation in rat adipocytes. J. Clin. Invest 1992, 90, 1254–1262. [Google Scholar]
- Reusch, J.E.; Begum, N.; Sussman, K.E.; Draznin, B. Regulation of glut-4 phosphorylation by intracellular calcium in adipocytes. Endocrinology 1991, 129, 3269–3273. [Google Scholar]
- Haga, Y.; Ishii, K.; Hibino, K.; Sako, Y.; Ito, Y.; Taniguchi, N.; Suzuki, T. Visualizing specific protein glycoforms by transmembrane fluorescence resonance energy transfer. Nat. Commun. 2012. [Google Scholar] [CrossRef]
- Marsh, B.J.; Martin, S.; Melvin, D.R.; Martin, L.B.; Alm, R.A.; Gould, G.W.; James, D.E. Mutational analysis of the carboxy-terminal phosphorylation site of glut-4 in 3t3-l1 adipocytes.
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sadler, J.B.A.; Bryant, N.J.; Gould, G.W.; Welburn, C.R. Posttranslational Modifications of GLUT4 Affect Its Subcellular Localization and Translocation. Int. J. Mol. Sci. 2013, 14, 9963-9978. https://doi.org/10.3390/ijms14059963
Sadler JBA, Bryant NJ, Gould GW, Welburn CR. Posttranslational Modifications of GLUT4 Affect Its Subcellular Localization and Translocation. International Journal of Molecular Sciences. 2013; 14(5):9963-9978. https://doi.org/10.3390/ijms14059963
Chicago/Turabian StyleSadler, Jessica B. A., Nia J. Bryant, Gwyn W. Gould, and Cassie R. Welburn. 2013. "Posttranslational Modifications of GLUT4 Affect Its Subcellular Localization and Translocation" International Journal of Molecular Sciences 14, no. 5: 9963-9978. https://doi.org/10.3390/ijms14059963