Structural Alterations of Human Serum Albumin Caused by Glycative and Oxidative Stressors Revealed by Circular Dichroism Analysis

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
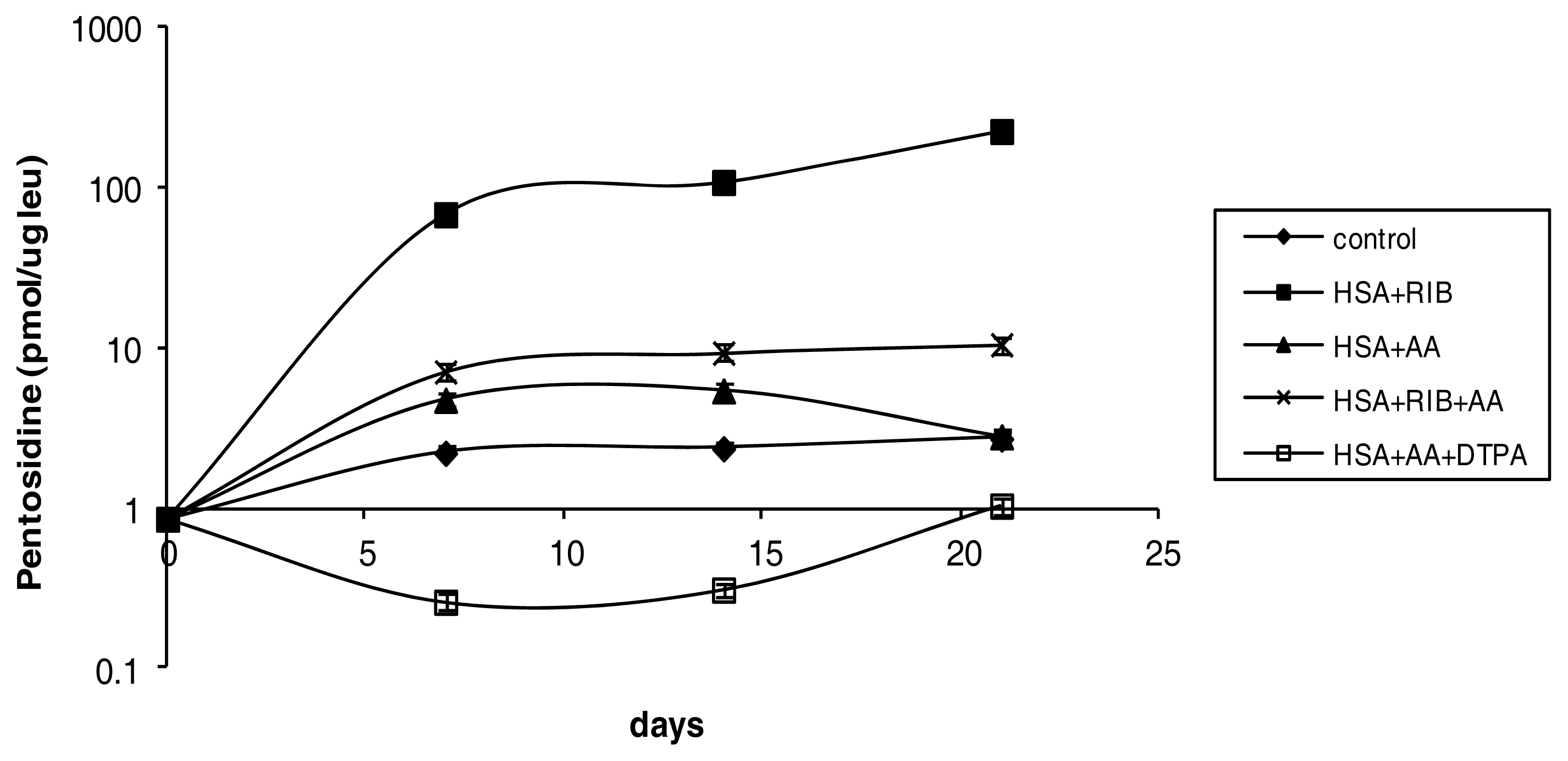
2.1. Pentosidine
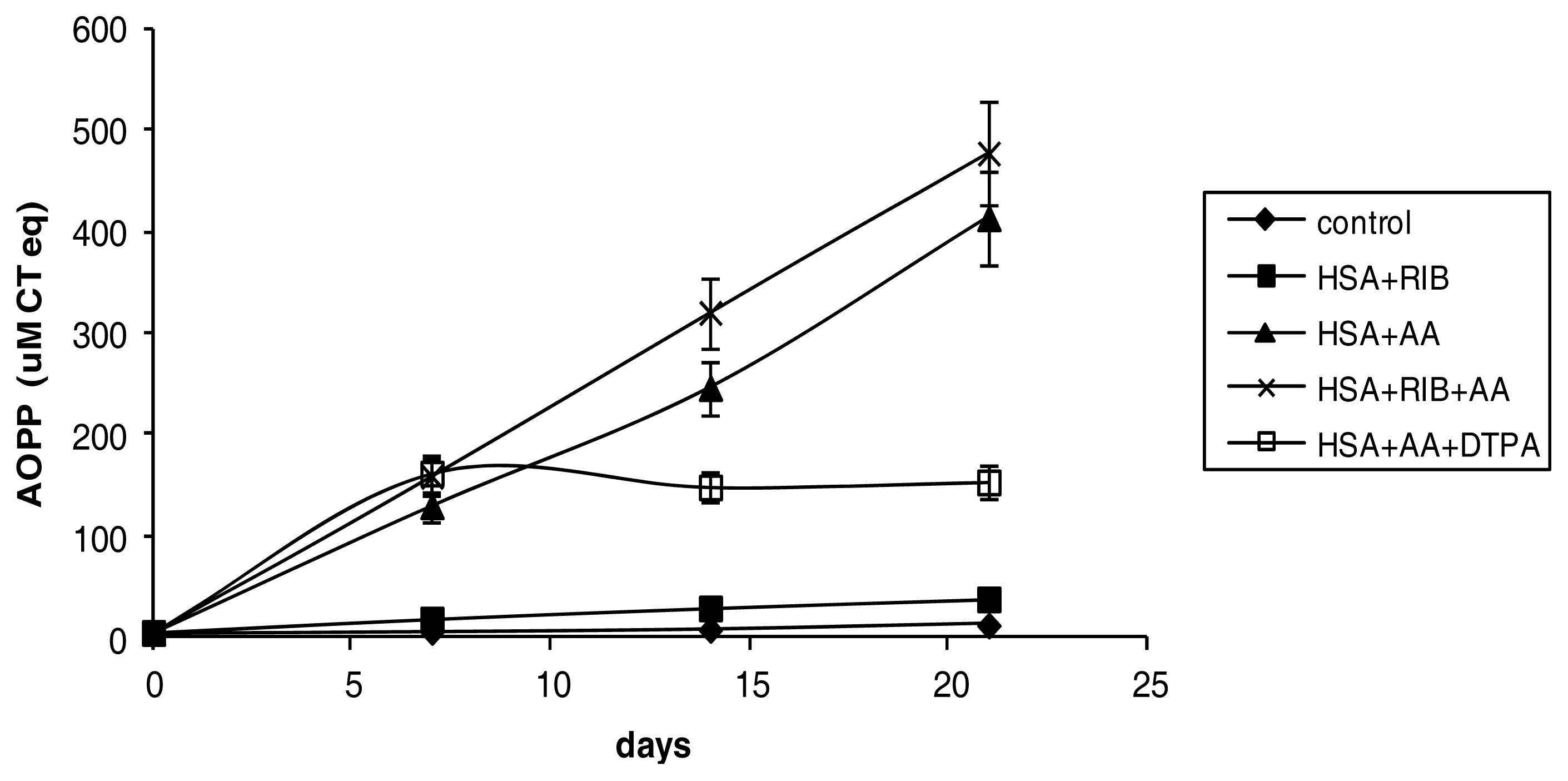
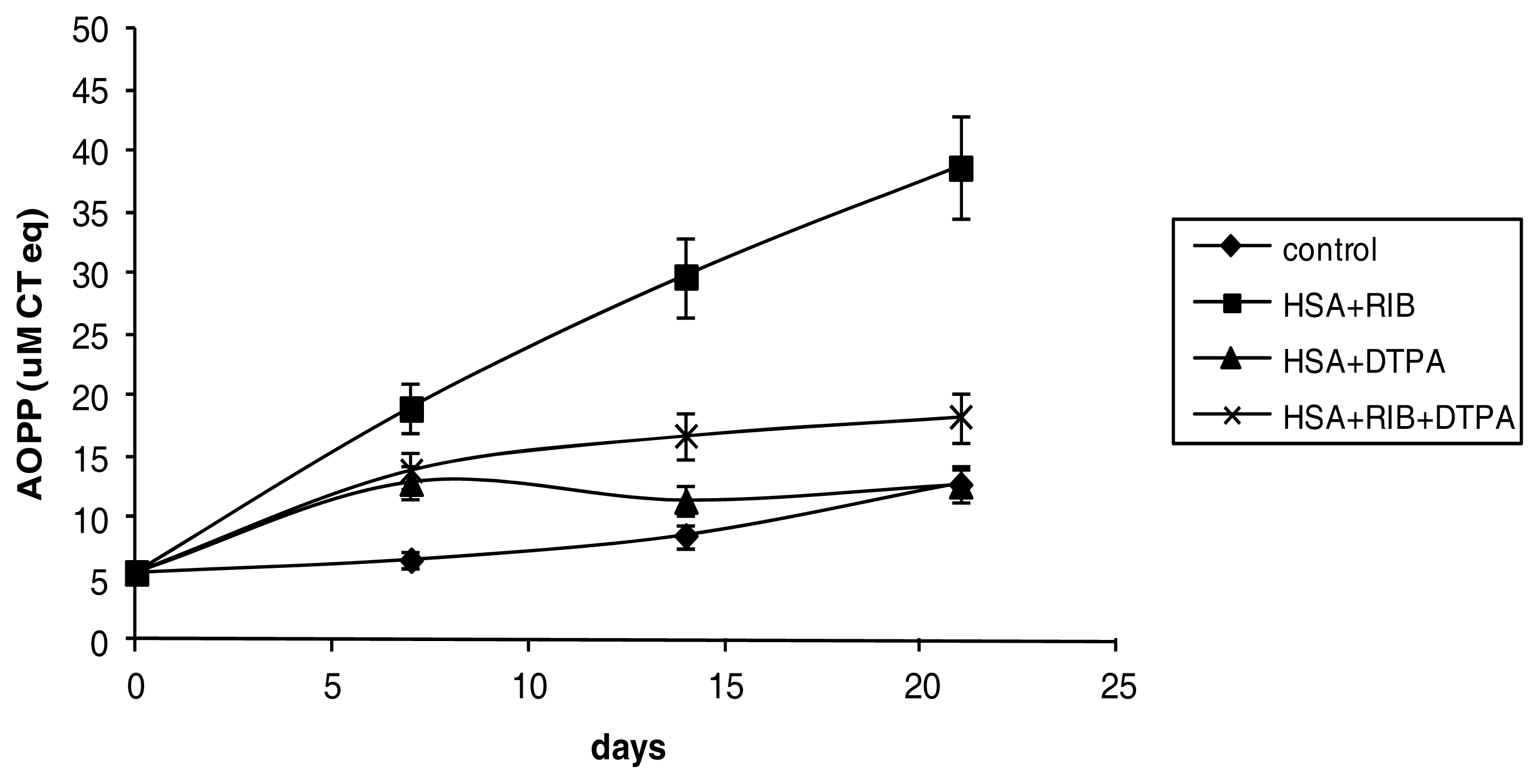
2.2. AOPP
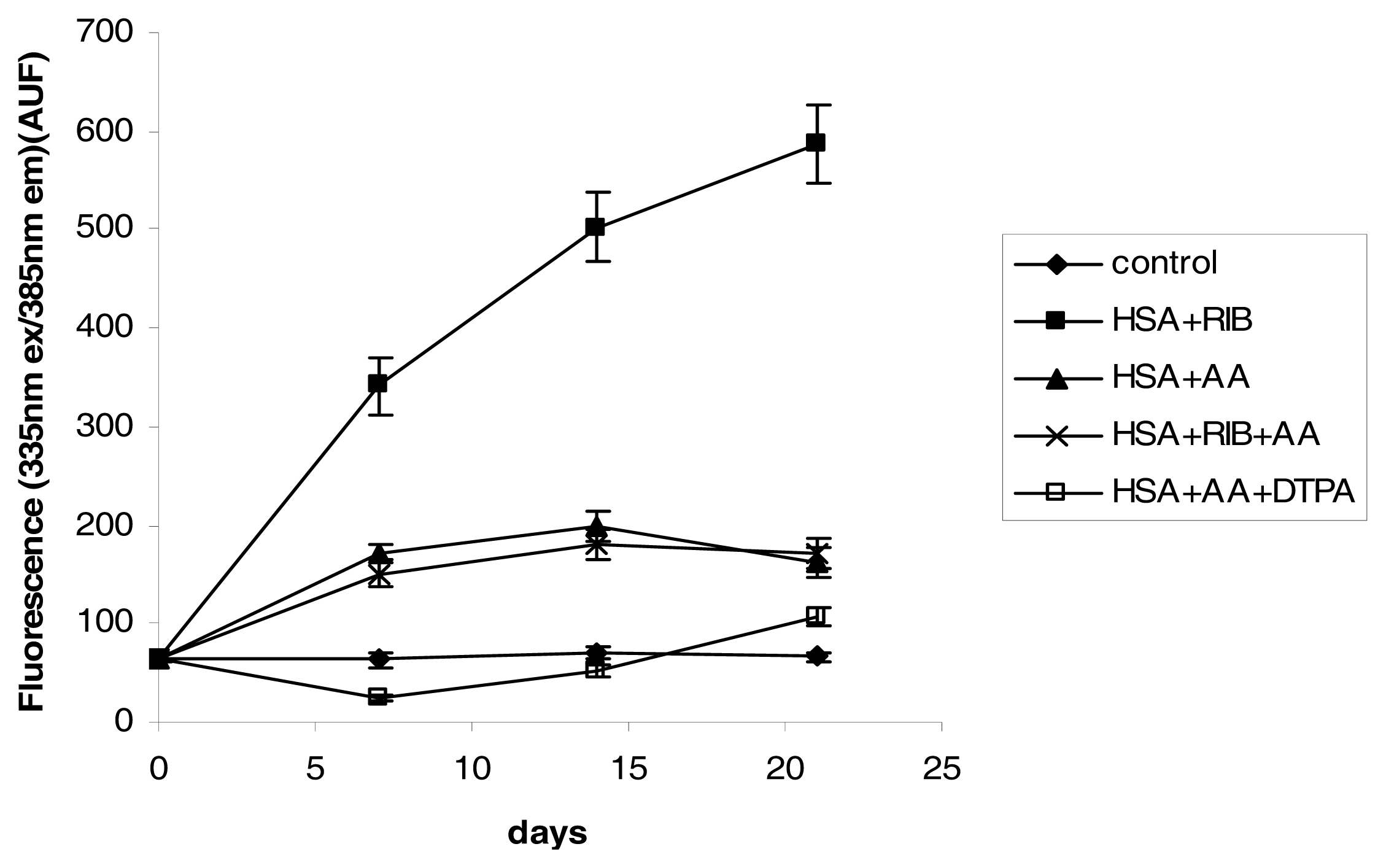
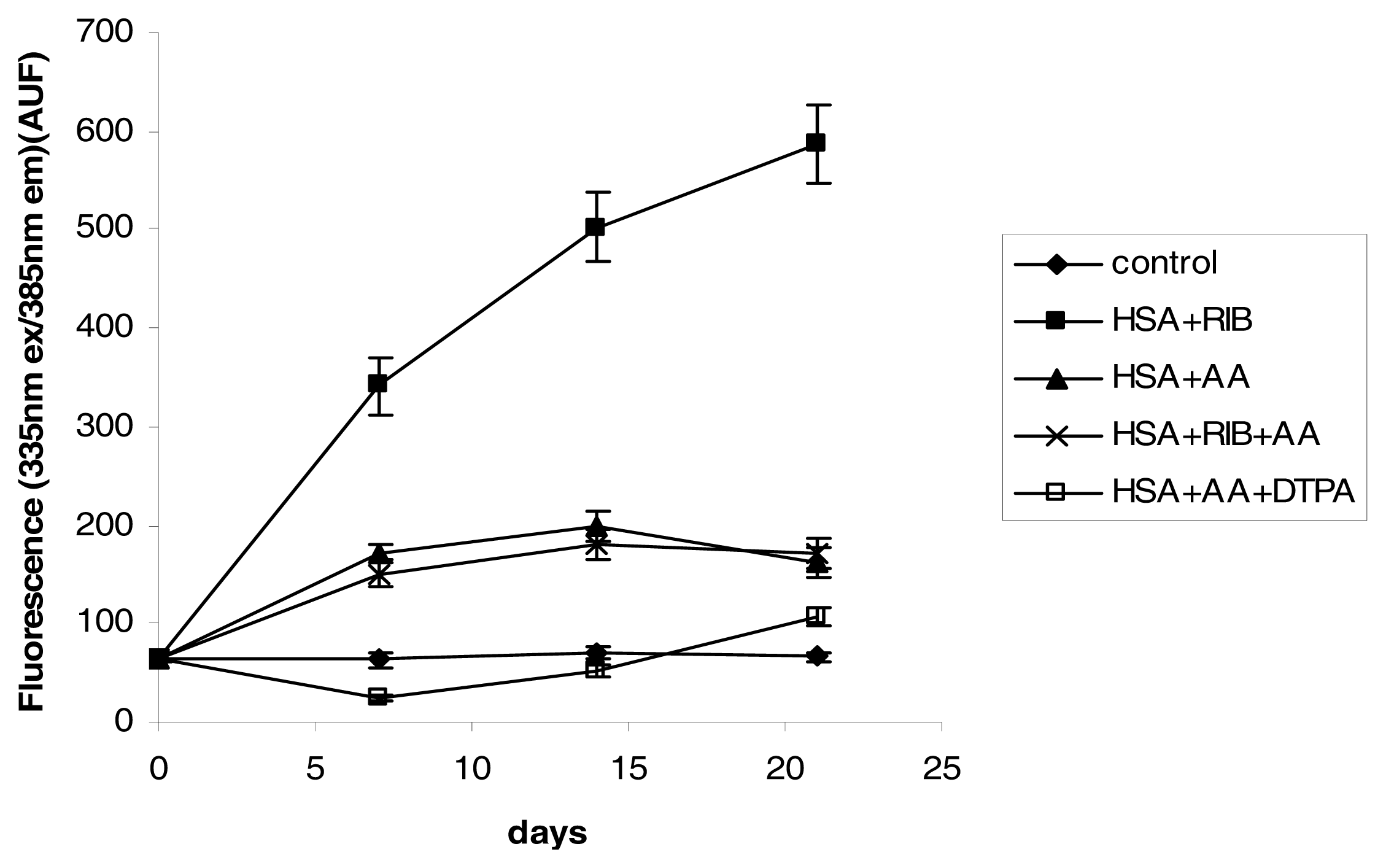
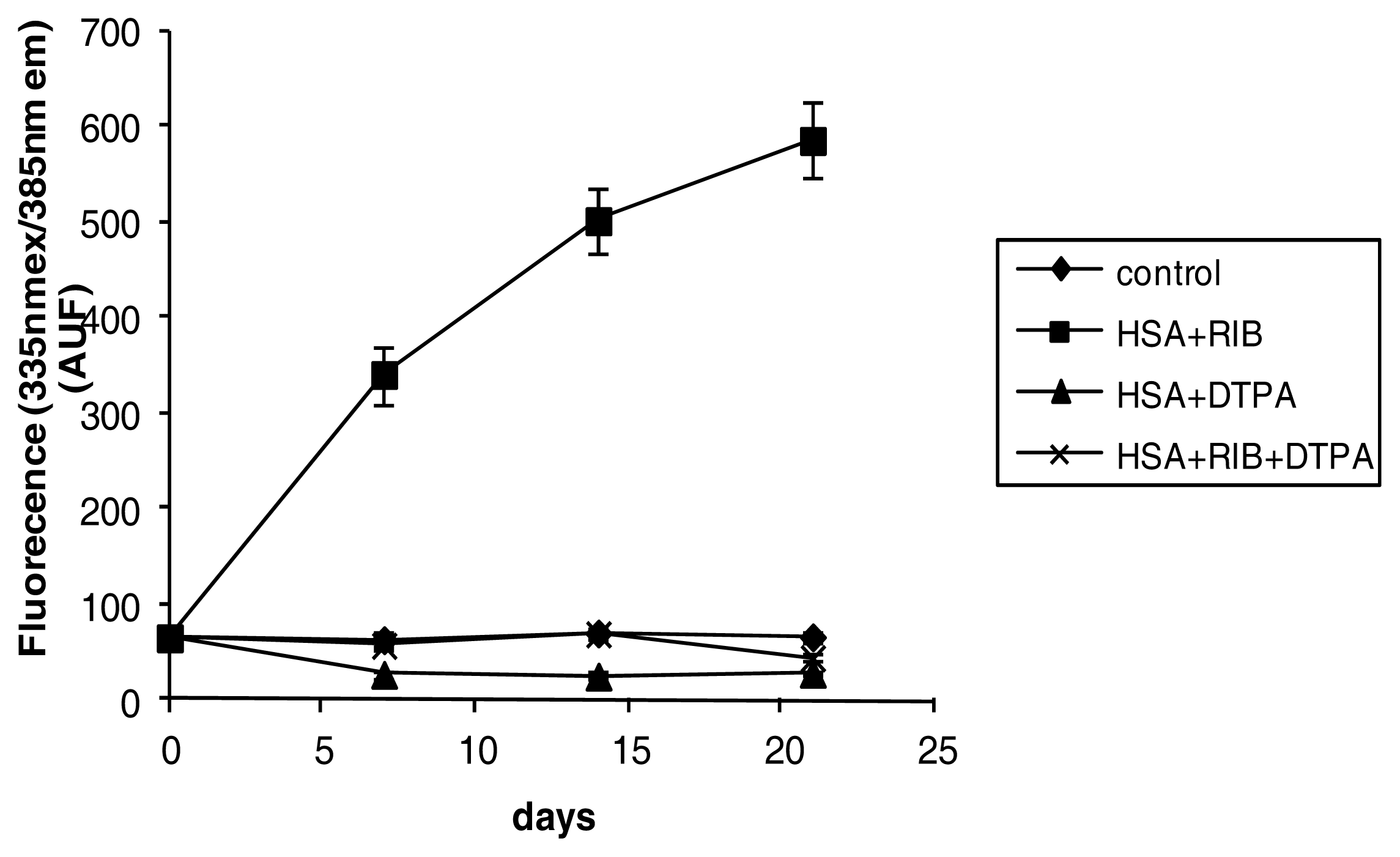
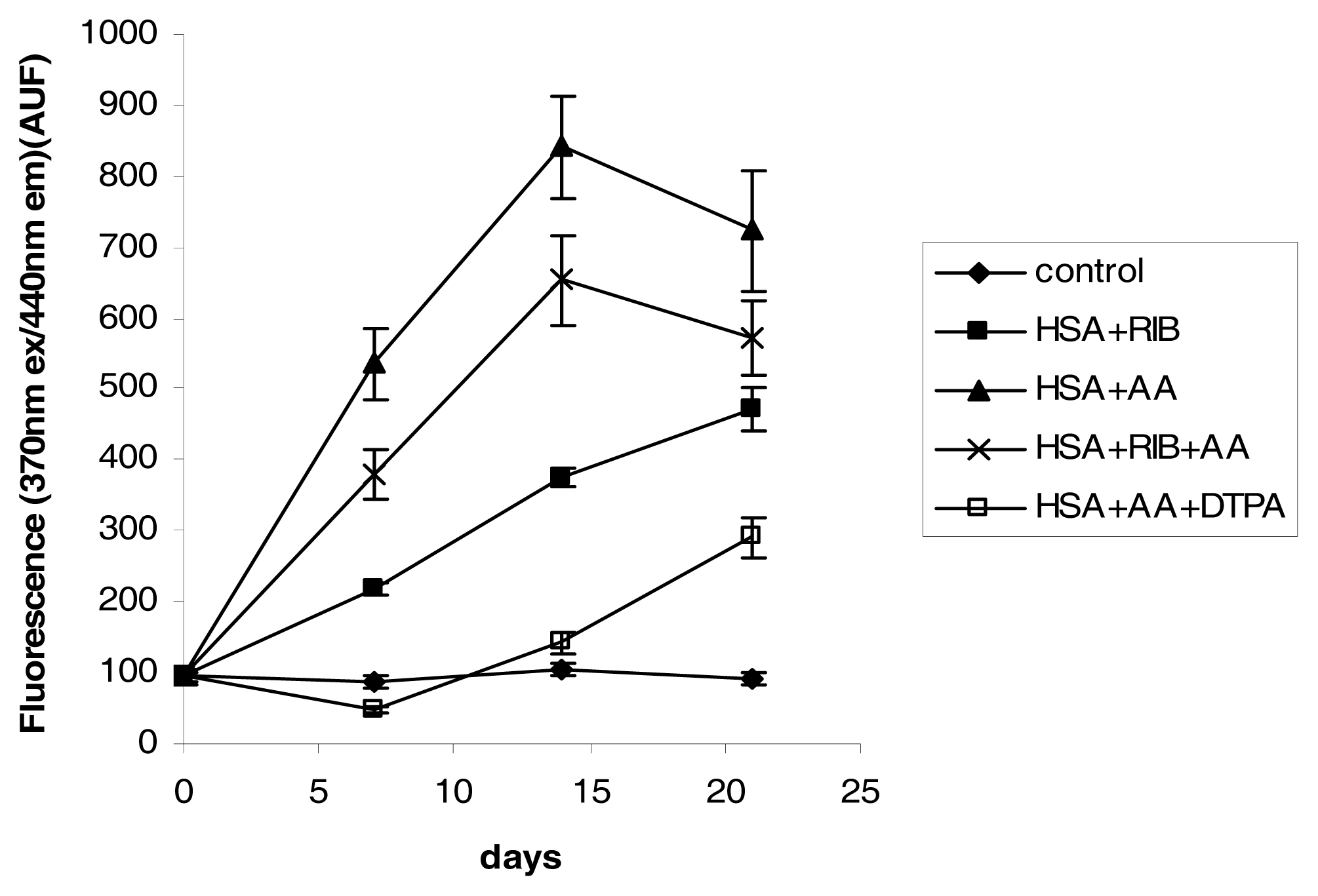
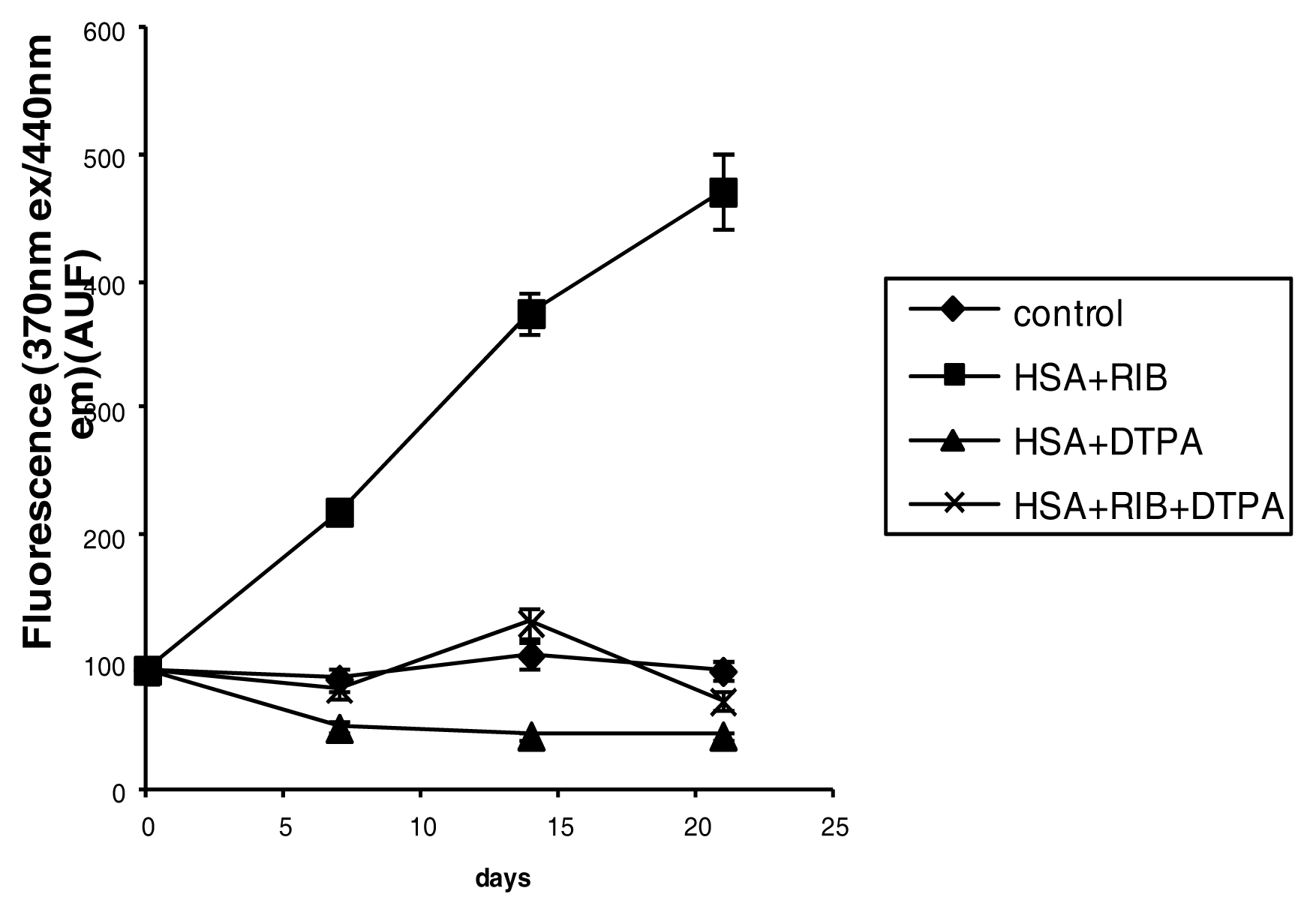
2.3. Fluorescence
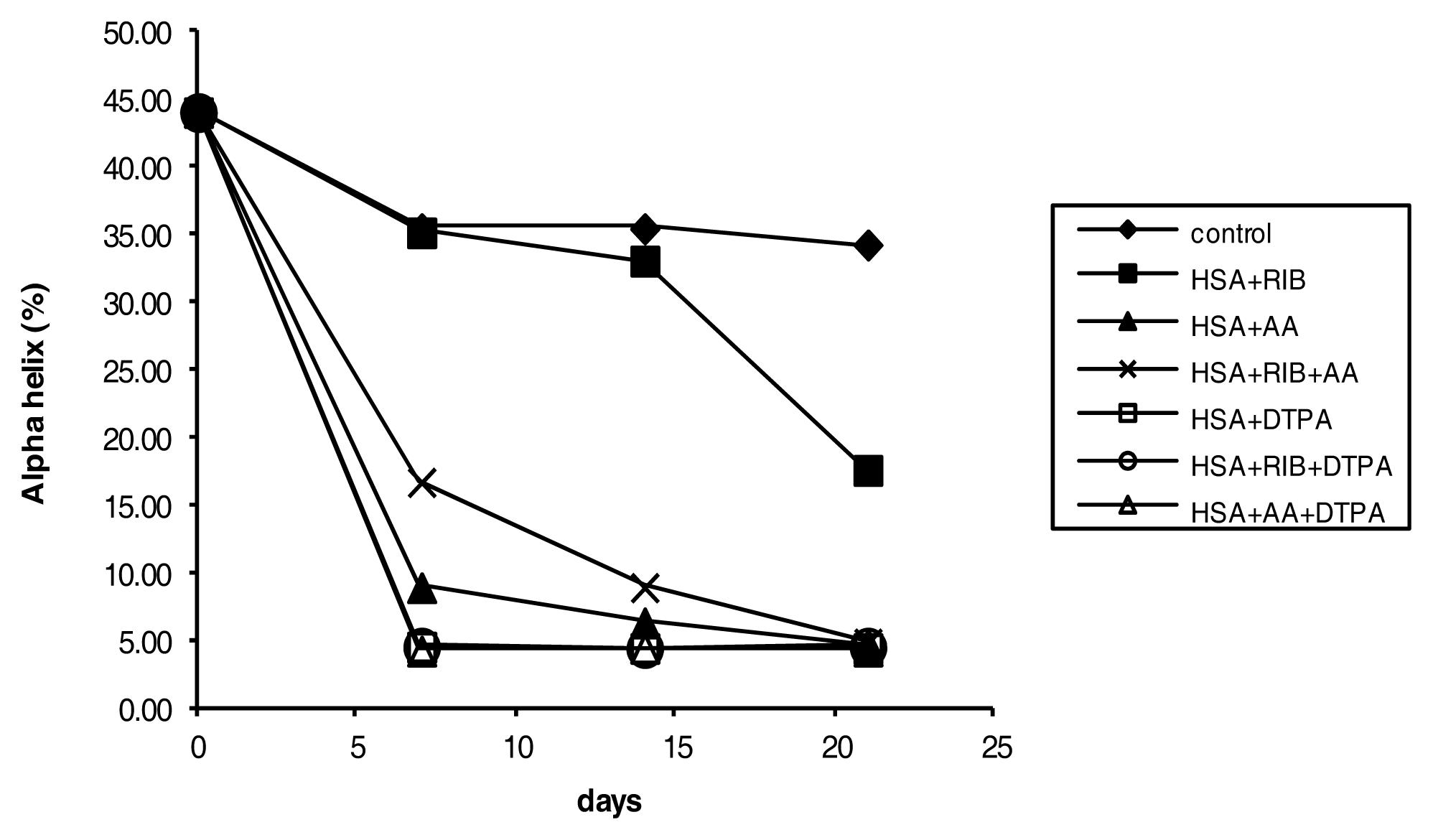
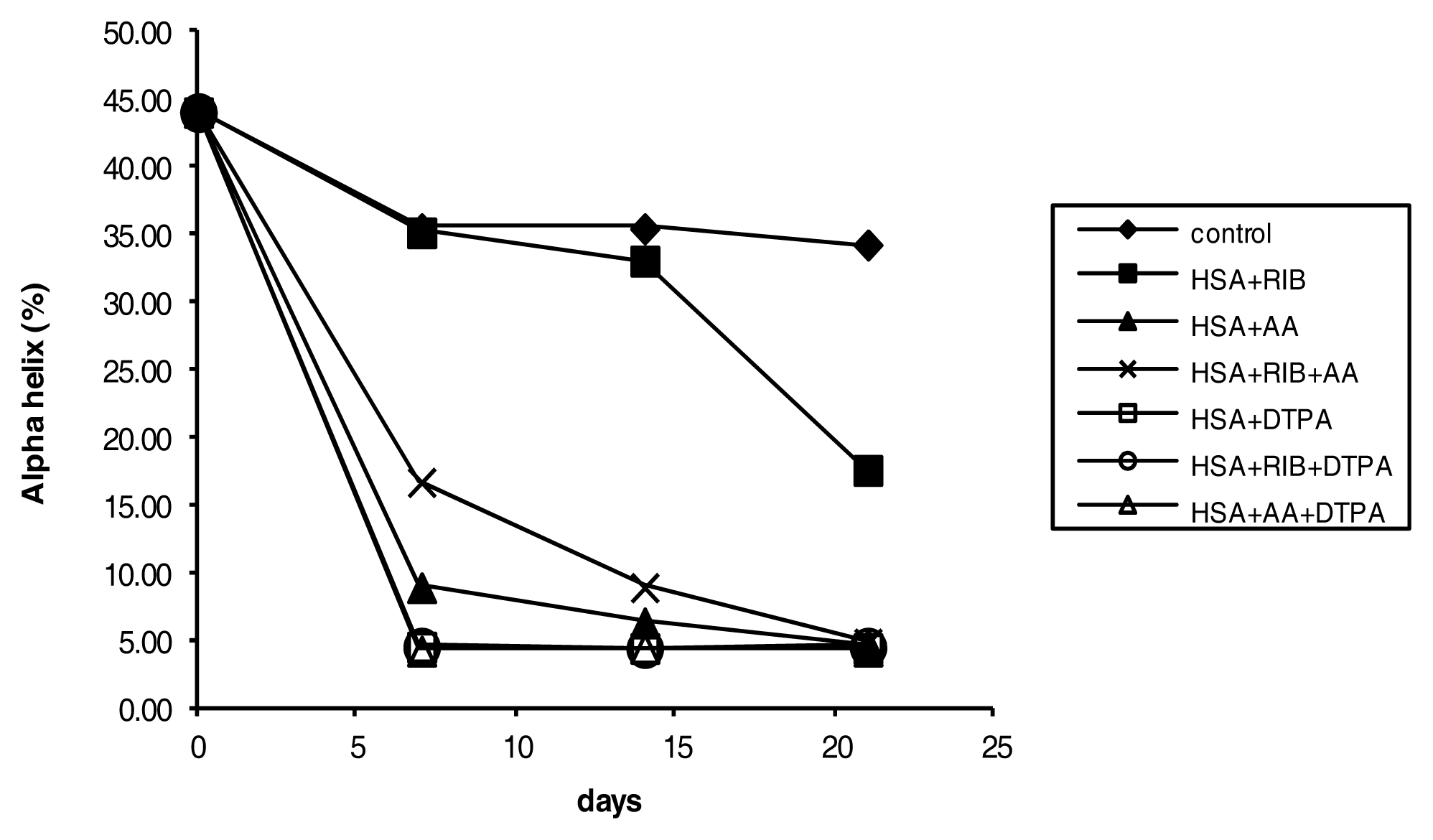
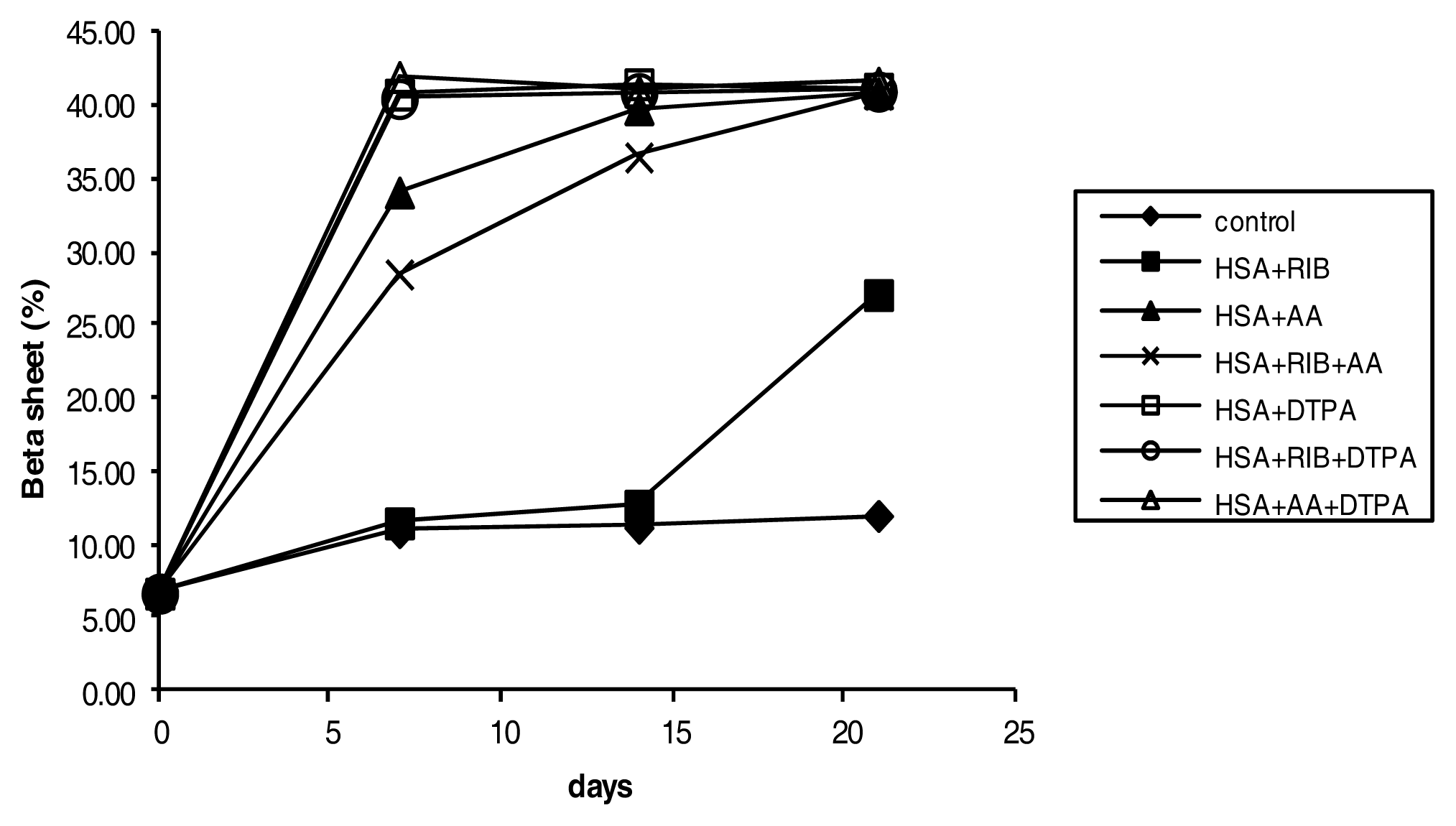
2.4. Circular Dichroism
2.5. Discussion
3. Materials and Methods
3.1. Glycation of HSA
3.2. Determination of Pentosidine by HPLC
3.3. AOPP Assay
3.4. Fluorescence Measurements
3.5. Amino Acid Estimation
3.6. CD Measurements
3.7. Statistical Analysis
4. Conclusions
Abbreviations
| HSA | human serum albumin |
| DTPA | diethylenetriamine pentacetate |
| AOPP | advanced oxidation protein product |
| AGE | advanced glycation end-product |
| HFBA | heptafluorobutyric acid |
| CT | chloramine-T |
| AUF | arbitrary units of fluorescence |
| CD | circular dichroism |
| AA | ascorbic acid |
Conflict of Interest
References
- Monnier, V.M. Nonenzymatic glycosylation, the maillard reaction and the aging process. J. Gerontol 1990, 45, B105–B111. [Google Scholar]
- Davies, K.J. Protein damage and degradation by oxygen radicals. I. General aspects. J. Biol. Chem 1987, 262, 9895–9901. [Google Scholar]
- Esterbauer, H.; Cheeseman, K.H. Determination of aldehydic lipid peroxidation products: Malonaldehyde and 4-hydroxynonenal. Methods Enzymol 1990, 186, 407–421. [Google Scholar]
- Du, J.; Cullen, J.J.; Buettner, G.R. Ascorbic acid: Chemistry, biology and the treatment of cancer. Biochim. Biophys. Acta 2012, 1826, 443–457. [Google Scholar]
- Koppenol, W.H. The haber-weiss cycle—70 years later. Redox Rep 2001, 6, 229–234. [Google Scholar]
- Vlassara, H.; Palace, M.R. Glycoxidation: The menace of diabetes and aging. Mt. Sinai J. Med 2003, 70, 232–241. [Google Scholar]
- Graham, L. A comprehensive survey of the acid-stable fluorescent cross-links formed by ribose with basic amino acids, and partial characterization of a novel maillard cross-link. Biochim. Biophys. Acta 1996, 1297, 9–16. [Google Scholar]
- Sell, D.R.; Lapolla, A.; Odetti, P.; Fogarty, J.; Monnier, V.M. Pentosidine formation in skin correlates with severity of complications in individuals with long-standing iddm. Diabetes 1992, 41, 1286–1292. [Google Scholar]
- Odetti, P.; Fogarty, J.; Sell, D.R.; Monnier, V.M. Chromatographic quantitation of plasma and erythrocyte pentosidine in diabetic and uremic subjects. Diabetes 1992, 41, 153–159. [Google Scholar]
- Nagaraj, R.H.; Sell, D.R.; Prabhakaram, M.; Ortwerth, B.J.; Monnier, V.M. High correlation between pentosidine protein crosslinks and pigmentation implicates ascorbate oxidation in human lens senescence and cataractogenesis. Proc. Natl. Acad. Sci. USA 1991, 88, 10257–10261. [Google Scholar]
- Horie, K.; Miyata, T.; Yasuda, T.; Takeda, A.; Yasuda, Y.; Maeda, K.; Sobue, G.; Kurokawa, K. Immunohistochemical localization of advanced glycation end products, pentosidine, and carboxymethyllysine in lipofuscin pigments of Alzheimer’s disease and aged neurons. Biochem. Biophys. Res. Commun 1997, 236, 327–332. [Google Scholar]
- Baynes, J.W. Role of oxidative stress in development of complications in diabetes. Diabetes 1991, 40, 405–412. [Google Scholar]
- Blakytny, R.; Carver, J.A.; Harding, J.J.; Kilby, G.W.; Sheil, M.M. A spectroscopic study of glycated bovine alpha-crystallin: Investigation of flexibility of the C-terminal extension, chaperone activity and evidence for diglycation. Biochim. Biophys. Acta 1997, 1343, 299–315. [Google Scholar]
- Howard, M.J.; Smales, C.M. Nmr analysis of synthetic human serum albumin alpha-helix 28 identifies structural distortion upon amadori modification. J. Biol. Chem 2005, 280, 22582–22589. [Google Scholar]
- Sell, D.R.; Monnier, V.M. Conversion of arginine into ornithine by advanced glycation in senescent human collagen and lens crystallins. J. Biol. Chem 2004, 279, 54173–54184. [Google Scholar]
- Schnider, S.L.; Kohn, R.R. Effects of age and diabetes mellitus on the solubility and nonenzymatic glucosylation of human skin collagen. J. Clin. Invest 1981, 67, 1630–1635. [Google Scholar]
- Snowden, J.M.; Eyre, D.R.; Swann, D.A. Vitreous structure. VI. Age-related changes in the thermal stability and crosslinks of vitreous, articular cartilage and tendon collagens. Biochim. Biophys. Acta 1982, 706, 153–157. [Google Scholar]
- Grandhee, S.K.; Monnier, V.M. Mechanism of formation of the maillard protein cross-link pentosidine. Glucose, fructose, and ascorbate as pentosidine precursors. J. Biol. Chem 1991, 266, 11649–11653. [Google Scholar]
- Halliwell, B. Oxidative stress and cancer: Have we moved forward? Biochem. J 2007, 401, 1–11. [Google Scholar]
- Dyer, D.G.; Blackledge, J.A.; Thorpe, S.R.; Baynes, J.W. Formation of pentosidine during nonenzymatic browning of proteins by glucose. Identification of glucose and other carbohydrates as possible precursors of pentosidine in vivo. J. Biol. Chem 1991, 266, 11654–11660. [Google Scholar]
- Khan, M.M.; Martell, A.E. Metal ion and metal chelate catalyzed oxidation of ascorbic acid by molecular oxygen. I. Cupric and ferric ion catalyzed oxidation. J. Am. Chem. Soc 1967, 89, 4176–4185. [Google Scholar]
- Wei, Y.; Chen, L.; Chen, J.; Ge, L.; He, R.Q. Rapid glycation with d-ribose induces globular amyloid-like aggregations of bsa with high cytotoxicity to sh-sy5y cells. BMC Cell Biol 2009, 10, 10. [Google Scholar]
- Murphy, M.E. Ascorbate and dehydroascorbate modulate nitric oxide-induced vasodilations of rat coronary arteries. J. Cardiovasc. Pharmacol 1999, 34, 295–303. [Google Scholar]
- Carr, A.C.; Frei, B. Toward a new recommended dietary allowance for vitamin C based on antioxidant and health effects in humans. Am. J. Clin. Nutr 1999, 69, 1086–1107. [Google Scholar]
- Cheng, R.; Lin, B.; Lee, K.W.; Ortwerth, B.J. Similarity of the yellow chromophores isolated from human cataracts with those from ascorbic acid-modified calf lens proteins: Evidence for ascorbic acid glycation during cataract formation. Biochim. Biophys. Acta 2001, 1537, 14–26. [Google Scholar]
- Davie, S.J.; Gould, B.J.; Yudkin, J.S. Effect of vitamin c on glycosylation of proteins. Diabetes 1992, 41, 167–173. [Google Scholar]
- Rondeau, P.; Bourdon, E. The glycation of albumin: Structural and functional impacts. Biochimie 2011, 93, 645–658. [Google Scholar]
- Whitmore, L.; Wallace, B.A. Protein secondary structure analyses from circular dichroism spectroscopy: Methods and reference databases. Biopolymers 2008, 89, 392–400. [Google Scholar]
- Seneviratne, C.; Narayanan, R.; Liu, W.; Dain, J.A. The in vitro inhibition effect of 2 nm gold nanoparticles on non-enzymatic glycation of human serum albumin. Biochem. Biophys. Res. Commun 2012, 422, 447–454. [Google Scholar]
- Vetter, S.W.; Indurthi, V.S. Moderate glycation of serum albumin affects folding, stability, and ligand binding. Clin. Chim. Acta 2011, 412, 2105–2116. [Google Scholar]
- Sharifi, E.; Sattarahmady, N.; Habibi-Rezaei, M.; Farhadi, M.; Sheibani, N.; Ahmad, F.; Moosavi-Movahedi, A.A. Inhibitory effects of beta-cyclodextrin and trehalose on nanofibril and age formation during glycation of human serum albumin. Protein Pept. Lett 2009, 16, 653–659. [Google Scholar]
- Luthra, M.; Balasubramanian, D. Nonenzymatic glycation alters protein structure and stability. A study of two eye lens crystallins. J. Biol. Chem 1993, 268, 18119–18127. [Google Scholar]
- Sattarahmady, N.; Moosavi-Movahedi, A.A.; Ahmad, F.; Hakimelahi, G.H.; Habibi-Rezaei, M.; Saboury, A.A.; Sheibani, N. Formation of the molten globule-like state during prolonged glycation of human serum albumin. Biochim. Biophys. Acta 2007, 1770, 933–942. [Google Scholar]
- Barzegar, A.; Moosavi-Movahedi, A.A.; Sattarahmady, N.; Hosseinpour-Faizi, M.A.; Aminbakhsh, M.; Ahmad, F.; Saboury, A.A.; Ganjali, M.R.; Norouzi, P. Spectroscopic studies of the effects of glycation of human serum albumin on l-trp binding. Protein Pept. Lett 2007, 14, 13–18. [Google Scholar]
- GhoshMoulick, R.; Bhattacharya, J.; Roy, S.; Basak, S.; Dasgupta, A.K. Compensatory secondary structure alterations in protein glycation. Biochim. Biophys. Acta 2007, 1774, 233–242. [Google Scholar]
- Bouma, B.; Kroon-Batenburg, L.M.; Wu, Y.P.; Brunjes, B.; Posthuma, G.; Kranenburg, O.; de Groot, P.G.; Voest, E.E.; Gebbink, M.F. Glycation induces formation of amyloid cross-beta structure in albumin. J. Biol. Chem 2003, 278, 41810–41819. [Google Scholar]
- Dickerson, J.E., Jr; Lou, M.F.; Gracy, R.W. Ascorbic acid mediated alteration of alpha-crystallin secondary structure. Curr. Eye Res. 1995, 14, 163–166. [Google Scholar]
- Mendez, D.L.; Jensen, R.A.; McElroy, L.A.; Pena, J.M.; Esquerra, R.M. The effect of non-enzymatic glycation on the unfolding of human serum albumin. Arch. Biochem. Biophys 2005, 444, 92–99. [Google Scholar]
- Mohamadi-Nejad, A.; Moosavi-Movahedi, A.A.; Hakimelahi, G.H.; Sheibani, N. Thermodynamic analysis of human serum albumin interactions with glucose: Insights into the diabetic range of glucose concentration. Int. J. Biochem. Cell Biol 2002, 34, 1115–1124. [Google Scholar]
- Paris, G.; Kraszewski, S.; Ramseyer, C.; Enescu, M. About the structural role of disulfide bridges in serum albumins: Evidence from protein simulated unfolding. Biopolymers 2012, 97, 889–898. [Google Scholar]
- Meucci, E.; Mordente, A.; Martorana, G.E. Metal-catalyzed oxidation of human serum albumin: Conformational and functional changes. Implications in protein aging. J. Biol. Chem 1991, 266, 4692–4699. [Google Scholar]
- Fan, X.; Xiaoqin, L.; Potts, B.; Strauch, C.M.; Nemet, I.; Monnier, V.M. Topical application of l-arginine blocks advanced glycation by ascorbic acid in the lens of hsvct2 transgenic mice. Mol. Vis 2011, 17, 2221–2227. [Google Scholar]
- Witko-Sarsat, V.; Friedlander, M.; Capeillere-Blandin, C.; Nguyen-Khoa, T.; Nguyen, A.T.; Zingraff, J.; Jungers, P.; Descamps-Latscha, B. Advanced oxidation protein products as a novel marker of oxidative stress in uremia. Kidney Int 1996, 49, 1304–1313. [Google Scholar]
- Monnier, V.M.; Kohn, R.R.; Cerami, A. Accelerated age-related browning of human collagen in diabetes mellitus. Proc. Natl. Acad. Sci. USA 1984, 81, 583–587. [Google Scholar]
- Moore, S.; Stein, W.H. A modified ninhydrin reagent for the photometric determination of amino acids and related compounds. J. Biol. Chem 1954, 211, 907–913. [Google Scholar]
- Sreerama, N.; Woody, R.W. Estimation of protein secondary structure from circular dichroism spectra: Comparison of contin, selcon, and cdsstr methods with an expanded reference set. Anal. Biochem 2000, 287, 252–260. [Google Scholar]


© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Monacelli, F.; Storace, D.; D'Arrigo, C.; Sanguineti, R.; Borghi, R.; Pacini, D.; Furfaro, A.L.; Pronzato, M.A.; Odetti, P.; Traverso, N. Structural Alterations of Human Serum Albumin Caused by Glycative and Oxidative Stressors Revealed by Circular Dichroism Analysis. Int. J. Mol. Sci. 2013, 14, 10694-10709. https://doi.org/10.3390/ijms140610694
Monacelli F, Storace D, D'Arrigo C, Sanguineti R, Borghi R, Pacini D, Furfaro AL, Pronzato MA, Odetti P, Traverso N. Structural Alterations of Human Serum Albumin Caused by Glycative and Oxidative Stressors Revealed by Circular Dichroism Analysis. International Journal of Molecular Sciences. 2013; 14(6):10694-10709. https://doi.org/10.3390/ijms140610694
Chicago/Turabian StyleMonacelli, Fiammetta, Daniela Storace, Cristina D'Arrigo, Roberta Sanguineti, Roberta Borghi, Davide Pacini, Anna L. Furfaro, Maria A. Pronzato, Patrizio Odetti, and Nicola Traverso. 2013. "Structural Alterations of Human Serum Albumin Caused by Glycative and Oxidative Stressors Revealed by Circular Dichroism Analysis" International Journal of Molecular Sciences 14, no. 6: 10694-10709. https://doi.org/10.3390/ijms140610694