Danger Control Programs Cause Tissue Injury and Remodeling

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Injuries Trigger a Series of Host Response Programs
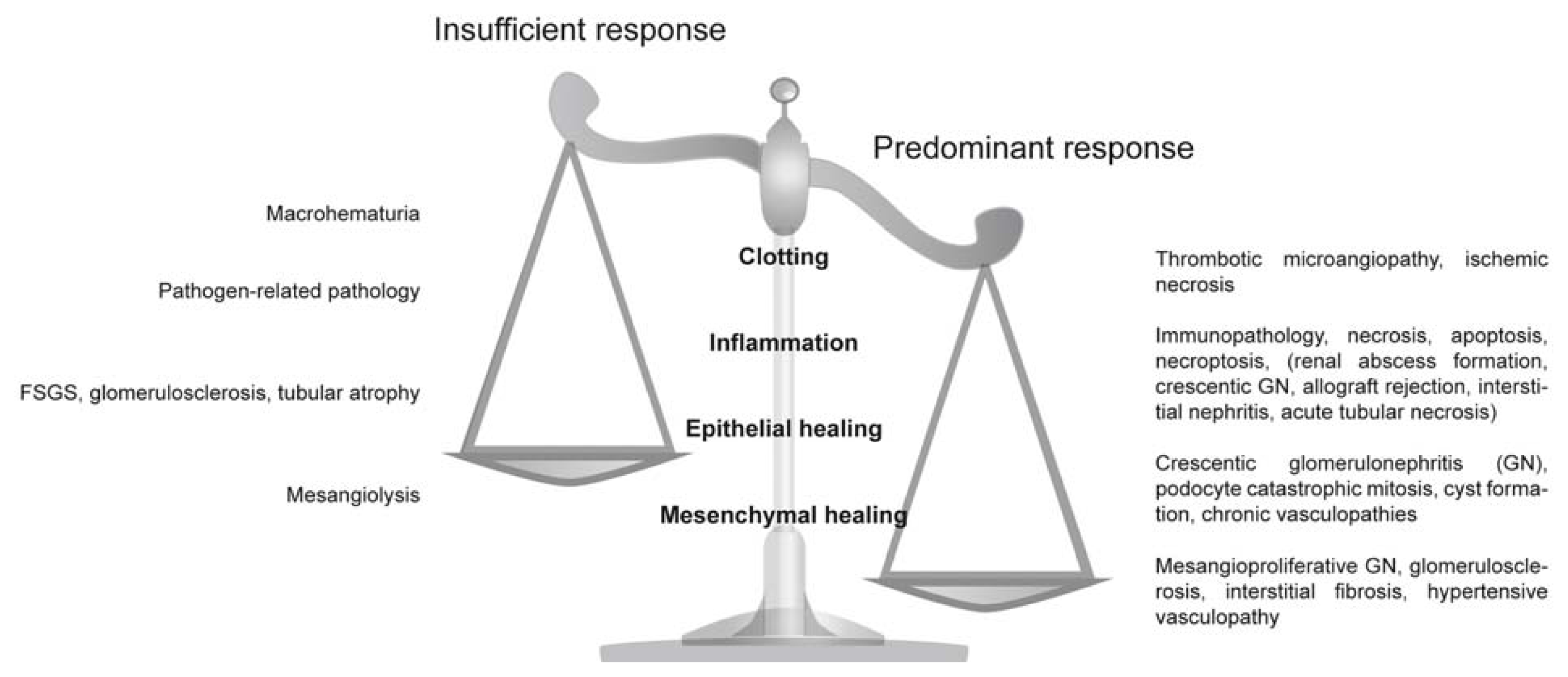
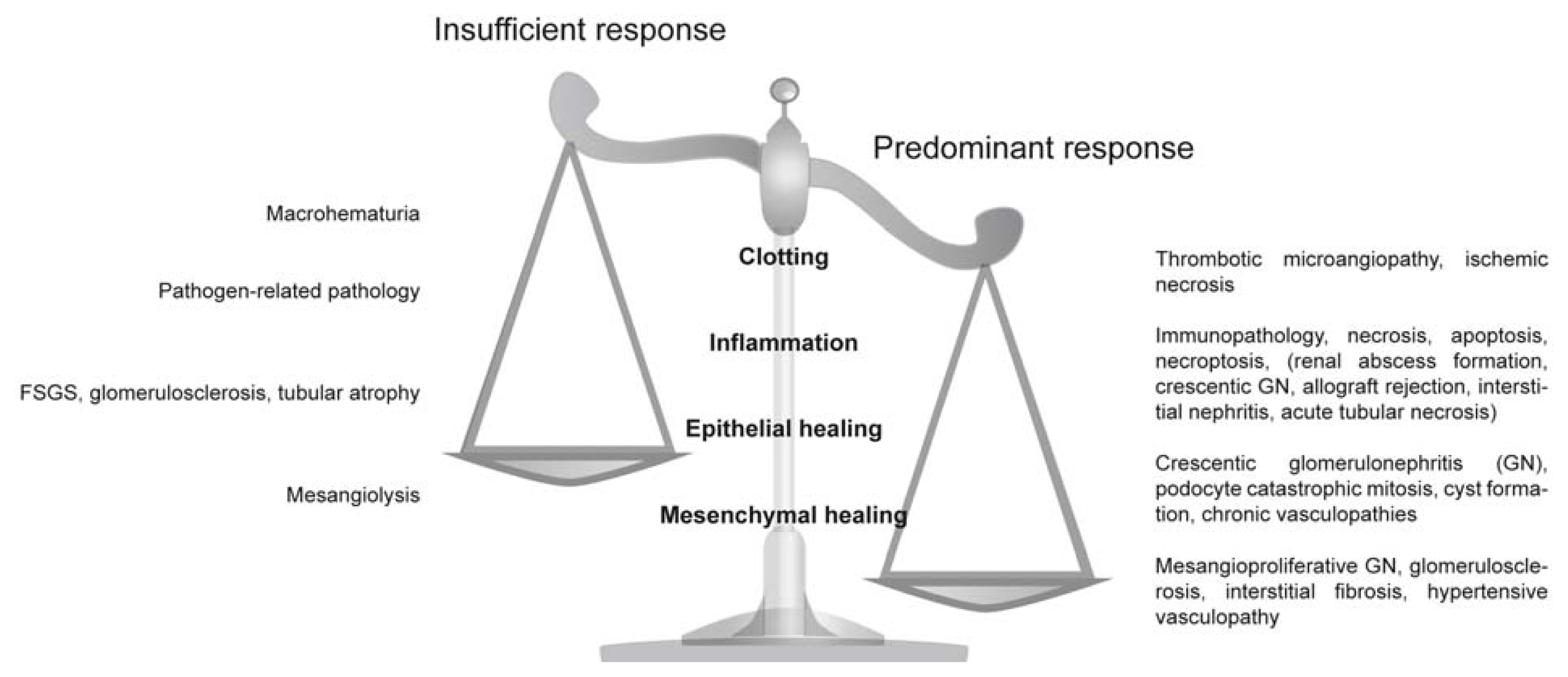
2.1. Clotting Addresses the Risk of Potentially Fatal Bleeding
2.2. Inflammation Addresses the Risk of Fatal Sepsis
2.3. Epithelial Regeneration Restores Barrier Functions
2.4. Mesenchymal Repair Restores Tissue Stability
3. Clotting
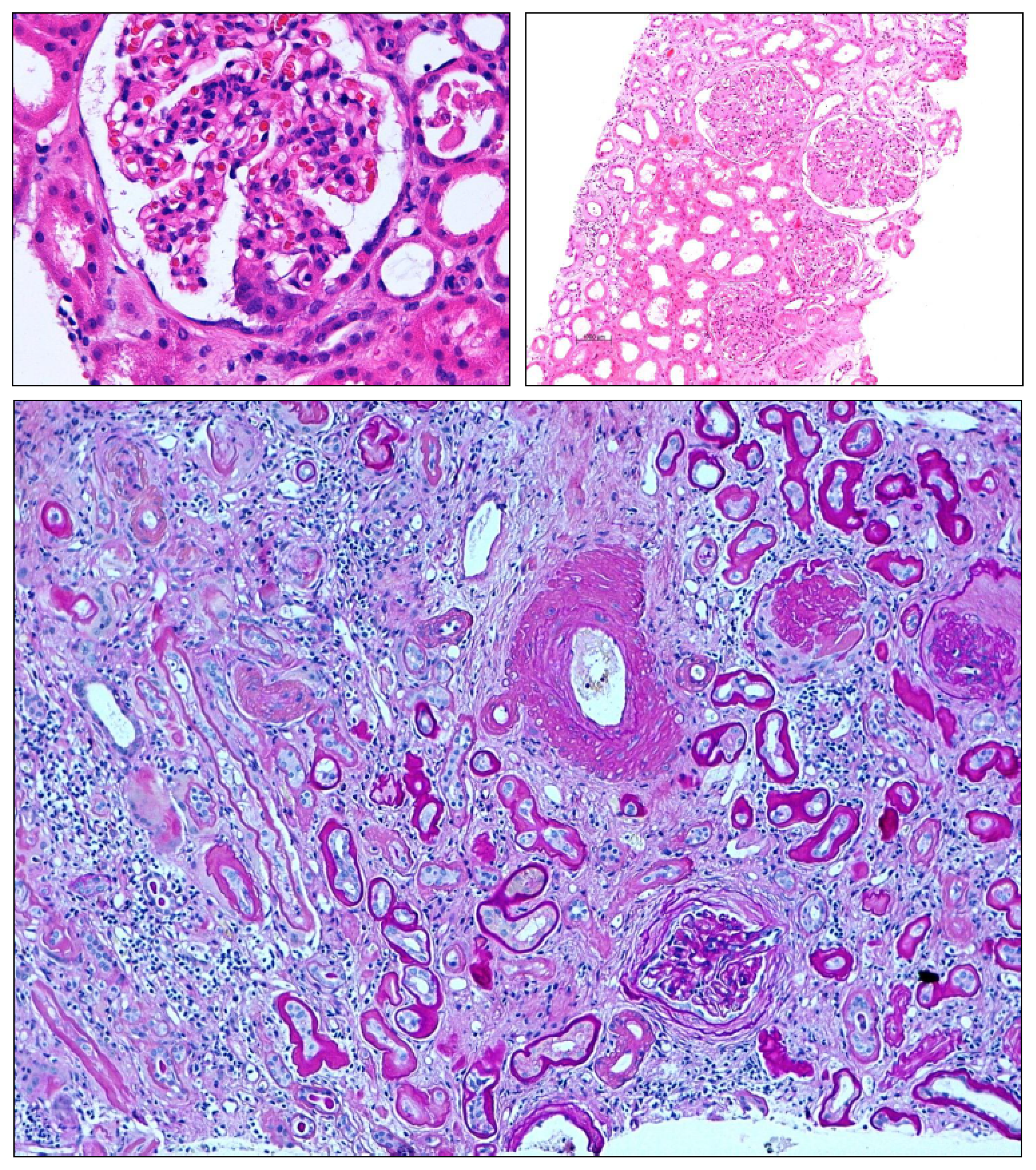
3.1. Overshooting Clotting in the Kidney
3.2. Insufficient Clotting in the Kidney
4. Inflammation
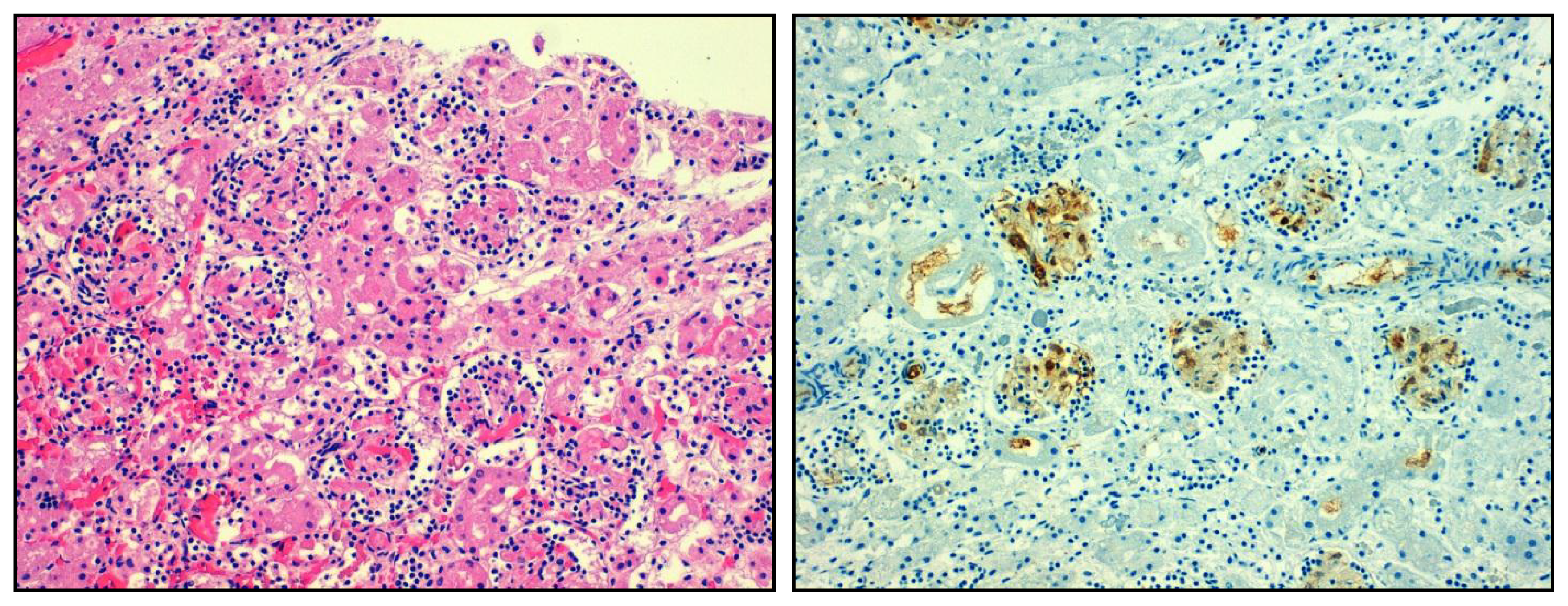
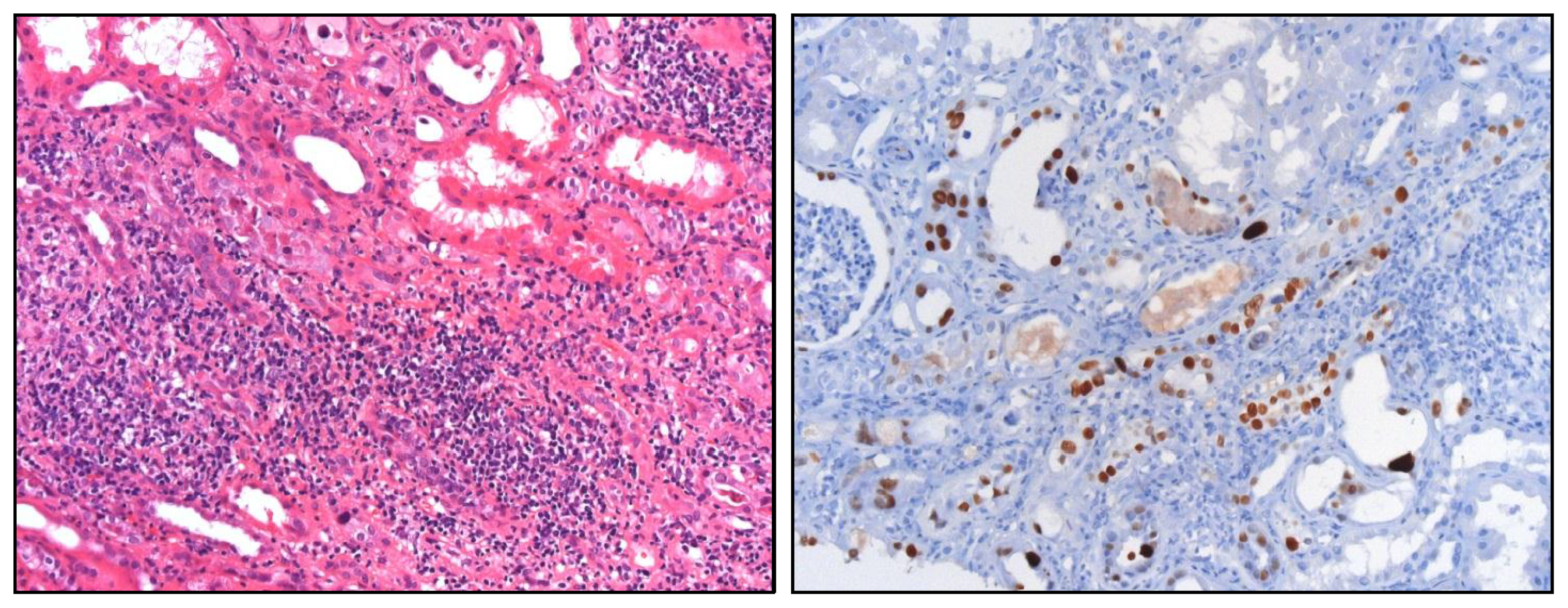
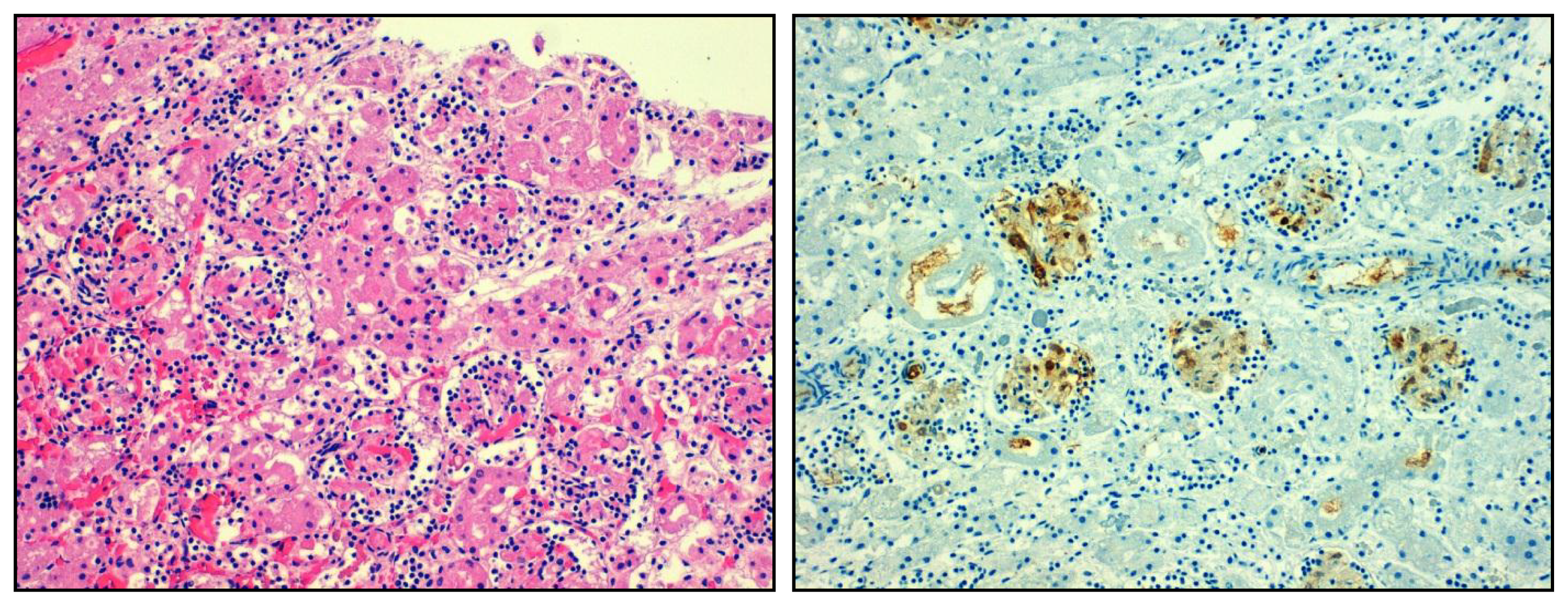
4.1. Overshooting Inflammation in the Kidney
4.2. Insufficient Inflammation in the Kidney
5. Epithelial Regeneration
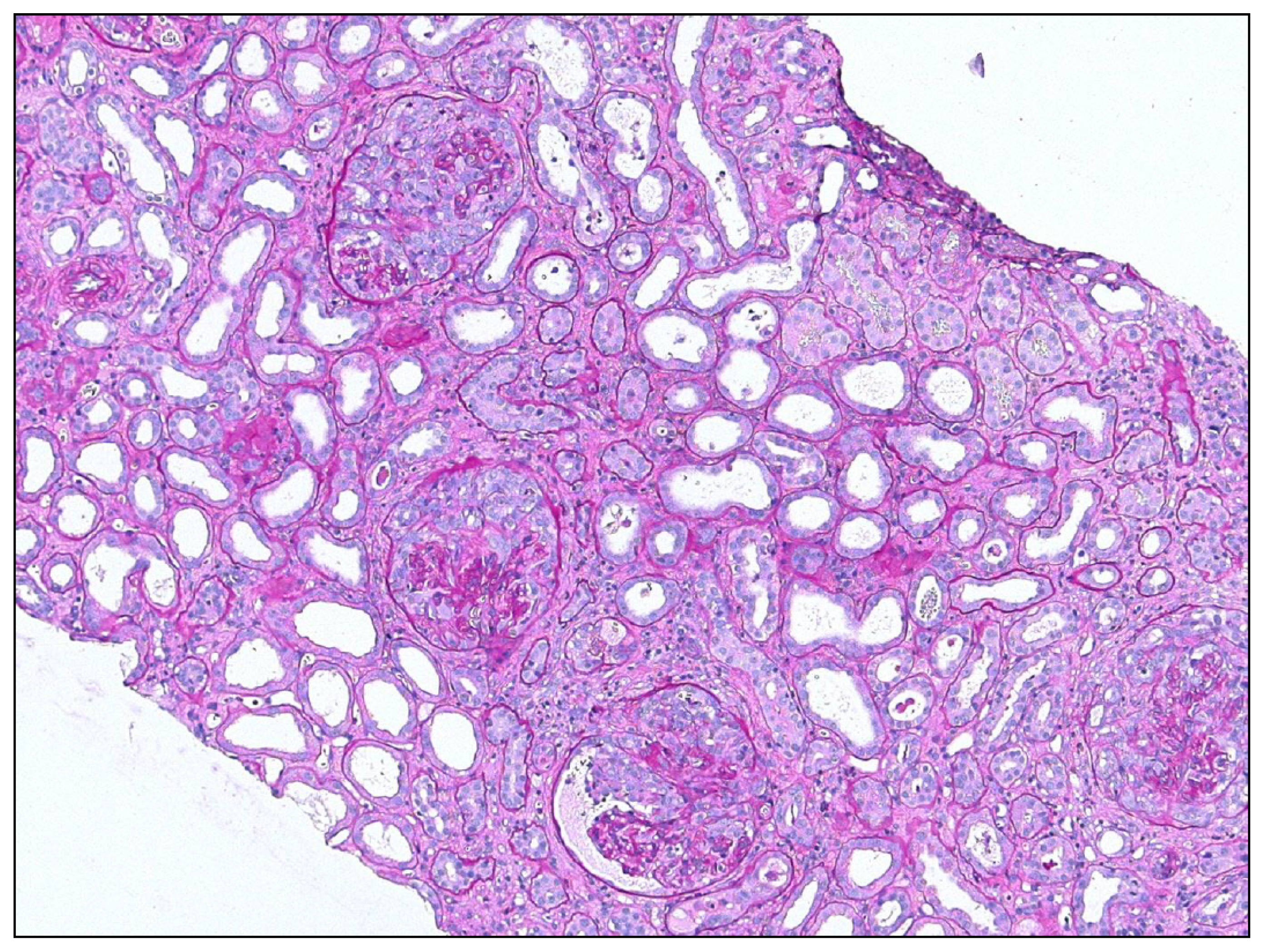
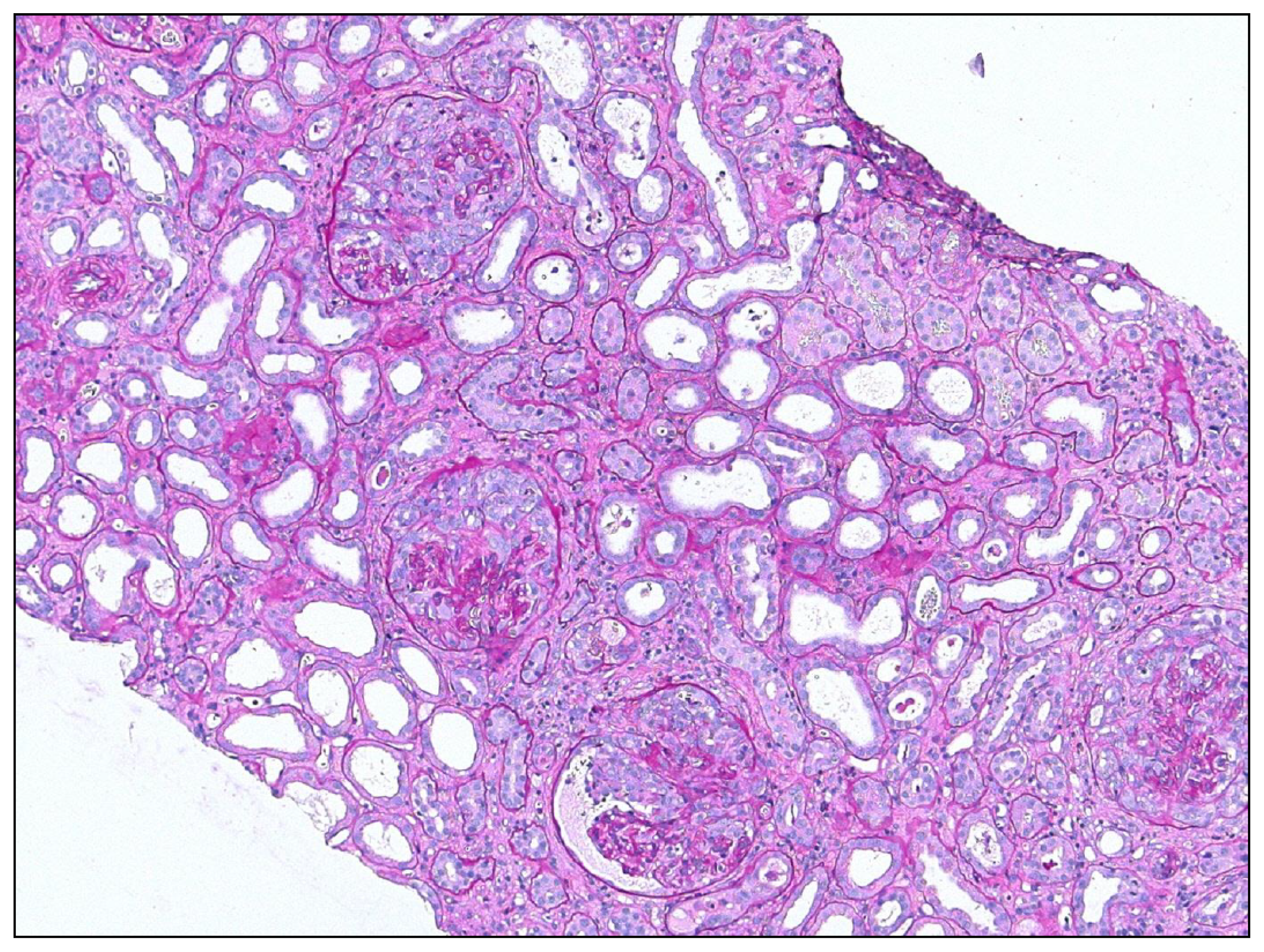
5.1. Overshooting Epithelial Regeneration in the Kidney
5.2. Insufficient Epithelial Regeneration in the Kidney
6. Mesenchymal Repair
6.1. Insufficient Mesenchymal Repair in the Kidney
6.2. Overshooting Mesenchymal Repair in the Kidney
7. Summary
Acknowledgments
Conflict of Interest
References
- Schilmiller, A.L.; Howe, G.A. Systemic signaling in the wound response. Curr. Opin. Plant Biol 2005, 8, 369–377. [Google Scholar]
- Gurtner, G.C.; Werner, S.; Barrandon, Y.; Longaker, M.T. Wound repair and regeneration. Nature 2008, 453, 314–321. [Google Scholar]
- Singer, A.J.; Clark, R.A. Cutaneous wound healing. N. Engl. J. Med 1999, 341, 738–746. [Google Scholar]
- Clark, R.A. Cutaneous tissue repair: Basic biologic considerations. I. J. Am. Acad. Dermatol 1985, 13, 701–725. [Google Scholar]
- Martin, P. Wound healing—Aiming for perfect skin regeneration. Science 1997, 276, 75–81. [Google Scholar]
- Engelmann, B.; Massberg, S. Thrombosis as an intravascular effector of innate immunity. Nat. Rev. Immunol 2013, 13, 34–45. [Google Scholar]
- Nurden, A.T. Platelets, inflammation and tissue regeneration. Thromb. Haemost 2011, 105, S13–S33. [Google Scholar]
- Furie, B.; Furie, B.C. Mechanisms of thrombus formation. N. Engl. J. Med 2008, 359, 938–949. [Google Scholar]
- Aird, W.C. The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood 2003, 101, 3765–3777. [Google Scholar]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar]
- Messier-Solek, C.; Buckley, K.M.; Rast, J.P. Highly diversified innate receptor systems and new forms of animal immunity. Semin. Immunol 2010, 22, 39–47. [Google Scholar]
- Gauthier, M.E.; Du Pasquier, L.; Degnan, B.M. The genome of the sponge Amphimedon queenslandica provides new perspectives into the origin of Toll-like and interleukin 1 receptor pathways. Evol. Dev 2010, 12, 519–533. [Google Scholar]
- Wiens, M.; Korzhev, M.; Perovic-Ottstadt, S.; Luthringer, B.; Brandt, D.; Klein, S.; Muller, W.E. Toll-like receptors are part of the innate immune defense system of sponges (demospongiae: Porifera). Mol. Biol. Evol 2007, 24, 792–804. [Google Scholar]
- Stearns-Kurosawa, D.J.; Osuchowski, M.F.; Valentine, C.; Kurosawa, S.; Remick, D.G. The pathogenesis of sepsis. Annu. Rev. Pathol 2011, 6, 19–48. [Google Scholar]
- Hickey, M.J.; Kubes, P. Intravascular immunity: The host-pathogen encounter in blood vessels. Nat. Rev. Immunol 2009, 9, 364–375. [Google Scholar]
- Chan, J.K.; Roth, J.; Oppenheim, J.J.; Tracey, K.J.; Vogl, T.; Feldmann, M.; Horwood, N.; Nanchahal, J. Alarmins: awaiting a clinical response. J. Clin. Invest 2012, 122, 2711–2719. [Google Scholar]
- Rock, K.L.; Latz, E.; Ontiveros, F.; Kono, H. The sterile inflammatory response. Annu. Rev. Immunol 2010, 28, 321–342. [Google Scholar]
- Anders, H.J. Toll-like receptors and danger signaling in kidney injury. J. Am. Soc. Nephrol 2010, 21, 1270–1274. [Google Scholar]
- Mulay, S.R.; Kulkarni, O.P.; Rupanagudi, K.V.; Migliorini, A.; Darisipudi, M.N.; Vilaysane, A.; Muruve, D.; Shi, Y.; Munro, F.; Liapis, H.; et al. Calcium oxalate crystals induce renal inflammation by NLRP3-mediated IL-1beta secretion. J. Clin. Invest 2013, 123, 236–246. [Google Scholar]
- Kannemeier, C.; Shibamiya, A.; Nakazawa, F.; Trusheim, H.; Ruppert, C.; Markart, P.; Song, Y.; Tzima, E.; Kennerknecht, E.; Niepmann, M.; et al. Extracellular RNA constitutes a natural procoagulant cofactor in blood coagulation. Proc. Natl. Acad. Sci. USA 2007, 104, 6388–6393. [Google Scholar]
- Semeraro, F.; Ammollo, C.T.; Morrissey, J.H.; Dale, G.L.; Friese, P.; Esmon, N.L.; Esmon, C.T. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: Involvement of platelet TLR2 and TLR4. Blood 2011, 118, 1952–1961. [Google Scholar]
- Delvaeye, M.; Conway, E.M. Coagulation and innate immune responses: Can we view them separately? Blood 2009, 114, 2367–2374. [Google Scholar]
- Niessen, F.; Schaffner, F.; Furlan-Freguia, C.; Pawlinski, R.; Bhattacharjee, G.; Chun, J.; Derian, C.K.; Andrade-Gordon, P.; Rosen, H.; Ruf, W. Dendritic cell PAR1-S1P3 signalling couples coagulation and inflammation. Nature 2008, 452, 654–658. [Google Scholar]
- Van der Poll, T.; de Boer, J.D.; Levi, M. The effect of inflammation on coagulation and vice versa. Curr. Opin. Infect. Dis 2011, 24, 273–278. [Google Scholar]
- Semple, J.W.; Italiano, J.E., Jr; Freedman, J. Platelets and the immune continuum. Nat. Rev. Immunol. 2011, 11, 264–274. [Google Scholar]
- Palabrica, T.; Lobb, R.; Furie, B.C.; Aronovitz, M.; Benjamin, C.; Hsu, Y.M.; Sajer, S.A.; Furie, B. Leukocyte accumulation promoting fibrin deposition is mediated in vivo by P-selectin on adherent platelets. Nature 1992, 359, 848–851. [Google Scholar]
- Ahronowitz, I.; Harp, J.; Shinkai, K. Etiology and management of pyoderma gangrenosum: A comprehensive review. Am. J. Clin. Dermatol 2012, 13, 191–211. [Google Scholar]
- Bonventre, J.V.; Zuk, A. Ischemic acute renal failure: An inflammatory disease? Kidney Int 2004, 66, 480–485. [Google Scholar]
- Hotchkiss, R.S.; Coopersmith, C.M.; McDunn, J.E.; Ferguson, T.A. The sepsis seesaw: Tilting toward immunosuppression. Nat. Med 2009, 15, 496–497. [Google Scholar]
- Romagnani, P. From proteus to prometheus: Learning from fish to modulate regeneration. J. Am. Soc. Nephrol 2010, 21, 726–728. [Google Scholar]
- Werner, S.; Grose, R. Regulation of wound healing by growth factors and cytokines. Physiol. Rev 2003, 83, 835–870. [Google Scholar]
- Sopova, K.; Tatsidou, P.; Stellos, K. Platelets and platelet interaction with progenitor cells in vascular homeostasis and inflammation. Curr. Vasc. Pharmacol 2012, 10, 555–562. [Google Scholar]
- Braun, R.K.; Ferrick, C.; Neubauer, P.; Sjoding, M.; Sterner-Kock, A.; Kock, M.; Putney, L.; Ferrick, D.A.; Hyde, D.M.; Love, R.B. IL-17 producing gammadelta T cells are required for a controlled inflammatory response after bleomycin-induced lung injury. Inflammation 2008, 31, 167–179. [Google Scholar]
- Jiang, G.X.; Zhong, X.Y.; Cui, Y.F.; Liu, W.; Tai, S.; Wang, Z.D.; Shi, Y.G.; Zhao, S.Y.; Li, C.L. IL-6/STAT3/TFF3 signaling regulates human biliary epithelial cell migration and wound healing in vitro. Mol. Biol. Rep 2010, 37, 3813–3818. [Google Scholar]
- Mizoguchi, A. Healing of intestinal inflammation by IL-22. Inflamm. Bowel. Dis 2012, 18, 1777–1784. [Google Scholar]
- Nishida, T.; Nakamura, M.; Mishima, H.; Otori, T. Interleukin 6 promotes epithelial migration by a fibronectin-dependent mechanism. J. Cell. Physiol 1992, 153, 1–5. [Google Scholar]
- Pickert, G.; Neufert, C.; Leppkes, M.; Zheng, Y.; Wittkopf, N.; Warntjen, M.; Lehr, H.A.; Hirth, S.; Weigmann, B.; Wirtz, S.; et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J. Exp. Med 2009, 206, 1465–1472. [Google Scholar]
- Sugawara, T.; Gallucci, R.M.; Simeonova, P.P.; Luster, M.I. Regulation and role of interleukin 6 in wounded human epithelial keratinocytes. Cytokine 2001, 15, 328–336. [Google Scholar]
- Zenewicz, L.A.; Flavell, R.A. Recent advances in IL-22 biology. Int. Immunol 2011, 23, 159–163. [Google Scholar]
- Sallustio, F.; Costantino, V.; Cox, S.N.; Loverre, A.; Divella, C.; Rizzi, M.; Schena, F.P. Human renal stem/progenitor cells repair tubular epithelial cell injury through TLR2-driven inhibin-A and microvesicle-shuttled decorin. Kidney Int 2013, 83, 392–403. [Google Scholar]
- Brockes, J.P. Amphibian limb regeneration: Rebuilding a complex structure. Science 1997, 276, 81–87. [Google Scholar]
- Sipos, F.; Valcz, G.; Molnar, B. Physiological and pathological role of local and immigrating colonic stem cells. World J. Gastroenterol 2012, 18, 295–301. [Google Scholar]
- Ryu, M.; Migliorini, A.; Miosge, N.; Gross, O.; Shankland, S.; Brinkkoetter, P.T.; Hagmann, H.; Romagnani, P.; Liapis, H.; Anders, H.J. Plasma leakage through glomerular basement membrane ruptures triggers the proliferation of parietal epithelial cells and crescent formation in non-inflammatory glomerular injury. J. Pathol 2012, 228, 482–494. [Google Scholar]
- Yang, L.; Besschetnova, T.Y.; Brooks, C.R.; Shah, J.V.; Bonventre, J.V. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat. Med 2010, 16, 535–543. [Google Scholar]
- Liu, Y. New insights into epithelial-mesenchymal transition in kidney fibrosis. J. Am. Soc. Nephrol 2010, 21, 212–222. [Google Scholar]
- Kalluri, R.; Neilson, E.G. Epithelial-mesenchymal transition and its implications for fibrosis. J. Clin. Invest 2003, 112, 1776–1784. [Google Scholar]
- Niedermeier, M.; Reich, B.; Rodriguez Gomez, M.; Denzel, A.; Schmidbauer, K.; Gobel, N.; Talke, Y.; Schweda, F.; Mack, M. CD4+ T cells control the differentiation of Gr1+ monocytes into fibrocytes. Proc. Natl. Acad. Sci. USA 2009, 106, 17892–17897. [Google Scholar]
- Humphreys, B.D.; Lin, S.L.; Kobayashi, A.; Hudson, T.E.; Nowlin, B.T.; Bonventre, J.V.; Valerius, M.T.; McMahon, A.P.; Duffield, J.S. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am. J. Pathol 2010, 176, 85–97. [Google Scholar]
- Zeisberg, M.; Neilson, E.G. Mechanisms of tubulointerstitial fibrosis. J. Am. Soc. Nephrol 2010, 21, 1819–1834. [Google Scholar]
- Chapman, K.; Seldon, M.; Richards, R. Thrombotic microangiopathies, thrombotic thrombocytopenic purpura, and ADAMTS-13. Semin. Thromb. Hemost 2012, 38, 47–54. [Google Scholar]
- Amengual, O.; Atsumi, T.; Koike, T. Pathophysiology of thrombosis and potential targeted therapies in antiphospholipid syndrome. Curr. Vasc. Pharmacol 2011, 9, 606–618. [Google Scholar]
- Bonsib, S.M. Glomerular basement membrane discontinuities. Scanning electron microscopic study of acellular glomeruli. Am. J. Pathol 1985, 119, 357–360. [Google Scholar]
- Sorensen, I.; Susnik, N.; Inhester, T.; Degen, J.L.; Melk, A.; Haller, H.; Schmitt, R. Fibrinogen, acting as a mitogen for tubulointerstitial fibroblasts, promotes renal fibrosis. Kidney Int 2011, 80, 1035–1044. [Google Scholar]
- Drew, A.F.; Tucker, H.L.; Liu, H.; Witte, D.P.; Degen, J.L.; Tipping, P.G. Crescentic glomerulonephritis is diminished in fibrinogen-deficient mice. Am. J. Physiol. Renal. Physiol 2001, 281, F1157–F1163. [Google Scholar]
- Downing, L.J.; Wakefield, T.W.; Strieter, R.M.; Prince, M.R.; Londy, F.J.; Fowlkes, J.B.; Hulin, M.S.; Kadell, A.M.; Wilke, C.A.; Brown, S.L.; et al. Anti-P-selectin antibody decreases inflammation and thrombus formation in venous thrombosis. J. Vasc. Surg 1997, 25, 816–827, ; discussion 828.. [Google Scholar]
- Esmon, C.T. The interactions between inflammation and coagulation. Br. J. Haematol 2005, 131, 417–430. [Google Scholar]
- Loof, T.G.; Morgelin, M.; Johansson, L.; Oehmcke, S.; Olin, A.I.; Dickneite, G.; Norrby-Teglund, A.; Theopold, U.; Herwald, H. Coagulation, an ancestral serine protease cascade, exerts a novel function in early immune defense. Blood 2011, 118, 2589–2598. [Google Scholar]
- Loof, T.G.; Schmidt, O.; Herwald, H.; Theopold, U. Coagulation systems of invertebrates and vertebrates and their roles in innate immunity: The same side of two coins? J. Innate Immun 2011, 3, 34–40. [Google Scholar]
- Rivers, R.P.; Hathaway, W.E.; Weston, W.L. The endotoxin-induced coagulant activity of human monocytes. Br. J. Haematol 1975, 30, 311–316. [Google Scholar]
- Cleary, C.M.; Moreno, J.A.; Fernández, B.; Ortiz, A.; Parra, E.G.; Gracia, C.; Blanco-Colio, L.M.; Barat, A.; Egido, J. Glomerular haematuria, renal interstitial haemorrhage and acute kidney injury. Nephrol. Dial. Transpl 2010, 25, 4103–4106. [Google Scholar]
- Degen, J.L.; Bugge, T.H.; Goguen, J.D. Fibrin and fibrinolysis in infection and host defense. J. Thromb. Haemost 2007, 5, 24–31. [Google Scholar]
- Nelson, P.J.; Rees, A.J.; Griffin, M.D.; Hughes, J.; Kurts, C.; Duffield, J. The renal mononuclear phagocytic system. J. Am. Soc. Nephrol 2012, 23, 194–203. [Google Scholar]
- Lech, M.; Avila-Ferrufino, A.; Skuginna, V.; Susanti, H.E.; Anders, H.J. Quantitative expression of RIG-like helicase, NOD-like receptor and inflammasome-related mRNAs in humans and mice. Int. Immunol 2010, 22, 717–728. [Google Scholar]
- Anders, H.J.; Banas, B.; Schlondorff, D. Signaling danger: Toll-Like receptors and their potential roles in kidney disease. J. Am. Soc. Nephrol 2004, 15, 854–867. [Google Scholar]
- Patole, P.S.; Pawar, R.D.; Lech, M.; Zecher, D.; Schmidt, H.; Segerer, S.; Ellwart, A.; Henger, A.; Kretzler, M.; Anders, H.J. Expression and regulation of Toll-like receptors in lupus-like immune complex glomerulonephritis of MRL-Fas(lpr) mice. Nephrol. Dial. Transplant 2006, 21, 3062–3073. [Google Scholar]
- Anders, H.J.; Schlondorff, D. Toll-Like receptors: Emerging concepts in kidney disease. Curr. Opin. Nephrol. Hypertens 2007, 16, 177–183. [Google Scholar]
- Anders, H.J.; Muruve, D.A. The inflammasomes in kidney disease. J. Am. Soc. Nephrol 2011, 22, 1007–1018. [Google Scholar]
- Pawar, R.D.; Castrezana-Lopez, L.; Allam, R.; Kulkarni, O.P.; Segerer, S.; Radomska, E.; Meyer, T.N.; Schwesinger, C.M.; Akis, N.; Grone, H.J.; et al. Bacterial lipopeptide triggers massive albuminuria in murine lupus nephritis by activating Toll-like receptor 2 at the glomerular filtration barrier. Immunology 2009, 128, e206–e221. [Google Scholar]
- Patole, P.S.; Grone, H.J.; Segerer, S.; Ciubar, R.; Belemezova, E.; Henger, A.; Kretzler, M.; Schlondorff, D.; Anders, H.J. Viral double-stranded RNA aggravates lupus nephritis through Toll-like receptor 3 on glomerular mesangial cells and antigen-presenting cells. J. Am. Soc. Nephrol 2005, 16, 1326–1338. [Google Scholar]
- Anders, H.J.; Banas, B.; Linde, Y.; Weller, L.; Cohen, C.D.; Kretzler, M.; Martin, S.; Vielhauer, V.; Schlondorff, D.; Grone, H.J. Bacterial CpG-DNA aggravates immune complex glomerulonephritis: Role of TLR9-mediated expression of chemokines and chemokine receptors. J. Am. Soc. Nephrol 2003, 14, 317–326. [Google Scholar]
- Anders, H.J.; Vielhauer, V.; Eis, V.; Linde, Y.; Kretzler, M.; Perez de Lema, G.; Strutz, F.; Bauer, S.; Rutz, M.; Wagner, H.; et al. Activation of toll-like receptor-9 induces progression of renal disease in MRL-Fas(lpr) mice. FASEB J 2004, 18, 534–536. [Google Scholar]
- Allam, R.; Pawar, R.D.; Kulkarni, O.P.; Hornung, V.; Hartmann, G.; Segerer, S.; Akira, S.; Endres, S.; Anders, H.J. Viral 5′-triphosphate RNA and non-CpG DNA aggravate autoimmunity and lupus nephritis via distinct TLR-independent immune responses. Eur. J. Immunol 2008, 38, 3487–3498. [Google Scholar]
- Brown, H.J.; Lock, H.R.; Sacks, S.H.; Robson, M.G. TLR2 stimulation of intrinsic renal cells in the induction of immune-mediated glomerulonephritis. J. Immunol 2006, 177, 1925–1931. [Google Scholar]
- Brown, H.J.; Lock, H.R.; Wolfs, T.G.; Buurman, W.A.; Sacks, S.H.; Robson, M.G. Toll-like receptor 4 ligation on intrinsic renal cells contributes to the induction of antibody-mediated glomerulonephritis via CXCL1 and CXCL2. J. Am. Soc. Nephrol 2007, 18, 1732–1739. [Google Scholar]
- Brown, H.J.; Sacks, S.H.; Robson, M.G. Toll-like receptor 2 agonists exacerbate accelerated nephrotoxic nephritis. J. Am. Soc. Nephrol 2006, 17, 1931–1939. [Google Scholar]
- Wornle, M.; Schmid, H.; Banas, B.; Merkle, M.; Henger, A.; Roeder, M.; Blattner, S.; Bock, E.; Kretzler, M.; Grone, H.J.; et al. Novel role of toll-like receptor 3 in hepatitis C-associated glomerulonephritis. Am. J. Pathol 2006, 168, 370–385. [Google Scholar]
- Lichtnekert, J.; Vielhauer, V.; Zecher, D.; Kulkarni, O.P.; Clauss, S.; Segerer, S.; Hornung, V.; Mayadas, T.N.; Beutler, B.; Akira, S.; et al. Trif is not required for immune complex glomerulonephritis: Dying cells activate mesangial cells via Tlr2/Myd88 rather than Tlr3/Trif. Am. J. Physiol. Renal. Physiol 2009, 296, F867–F874. [Google Scholar]
- Ryu, M.; Kulkarni, O.P.; Radomska, E.; Miosge, N.; Gross, O.; Anders, H.J. Bacterial CpG-DNA accelerates Alport glomerulosclerosis by inducing an M1 macrophage phenotype and tumor necrosis factor-alpha-mediated podocyte loss. Kidney Int 2011, 79, 189–198. [Google Scholar]
- Brahler, S.; Ising, C.; Hagmann, H.; Rasmus, M.; Hoehne, M.; Kurschat, C.; Kisner, T.; Goebel, H.; Shankland, S.; Addicks, K.; et al. Intrinsic proinflammatory signaling in podocytes contributes to podocyte damage and prolonged proteinuria. Am. J. Physiol. Renal. Physiol 2012, 303, F1473–F1485. [Google Scholar]
- Lasagni, L.; Romagnani, P. Glomerular epithelial stem cells: The good, the bad, and the ugly. J. Am. Soc. Nephrol 2010, 21, 1612–1619. [Google Scholar]
- Pawar, R.D.; Patole, P.S.; Wornle, M.; Anders, H.J. Microbial nucleic acids pay a Toll in kidney disease. Am. J. Physiol. Renal. Physiol 2006, 291, F509–F516. [Google Scholar]
- Marshak-Rothstein, A.; Rifkin, I.R. Immunologically active autoantigens: The role of toll-like receptors in the development of chronic inflammatory disease. Annu. Rev. Immunol 2007, 25, 419–441. [Google Scholar]
- Leadbetter, E.A.; Rifkin, I.R.; Hohlbaum, A.M.; Beaudette, B.C.; Shlomchik, M.J.; Marshak-Rothstein, A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature 2002, 416, 603–607. [Google Scholar]
- Steinman, R.M.; Banchereau, J. Taking dendritic cells into medicine. Nature 2007, 449, 419–426. [Google Scholar]
- Migliorini, A.; Anders, H.J. A novel pathogenetic concept-antiviral immunity in lupus nephritis. Nat. Rev. Nephrol 2012, 8, 183–189. [Google Scholar]
- Anders, H.J. Pseudoviral immunity—A novel concept for lupus. Trends Mol. Med 2009, 15, 553–561. [Google Scholar]
- Anders, H.J.; Lichtnekert, J.; Allam, R. Interferon-alpha and -beta in kidney inflammation. Kidney Int 2010, 77, 848–854. [Google Scholar]
- Flur, K.; Allam, R.; Zecher, D.; Kulkarni, O.P.; Lichtnekert, J.; Schwarz, M.; Beutler, B.; Vielhauer, V.; Anders, H.J. Viral RNA induces type I interferon-dependent cytokine release and cell death in mesangial cells via melanoma-differentiation-associated gene-5: Implications for viral infection-associated glomerulonephritis. Am. J. Pathol 2009, 175, 2014–2022. [Google Scholar]
- Hagele, H.; Allam, R.; Pawar, R.D.; Anders, H.J. Double-stranded RNA activates type I interferon secretion in glomerular endothelial cells via retinoic acid-inducible gene (RIG)-1. Nephrol. Dial. Transplant 2009, 24, 3312–3318. [Google Scholar]
- Hagele, H.; Allam, R.; Pawar, R.D.; Reichel, C.A.; Krombach, F.; Anders, H.J. Double-stranded DNA activates glomerular endothelial cells and enhances albumin permeability via a toll-like receptor-independent cytosolic DNA recognition pathway. Am. J. Pathol 2009, 175, 1896–1904. [Google Scholar]
- Allam, R.; Lichtnekert, J.; Moll, A.; Taubitz, A.; Vielhauer, V.; Anders, H.J. Viral RNA and DNA sense common antiviral responses including type I interferons in mesangial cells. J. Am. Soc. Nephrol 2009, 20, 1986–1996. [Google Scholar]
- Fairhurst, A.M.; Mathian, A.; Connolly, J.E.; Wang, A.; Gray, H.F.; George, T.A.; Boudreaux, C.D.; Zhou, X.J.; Li, Q.Z.; Koutouzov, S.; et al. Systemic IFN-alpha drives kidney nephritis in B6.Sle123 mice. Eur. J. Immunol 2008, 38, 1948–1960. [Google Scholar]
- Fairhurst, A.M.; Xie, C.; Fu, Y.; Wang, A.; Boudreaux, C.; Zhou, X.J.; Cibotti, R.; Coyle, A.; Connolly, J.E.; Wakeland, E.K.; et al. Type I interferons produced by resident renal cells may promote end-organ disease in autoantibody-mediated glomerulonephritis. J. Immunol 2009, 183, 6831–6838. [Google Scholar]
- Pawar, R.D.; Ramanjaneyulu, A.; Kulkarni, O.P.; Lech, M.; Segerer, S.; Anders, H.J. Inhibition of Toll-like receptor-7 (TLR-7) or TLR-7 plus TLR-9 attenuates glomerulonephritis and lung injury in experimental lupus. J. Am. Soc. Nephrol 2007, 18, 1721–1731. [Google Scholar]
- Wu, H.; Ma, J.; Wang, P.; Corpuz, T.M.; Panchapakesan, U.; Wyburn, K.R.; Chadban, S.J. HMGB1 contributes to kidney ischemia reperfusion injury. J. Am. Soc. Nephrol 2010, 21, 1878–1890. [Google Scholar]
- Shigeoka, A.A.; Holscher, T.D.; King, A.J.; Hall, F.W.; Kiosses, W.B.; Tobias, P.S.; Mackman, N.; McKay, D.B. TLR2 is constitutively expressed within the kidney and participates in ischemic renal injury through both MyD88-dependent and -independent pathways. J. Immunol 2007, 178, 6252–6258. [Google Scholar]
- Leemans, J.C.; Stokman, G.; Claessen, N.; Rouschop, K.M.; Teske, G.J.; Kirschning, C.J.; Akira, S.; van der Poll, T.; Weening, J.J.; Florquin, S. Renal-associated TLR2 mediates ischemia/reperfusion injury in the kidney. J. Clin. Invest 2005, 115, 2894–2903. [Google Scholar]
- Wu, H.; Chen, G.; Wyburn, K.R.; Yin, J.; Bertolino, P.; Eris, J.M.; Alexander, S.I.; Sharland, A.F.; Chadban, S.J. TLR4 activation mediates kidney ischemia/reperfusion injury. J. Clin. Invest 2007, 117, 2847–2859. [Google Scholar]
- Allam, R.; Scherbaum, C.R.; Darisipudi, M.N.; Mulay, S.R.; Hagele, H.; Lichtnekert, J.; Hagemann, J.H.; Rupanagudi, K.V.; Ryu, M.; Schwarzenberger, C.; et al. Histones from dying renal cells aggravate kidney injury via TLR2 and TLR4. J. Am. Soc. Nephrol 2012, 23, 1375–1388. [Google Scholar]
- Saemann, M.D.; Weichhart, T.; Zeyda, M.; Staffler, G.; Schunn, M.; Stuhlmeier, K.M.; Sobanov, Y.; Stulnig, T.M.; Akira, S.; von Gabain, A.; et al. Tamm-Horsfall glycoprotein links innate immune cell activation with adaptive immunity via a Toll-like receptor-4-dependent mechanism. J. Clin. Invest 2005, 115, 468–475. [Google Scholar]
- Darisipudi, M.N.; Thomasova, D.; Mulay, S.R.; Brech, D.; Noessner, E.; Liapis, H.; Anders, H.J. Uromodulin triggers IL-1beta-dependent innate immunity via the NLRP3 inflammasome. J. Am. Soc. Nephrol 2012, 23, 1783–1789. [Google Scholar]
- Lech, M.; Garlanda, C.; Mantovani, A.; Kirschning, C.J.; Schlondorff, D.; Anders, H.J. Different roles of TiR8/Sigirr on toll-like receptor signaling in intrarenal antigen-presenting cells and tubular epithelial cells. Kidney Int 2007, 72, 182–192. [Google Scholar]
- Lassen, S.; Lech, M.; Rommele, C.; Mittruecker, H.W.; Mak, T.W.; Anders, H.J. Ischemia reperfusion induces IFN regulatory factor 4 in renal dendritic cells, which suppresses postischemic inflammation and prevents acute renal failure. J. Immunol 2010, 185, 1976–1983. [Google Scholar]
- Lech, M.; Avila-Ferrufino, A.; Allam, R.; Segerer, S.; Khandoga, A.; Krombach, F.; Garlanda, C.; Mantovani, A.; Anders, H.J. Resident dendritic cells prevent postischemic acute renal failure by help of single Ig IL-1 receptor-related protein. J. Immunol 2009, 183, 4109–4118. [Google Scholar]
- Gong, J.; Wei, T.; Stark, R.W.; Jamitzky, F.; Heckl, W.M.; Anders, H.J.; Lech, M.; Rossle, S.C. Inhibition of Toll-like receptors TLR4 and 7 signaling pathways by SIGIRR: A computational approach. J. Struct. Biol 2010, 169, 323–330. [Google Scholar]
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. [Google Scholar]
- John, R.; Nelson, P.J. Dendritic cells in the kidney. J. Am. Soc. Nephrol 2007, 18, 2628–2635. [Google Scholar]
- Kruger, T.; Benke, D.; Eitner, F.; Lang, A.; Wirtz, M.; Hamilton-Williams, E.E.; Engel, D.; Giese, B.; Muller-Newen, G.; Floege, J.; et al. Identification and functional characterization of dendritic cells in the healthy murine kidney and in experimental glomerulonephritis. J. Am. Soc. Nephrol 2004, 15, 613–621. [Google Scholar]
- Vilaysane, A.; Chun, J.; Seamone, M.E.; Wang, W.; Chin, R.; Hirota, S.; Li, Y.; Clark, S.A.; Tschopp, J.; Trpkov, K.; et al. The NLRP3 inflammasome promotes renal inflammation and contributes to CKD. J. Am. Soc. Nephrol 2010, 21, 1732–1744. [Google Scholar]
- Lichtnekert, J.; Kulkarni, O.P.; Mulay, S.R.; Rupanagudi, K.V.; Ryu, M.; Allam, R.; Vielhauer, V.; Muruve, D.; Lindenmeyer, M.T.; Cohen, C.D.; et al. Anti-GBM glomerulonephritis involves IL-1 but is independent of NLRP3/ASC inflammasome-mediated activation of caspase-1. PLoS One 2011, 6, e26778. [Google Scholar]
- Iyer, S.S.; Pulskens, W.P.; Sadler, J.J.; Butter, L.M.; Teske, G.J.; Ulland, T.K.; Eisenbarth, S.C.; Florquin, S.; Flavell, R.A.; Leemans, J.C.; et al. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc. Natl. Acad. Sci. USA 2009, 106, 20388–20393. [Google Scholar]
- Anders, H.J.; Vielhauer, V.; Schlondorff, D. Chemokines and chemokine receptors are involved in the resolution or progression of renal disease. Kidney Int 2003, 63, 401–415. [Google Scholar]
- Vielhauer, V.; Kulkarni, O.; Reichel, C.A.; Anders, H.J. Targeting the recruitment of monocytes and macrophages in renal disease. Semin. Nephrol 2010, 30, 318–333. [Google Scholar]
- Heller, F.; Lindenmeyer, M.T.; Cohen, C.D.; Brandt, U.; Draganovici, D.; Fischereder, M.; Kretzler, M.; Anders, H.J.; Sitter, T.; Mosberger, I.; et al. The contribution of B cells to renal interstitial inflammation. Am. J. Pathol 2007, 170, 457–468. [Google Scholar]
- Steinmetz, O.M.; Stahl, R.A.; Panzer, U. Chemokines and B cells in renal inflammation and allograft rejection. Front. Biosci. (Schol Ed.) 2009, 1, 13–22. [Google Scholar]
- Swaminathan, S.; Griffin, M.D. First responders: Understanding monocyte-lineage traffic in the acutely injured kidney. Kidney Int 2008, 74, 1509–1511. [Google Scholar]
- Ley, K.; Laudanna, C.; Cybulsky, M.I.; Nourshargh, S. Getting to the site of inflammation: The leukocyte adhesion cascade updated. Nat. Rev. Immunol 2007, 7, 678–689. [Google Scholar]
- Stroo, I.; Stokman, G.; Teske, G.J.; Raven, A.; Butter, L.M.; Florquin, S.; Leemans, J.C. Chemokine expression in renal ischemia/reperfusion injury is most profound during the reparative phase. Int. Immunol 2010, 22, 433–442. [Google Scholar]
- Segerer, S.; Nelson, P.J. Chemokines in renal diseases. ScientificWorldJournal 2005, 5, 835–844. [Google Scholar]
- Ishida, Y.; Gao, J.L.; Murphy, P.M. Chemokine receptor CX3CR1 mediates skin wound healing by promoting macrophage and fibroblast accumulation and function. J. Immunol 2008, 180, 569–579. [Google Scholar]
- Panzer, U.; Kurts, C. T cell cross-talk with kidney dendritic cells in glomerulonephritis. J. Mol. Med. (Berl. ) 2010, 88, 19–26. [Google Scholar]
- Anders, H.J.; Ryu, M. Renal microenvironments and macrophage phenotypes determine progression or resolution of renal inflammation and fibrosis. Kidney Int 2011, 80, 915–925. [Google Scholar]
- Lech, M.; Anders, H.J. Macrophages and fibrosis: How resident and infiltrating mononuclear phagocytes orchestrate all phases of tissue injury and repair. Biochim. Biophys. Acta 2012, 1832, 989–997. [Google Scholar]
- Lech, M.; Grobmayr, R.; Weidenbusch, M.; Anders, H.J. Tissues use resident dendritic cells and macrophages to maintain homeostasis and to regain homeostasis upon tissue injury: The immunoregulatory role of changing tissue environments. Mediators Inflamm 2012, 2012, 951390. [Google Scholar]
- Duffield, J.S. Macrophages and immunologic inflammation of the kidney. Semin. Nephrol 2010, 30, 234–254. [Google Scholar]
- Anders, H.J.; Frink, M.; Linde, Y.; Banas, B.; Wornle, M.; Cohen, C.D.; Vielhauer, V.; Nelson, P.J.; Grone, H.J.; Schlondorff, D. CC chemokine ligand 5/RANTES chemokine antagonists aggravate glomerulonephritis despite reduction of glomerular leukocyte infiltration. J. Immunol 2003, 170, 5658–5666. [Google Scholar]
- Pawar, R.D.; Patole, P.S.; Ellwart, A.; Lech, M.; Segerer, S.; Schlondorff, D.; Anders, H.J. Ligands to nucleic acid-specific toll-like receptors and the onset of lupus nephritis. J. Am. Soc. Nephrol 2006, 17, 3365–3373. [Google Scholar]
- Anders, H.J.; Zecher, D.; Pawar, R.D.; Patole, P.S. Molecular mechanisms of autoimmunity triggered by microbial infection. Arthritis Res. Ther 2005, 7, 215–224. [Google Scholar]
- Ble, A.; Mosca, M.; Di Loreto, G.; Guglielmotti, A.; Biondi, G.; Bombardieri, S.; Remuzzi, G.; Ruggenenti, P. Antiproteinuric effect of chemokine C-C motif ligand 2 inhibition in subjects with acute proliferative lupus nephritis. Am. J. Nephrol 2011, 34, 367–372. [Google Scholar]
- Kulkarni, O.; Pawar, R.D.; Purschke, W.; Eulberg, D.; Selve, N.; Buchner, K.; Ninichuk, V.; Segerer, S.; Vielhauer, V.; Klussmann, S.; et al. Spiegelmer inhibition of CCL2/MCP-1 ameliorates lupus nephritis in MRL-(Fas)lpr mice. J. Am. Soc. Nephrol 2007, 18, 2350–2358. [Google Scholar]
- Ninichuk, V.; Clauss, S.; Kulkarni, O.; Schmid, H.; Segerer, S.; Radomska, E.; Eulberg, D.; Buchner, K.; Selve, N.; Klussmann, S.; et al. Late onset of Ccl2 blockade with the Spiegelmer mNOX-E36–3′PEG prevents glomerulosclerosis and improves glomerular filtration rate in db/db mice. Am. J. Pathol 2008, 172, 628–637. [Google Scholar]
- Kulkarni, O.; Eulberg, D.; Selve, N.; Zollner, S.; Allam, R.; Pawar, R.D.; Pfeiffer, S.; Segerer, S.; Klussmann, S.; Anders, H.J. Anti-Ccl2 Spiegelmer permits 75% dose reduction of cyclophosphamide to control diffuse proliferative lupus nephritis and pneumonitis in MRL-Fas(lpr) mice. J. Pharmacol. Exp. Ther 2009, 328, 371–377. [Google Scholar]
- Sayyed, S.G.; Ryu, M.; Kulkarni, O.P.; Schmid, H.; Lichtnekert, J.; Gruner, S.; Green, L.; Mattei, P.; Hartmann, G.; Anders, H.J. An orally active chemokine receptor CCR2 antagonist prevents glomerulosclerosis and renal failure in type 2 diabetes. Kidney Int 2011, 80, 68–78. [Google Scholar]
- Clauss, S.; Gross, O.; Kulkarni, O.; Avila-Ferrufino, A.; Radomska, E.; Segerer, S.; Eulberg, D.; Klussmann, S.; Anders, H.J. Ccl2/Mcp-1 blockade reduces glomerular and interstitial macrophages but does not ameliorate renal pathology in collagen4A3-deficient mice with autosomal recessive Alport nephropathy. J. Pathol 2009, 218, 40–47. [Google Scholar]
- Arnold, L.; Henry, A.; Poron, F.; Baba-Amer, Y.; van Rooijen, N.; Plonquet, A.; Gherardi, R.K.; Chazaud, B. Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J. Exp. Med 2007, 204, 1057–1069. [Google Scholar]
- Gordon, S.; Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol 2005, 5, 953–964. [Google Scholar]
- Lucas, T.; Waisman, A.; Ranjan, R.; Roes, J.; Krieg, T.; Muller, W.; Roers, A.; Eming, S.A. Differential roles of macrophages in diverse phases of skin repair. J. Immunol 2010, 184, 3964–3977. [Google Scholar]
- Xu, J.; Zhang, X.; Monestier, M.; Esmon, N.L.; Esmon, C.T. Extracellular histones are mediators of death through TLR2 and TLR4 in mouse fatal liver injury. J. Immunol 2011, 187, 2626–2631. [Google Scholar]
- Xu, J.; Zhang, X.; Pelayo, R.; Monestier, M.; Ammollo, C.T.; Semeraro, F.; Taylor, F.B.; Esmon, N.L.; Lupu, F.; Esmon, C.T. Extracellular histones are major mediators of death in sepsis. Nat. Med 2009, 15, 1318–1321. [Google Scholar]
- Kulkarni, O.P.; Ryu, M.; Kantner, C.; Sardy, M.; Naylor, D.; Lambert, D.; Brown, R.; Anders, H.J. Recombinant chaperonin 10 suppresses cutaneous lupus and lupus nephritis in MRL-(Fas)lpr mice. Nephrol. Dial. Transplant 2011, 27, 1358–1367. [Google Scholar]
- Vandewalle, A. Toll-like receptors and renal bacterial infections. Chang. Gung Med. J 2008, 31, 525–537. [Google Scholar]
- Ribeiro, A.; Wornle, M.; Motamedi, N.; Anders, H.J.; Grone, E.F.; Nitschko, H.; Kurktschiev, P.; Debiec, H.; Kretzler, M.; Cohen, C.D.; et al. Activation of innate immune defense mechanisms contributes to polyomavirus BK-associated nephropathy. Kidney Int 2012, 81, 100–111. [Google Scholar]
- Babel, N.; Volk, H.D.; Reinke, P. BK polyomavirus infection and nephropathy: the virus-immune system interplay. Nat. Rev. Nephrol 2011, 7, 399–406. [Google Scholar]
- Anders, H.J.; Patole, P.S. Toll-like receptors recognize uropathogenic Escherichia coli and trigger inflammation in the urinary tract. Nephrol. Dial. Transplant 2005, 20, 1529–1532. [Google Scholar]
- Patole, P.S.; Schubert, S.; Hildinger, K.; Khandoga, S.; Khandoga, A.; Segerer, S.; Henger, A.; Kretzler, M.; Werner, M.; Krombach, F.; et al. Toll-like receptor-4: Renal cells and bone marrow cells signal for neutrophil recruitment during pyelonephritis. Kidney Int 2005, 68, 2582–2587. [Google Scholar]
- Yang, C.W.; Hung, C.C.; Wu, M.S.; Tian, Y.C.; Chang, C.T.; Pan, M.J.; Vandewalle, A. Toll-like receptor 2 mediates early inflammation by leptospiral outer membrane proteins in proximal tubule cells. Kidney Int 2006, 69, 815–822. [Google Scholar]
- Bonventre, J.V. Dedifferentiation and proliferation of surviving epithelial cells in acute renal failure. J. Am. Soc. Nephrol 2003, 14, S55–S61. [Google Scholar]
- Abuelo, J.G. Normotensive ischemic acute renal failure. N. Engl. J. Med 2007, 357, 797–805. [Google Scholar]
- Sugimoto, H.; Lebleu, V.S.; Bosukonda, D.; Keck, P.; Taduri, G.; Bechtel, W.; Okada, H.; Carlson, W.; Bey, P.; Rusckowski, M.; et al. Activin-like kinase 3 is important for kidney regeneration and reversal of fibrosis. Nat. Med 2012, 18, 396–404. [Google Scholar]
- Mulay, S.R.; Thomasova, D.; Ryu, M.; Anders, H.J. MDM2 (murine double minute-2) links inflammation and tubular cell healing during acute kidney injury in mice. Kidney Int 2012, 81, 1199–1211. [Google Scholar]
- Thomasova, D.; Mulay, S.R.; Bruns, H.; Anders, H.J. p53-Independent Roles of MDM2 in NF-kappaB signaling: Implications for cancer therapy, wound healing, and autoimmune diseases. Neoplasia 2012, 14, 1097–1101. [Google Scholar]
- Weidenbusch, M.; Anders, H.J. Tissue microenvironments define and get reinforced by macrophage phenotypes in homeostasis or during inflammation, repair and fibrosis. J. Innate Immun 2012, 4, 463–477. [Google Scholar]
- Ricardo, S.D.; van Goor, H.; Eddy, A.A. Macrophage diversity in renal injury and repair. J. Clin. Invest 2008, 118, 3522–3530. [Google Scholar]
- Duffield, J.S.; Park, K.M.; Hsiao, L.L.; Kelley, V.R.; Scadden, D.T.; Ichimura, T.; Bonventre, J.V. Restoration of tubular epithelial cells during repair of the postischemic kidney occurs independently of bone marrow-derived stem cells. J. Clin. Invest 2005, 115, 1743–1755. [Google Scholar]
- Lee, S.; Huen, S.; Nishio, H.; Nishio, S.; Lee, H.K.; Choi, B.S.; Ruhrberg, C.; Cantley, L.G. Distinct macrophage phenotypes contribute to kidney injury and repair. J. Am. Soc. Nephrol 2011, 22, 317–326. [Google Scholar]
- Zhang, M.Z.; Yao, B.; Yang, S.; Jiang, L.; Wang, S.; Fan, X.; Yin, H.; Wong, K.; Miyazawa, T.; Chen, J.; et al. CSF-1 signaling mediates recovery from acute kidney injury. J. Clin. Invest 2012, 122, 4519–4532. [Google Scholar]
- Duffield, J.S.; Forbes, S.J.; Constandinou, C.M.; Clay, S.; Partolina, M.; Vuthoori, S.; Wu, S.; Lang, R.; Iredale, J.P. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J. Clin. Invest 2005, 115, 56–65. [Google Scholar]
- Lee, S.B.; Kalluri, R. Mechanistic connection between inflammation and fibrosis. Kidney Int 2010, 78, S22–S26. [Google Scholar]
- Iwata, Y.; Bostrom, E.A.; Menke, J.; Rabacal, W.A.; Morel, L.; Wada, T.; Kelley, V.R. Aberrant macrophages mediate defective kidney repair that triggers nephritis in lupus-susceptible mice. J. Immunol 2012, 188, 4568–4580. [Google Scholar]
- Smeets, B.; Angelotti, M.L.; Rizzo, P.; Dijkman, H.; Lazzeri, E.; Mooren, F.; Ballerini, L.; Parente, E.; Sagrinati, C.; Mazzinghi, B.; et al. Renal progenitor cells contribute to hyperplastic lesions of podocytopathies and crescentic glomerulonephritis. J. Am. Soc. Nephrol 2009, 20, 2593–2603. [Google Scholar]
- Smeets, B.; Uhlig, S.; Fuss, A.; Mooren, F.; Wetzels, J.F.; Floege, J.; Moeller, M.J. Tracing the origin of glomerular extracapillary lesions from parietal epithelial cells. J. Am. Soc. Nephrol 2009, 20, 2604–2615. [Google Scholar]
- Atkins, R.C.; Nikolic-Paterson, D.J.; Song, Q.; Lan, H.Y. Modulators of crescentic glomerulonephritis. J. Am. Soc. Nephrol 1996, 7, 2271–2278. [Google Scholar]
- Tipping, P.G.; Kitching, P.R.; Holdsworth, S.R. The Formation of the Glomerular Crescent. In Immunologic Renal Diseases, 2nd ed.; Neilson, E.G., Couser, W.G., Eds.; Lippincott Williams & Wilkins Publishers: Philadelphia, PA, USA, 2001. [Google Scholar]
- Bollee, G.; Flamant, M.; Schordan, S.; Fligny, C.; Rumpel, E.; Milon, M.; Schordan, E.; Sabaa, N.; Vandermeersch, S.; Galaup, A.; et al. Epidermal growth factor receptor promotes glomerular injury and renal failure in rapidly progressive crescentic glomerulonephritis. Nat. Med 2011, 17, 1242–1250. [Google Scholar]
- Ohse, T.; Pippin, J.W.; Chang, A.M.; Krofft, R.D.; Miner, J.H.; Vaughan, M.R.; Shankland, S.J. The enigmatic parietal epithelial cell is finally getting noticed: A review. Kidney Int 2009, 76, 1225–1238. [Google Scholar]
- Shankland, S.J.; Anders, H.J.; Romagnani, P. Glomerular parietal epithelial cells in kidney physiology, pathology, and repair. Curr. Opin. Nephrol. Hypertens 2013, 22, 302–309. [Google Scholar]
- Lindgren, D.; Bostrom, A.K.; Nilsson, K.; Hansson, J.; Sjolund, J.; Moller, C.; Jirstrom, K.; Nilsson, E.; Landberg, G.; Axelson, H.; et al. Isolation and characterization of progenitor-like cells from human renal proximal tubules. Am. J. Pathol 2011, 178, 828–837. [Google Scholar]
- Romagnani, P. Family portrait: Renal progenitor of Bowman’s capsule and its tubular brothers. Am. J. Pathol 2011, 178, 490–493. [Google Scholar]
- Forbes, M.S.; Thornhill, B.A.; Chevalier, R.L. Proximal tubular injury and rapid formation of atubular glomeruli in mice with unilateral ureteral obstruction: A new look at an old model. Am. J. Physiol. Renal. Physiol 2011, 301, F110–F117. [Google Scholar]
- Chevalier, R.L.; Forbes, M.S. Generation and evolution of atubular glomeruli in the progression of renal disorders. J. Am. Soc. Nephrol 2008, 19, 197–206. [Google Scholar]
- D’Agati, V.D.; Kaskel, F.J.; Falk, R.J. Focal segmental glomerulosclerosis. N. Engl. J. Med 2011, 365, 2398–2411. [Google Scholar]
- Romagnani, P.; Remuzzi, G. Renal progenitors in non-diabetic and diabetic nephropathies. Trends Endocrinol. Metab 2013, 24, 13–20. [Google Scholar]
- Kriz, W.; Lemley, K.V. The role of the podocyte in glomerulosclerosis. Curr. Opin. Nephrol. Hypertens 1999, 8, 489–497. [Google Scholar]
- de Teixeira, V.P.; Blattner, S.M.; Li, M.; Anders, H.J.; Cohen, C.D.; Edenhofer, I.; Calvaresi, N.; Merkle, M.; Rastaldi, M.P.; Kretzler, M. Functional consequences of integrin-linked kinase activation in podocyte damage. Kidney Int 2005, 67, 514–523. [Google Scholar]
- Wharram, B.L.; Goyal, M.; Wiggins, J.E.; Sanden, S.K.; Hussain, S.; Filipiak, W.E.; Saunders, T.L.; Dysko, R.C.; Kohno, K.; Holzman, L.B.; et al. Podocyte depletion causes glomerulosclerosis: Diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. J. Am. Soc. Nephrol 2005, 16, 2941–2952. [Google Scholar]
- Sachs, N.; Sonnenberg, A. Cell-matrix adhesion of podocytes in physiology and disease. Nat. Rev. Nephrol 2013, 9, 200–210. [Google Scholar]
- Lasagni, L.; Lazzeri, E.; Shankland, S.J.; Anders, H.J.; Romagnani, P. Podocyte mitosis—A catastrophe. Curr. Mol. Med 2013, 13, 13–23. [Google Scholar]
- Lasagni, L.; Ballerini, L.; Angelotti, M.L.; Parente, E.; Sagrinati, C.; Mazzinghi, B.; Peired, A.; Ronconi, E.; Becherucci, F.; Bani, D.; et al. Notch activation differentially regulates renal progenitors proliferation and differentiation toward the podocyte lineage in glomerular disorders. Stem Cells 2010, 28, 1674–1685. [Google Scholar]
- Pippin, J.W.; Durvasula, R.; Petermann, A.; Hiromura, K.; Couser, W.G.; Shankland, S.J. DNA damage is a novel response to sublytic complement C5b-9-induced injury in podocytes. J. Clin. Invest 2003, 111, 877–885. [Google Scholar]
- Mulay, S.R.; Thomasova, D.; Ryu, M.; Kulkarni, O.P.; Migliorini, A.; Bruns, H.; Gröbmayr, R.; Lazzeri, E.; Lasagni, L.; Liapis, H.; Romagnani, P.; Anders, H.-J. Podocyte loss involves MDM2-driven mitotic catastrophe of podocytes. J. Pathol. 2013, in press. [Google Scholar]
- Sugimoto, H.; Mundel, T.M.; Sund, M.; Xie, L.; Cosgrove, D.; Kalluri, R. Bone-marrow-derived stem cells repair basement membrane collagen defects and reverse genetic kidney disease. Proc. Natl. Acad. Sci. USA 2006, 103, 7321–7326. [Google Scholar]
- LeBleu, V.; Sugimoto, H.; Mundel, T.M.; Gerami-Naini, B.; Finan, E.; Miller, C.A.; Gattone, V.H., II; Lu, L.; Shield, C.F., III; Folkman, J.; et al. Stem cell therapies benefit Alport syndrome. J. Am. Soc. Nephrol. 2009, 20, 2359–2370. [Google Scholar]
- Gross, O.; Borza, D.B.; Anders, H.J.; Licht, C.; Weber, M.; Segerer, S.; Torra, R.; Gubler, M.C.; Heidet, L.; Harvey, S.; et al. Stem cell therapy for Alport syndrome: The hope beyond the hype. Nephrol. Dial. Transplant 2009, 24, 731–734. [Google Scholar]
- Lazzeri, E.; Crescioli, C.; Ronconi, E.; Mazzinghi, B.; Sagrinati, C.; Netti, G.S.; Angelotti, M.L.; Parente, E.; Ballerini, L.; Cosmi, L.; et al. Regenerative potential of embryonic renal multipotent progenitors in acute renal failure. J. Am. Soc. Nephrol 2007, 18, 3128–3138. [Google Scholar]
- Ronconi, E.; Sagrinati, C.; Angelotti, M.L.; Lazzeri, E.; Mazzinghi, B.; Ballerini, L.; Parente, E.; Becherucci, F.; Gacci, M.; Carini, M.; et al. Regeneration of glomerular podocytes by human renal progenitors. J. Am. Soc. Nephrol 2009, 20, 322–332. [Google Scholar]
- Appel, D.; Kershaw, D.B.; Smeets, B.; Yuan, G.; Fuss, A.; Frye, B.; Elger, M.; Kriz, W.; Floege, J.; Moeller, M.J. Recruitment of podocytes from glomerular parietal epithelial cells. J. Am. Soc. Nephrol 2009, 20, 333–343. [Google Scholar]
- Sayyed, S.G.; Hagele, H.; Kulkarni, O.P.; Endlich, K.; Segerer, S.; Eulberg, D.; Klussmann, S.; Anders, H.J. Podocytes produce homeostatic chemokine stromal cell-derived factor-1/CXCL12, which contributes to glomerulosclerosis, podocyte loss and albuminuria in a mouse model of type 2 diabetes. Diabetologia 2009, 52, 2445–2454. [Google Scholar]
- Darisipudi, M.N.; Kulkarni, O.P.; Sayyed, S.G.; Ryu, M.; Migliorini, A.; Sagrinati, C.; Parente, E.; Vater, A.; Eulberg, D.; Klussmann, S.; et al. Dual blockade of the homeostatic chemokine CXCL12 and the proinflammatory chemokine CCL2 has additive protective effects on diabetic kidney disease. Am. J. Pathol 2011, 179, 116–124. [Google Scholar]
- Gaikwad, A.B.; Sayyed, S.G.; Lichtnekert, J.; Tikoo, K.; Anders, H.J. Renal failure increases cardiac histone h3 acetylation, dimethylation, and phosphorylation and the induction of cardiomyopathy-related genes in type 2 diabetes. Am. J. Pathol 2010, 176, 1079–1083. [Google Scholar]
- Sayyed, S.G.; Gaikwad, A.B.; Lichtnekert, J.; Kulkarni, O.; Eulberg, D.; Klussmann, S.; Tikoo, K.; Anders, H.J. Progressive glomerulosclerosis in type 2 diabetes is associated with renal histone H3K9 and H3K23 acetylation, H3K4 dimethylation and phosphorylation at serine 10. Nephrol. Dial. Transplant 2010, 25, 1811–1817. [Google Scholar]
- Angelotti, M.L.; Ronconi, E.; Ballerini, L.; Peired, A.; Mazzinghi, B.; Sagrinati, C.; Parente, E.; Gacci, M.; Carini, M.; Rotondi, M.; et al. Characterization of renal progenitors committed toward tubular lineage and their regenerative potential in renal tubular injury. Stem Cells 2012, 30, 1714–1725. [Google Scholar]
- Humphreys, B.D.; Czerniak, S.; DiRocco, D.P.; Hasnain, W.; Cheema, R.; Bonventre, J.V. Repair of injured proximal tubule does not involve specialized progenitors. Proc. Natl. Acad. Sci. USA 2011, 108, 9226–9231. [Google Scholar]
- Humphreys, B.D.; Valerius, M.T.; Kobayashi, A.; Mugford, J.W.; Soeung, S.; Duffield, J.S.; McMahon, A.P.; Bonventre, J.V. Intrinsic epithelial cells repair the kidney after injury. Cell Stem Cell 2008, 2, 284–291. [Google Scholar]
- Togel, F.E.; Westenfelder, C. Mesenchymal stem cells: A new therapeutic tool for AKI. Nat. Rev. Nephrol 2010, 6, 179–183. [Google Scholar]
- Higgins, D.F.; Lappin, D.W.; Kieran, N.E.; Anders, H.J.; Watson, R.W.; Strutz, F.; Schlondorff, D.; Haase, V.H.; Fitzpatrick, J.M.; Godson, C.; et al. DNA oligonucleotide microarray technology identifies fisp-12 among other potential fibrogenic genes following murine unilateral ureteral obstruction (UUO): Modulation during epithelial-mesenchymal transition. Kidney Int 2003, 64, 2079–2091. [Google Scholar]
- Ninichuk, V.; Gross, O.; Segerer, S.; Hoffmann, R.; Radomska, E.; Buchstaller, A.; Huss, R.; Akis, N.; Schlondorff, D.; Anders, H.J. Multipotent mesenchymal stem cells reduce interstitial fibrosis but do not delay progression of chronic kidney disease in collagen4A3-deficient mice. Kidney Int 2006, 70, 121–129. [Google Scholar]
- Migliorini, A.; Ebid, R.; Scherbaum, C.R.; Anders, H.J. The danger control concept in kidney disease: mesangial cells. J. Nephrol 2013, 26, 437–449. [Google Scholar]
- Johnson, R.J.; Raines, E.W.; Floege, J.; Yoshimura, A.; Pritzl, P.; Alpers, C.; Ross, R. Inhibition of mesangial cell proliferation and matrix expansion in glomerulonephritis in the rat by antibody to platelet-derived growth factor. J. Exp. Med 1992, 175, 1413–1416. [Google Scholar]
- Bomback, A.S.; Appel, G.B. Pathogenesis of the C3 glomerulopathies and reclassification of MPGN. Nat. Rev. Nephrol 2012, 8, 634–642. [Google Scholar]
- Hugo, C.; Shankland, S.J.; Bowen-Pope, D.F.; Couser, W.G.; Johnson, R.J. Extraglomerular origin of the mesangial cell after injury. A new role of the juxtaglomerular apparatus. J. Clin. Invest 1997, 100, 786–794. [Google Scholar]
- Imasawa, T.; Utsunomiya, Y.; Kawamura, T.; Zhong, Y.; Nagasawa, R.; Okabe, M.; Maruyama, N.; Hosoya, T.; Ohno, T. The potential of bone marrow-derived cells to differentiate to glomerular mesangial cells. J. Am. Soc. Nephrol 2001, 12, 1401–1409. [Google Scholar]
- Sethi, S.; Fervenza, F.C. Membranoproliferative glomerulonephritis—A new look at an old entity. N. Engl. J. Med 2012, 366, 1119–1131. [Google Scholar]
- Remuzzi, G.; Benigni, A.; Remuzzi, A. Mechanisms of progression and regression of renal lesions of chronic nephropathies and diabetes. J. Clin. Invest 2006, 116, 288–296. [Google Scholar]
- Smeets, B.; Kuppe, C.; Sicking, E.M.; Fuss, A.; Jirak, P.; van Kuppevelt, T.H.; Endlich, K.; Wetzels, J.F.; Grone, H.J.; Floege, J.; et al. Parietal epithelial cells participate in the formation of sclerotic lesions in focal segmental glomerulosclerosis. J. Am. Soc. Nephrol 2011, 22, 1262–1274. [Google Scholar]
- Helal, I.; Fick-Brosnahan, G.M.; Reed-Gitomer, B.; Schrier, R.W. Glomerular hyperfiltration: Definitions, mechanisms and clinical implications. Nat. Rev. Nephrol 2012, 8, 293–300. [Google Scholar]
- Bariety, J.; Hill, G.S.; Mandet, C.; Irinopoulou, T.; Jacquot, C.; Meyrier, A.; Bruneval, P. Glomerular epithelial-mesenchymal transdifferentiation in pauci-immune crescentic glomerulonephritis. Nephrol. Dial. Transplant 2003, 18, 1777–1784. [Google Scholar]
- Duffield, J.S. Epithelial to mesenchymal transition in injury of solid organs: Fact or artifact? Gastroenterology 2010, 139. [Google Scholar] [CrossRef]
- Zeisberg, M.; Duffield, J.S. Resolved: EMT produces fibroblasts in the kidney. J. Am. Soc. Nephrol 2010, 21, 1247–1253. [Google Scholar]
- Kriz, W.; Kaissling, B.; Le Hir, M. Epithelial-mesenchymal transition (EMT) in kidney fibrosis: Fact or fantasy? J. Clin. Invest 2011, 121, 468–474. [Google Scholar]
- Bohle, A.; Wehrmann, M.; Bogenschutz, O.; Batz, C.; Vogl, W.; Schmitt, H.; Muller, C.A.; Muller, G.A. The long-term prognosis of the primary glomerulonephritides. A morphological and clinical analysis of 1747 cases. Pathol. Res. Pract 1992, 188, 908–924. [Google Scholar]
- Famulski, K.S.; Reeve, J.; de Freitas, D.G.; Kreepala, C.; Chang, J.; Halloran, P.F. Kidney transplants with progressing chronic diseases express high levels of acute kidney injury transcripts. Am. J. Transplant 2013, 13, 634–644. [Google Scholar]
- Li, Y.; Liu, Z.; Guo, X.; Shu, J.; Chen, Z.; Li, L. Aristolochic acid I-induced DNA damage and cell cycle arrest in renal tubular epithelial cells in vitro. Arch. Toxicol 2006, 80, 524–532. [Google Scholar]
- Debelle, F.D.; Vanherweghem, J.L.; Nortier, J.L. Aristolochic acid nephropathy: A worldwide problem. Kidney Int 2008, 74, 158–169. [Google Scholar]
- Ninichuk, V.; Anders, H.J. Bone marrow-derived progenitor cells and renal fibrosis. Front. Biosci 2008, 13, 5163–5173. [Google Scholar]
- Vielhauer, V.; Anders, H.J.; Mack, M.; Cihak, J.; Strutz, F.; Stangassinger, M.; Luckow, B.; Grone, H.J.; Schlondorff, D. Obstructive nephropathy in the mouse: Progressive fibrosis correlates with tubulointerstitial chemokine expression and accumulation of CC chemokine receptor 2- and 5-positive leukocytes. J. Am. Soc. Nephrol 2001, 12, 1173–1187. [Google Scholar]
- Anders, H.J.; Vielhauer, V.; Kretzler, M.; Cohen, C.D.; Segerer, S.; Luckow, B.; Weller, L.; Grone, H.J.; Schlondorff, D. Chemokine and chemokine receptor expression during initiation and resolution of immune complex glomerulonephritis. J. Am. Soc. Nephrol 2001, 12, 919–931. [Google Scholar]
- Mayer, V.; Hudkins, K.L.; Heller, F.; Schmid, H.; Kretzler, M.; Brandt, U.; Anders, H.J.; Regele, H.; Nelson, P.J.; Alpers, C.E.; et al. Expression of the chemokine receptor CCR1 in human renal allografts. Nephrol. Dial. Transplant 2007, 22, 1720–1729. [Google Scholar]
- Vielhauer, V.; Anders, H.J. Chemokines and chemokine receptors as therapeutic targets in chronic kidney disease. Front. Biosci. (Schol Ed. ) 2009, 1, 1–12. [Google Scholar]
- Jedlicka, J.; Soleiman, A.; Draganovici, D.; Mandelbaum, J.; Ziegler, U.; Regele, H.; Wuthrich, R.P.; Gross, O.; Anders, H.J.; Segerer, S. Interstitial inflammation in Alport syndrome. Hum. Pathol 2010, 41, 582–593. [Google Scholar]
- Anders, H.J.; Ninichuk, V.; Schlondorff, D. Progression of kidney disease: Blocking leukocyte recruitment with chemokine receptor CCR1 antagonists. Kidney Int 2006, 69, 29–32. [Google Scholar]
- Eis, V.; Vielhauer, V.; Anders, H.J. Targeting the chemokine network in renal inflammation. Arch. Immunol. Ther. Exp. (Warsz) 2004, 52, 164–172. [Google Scholar]
- Eis, V.; Luckow, B.; Vielhauer, V.; Siveke, J.T.; Linde, Y.; Segerer, S.; Perez De Lema, G.; Cohen, C.D.; Kretzler, M.; Mack, M.; et al. Chemokine receptor CCR1 but not CCR5 mediates leukocyte recruitment and subsequent renal fibrosis after unilateral ureteral obstruction. J. Am. Soc. Nephrol 2004, 15, 337–347. [Google Scholar]
- Anders, H.J.; Vielhauer, V.; Frink, M.; Linde, Y.; Cohen, C.D.; Blattner, S.M.; Kretzler, M.; Strutz, F.; Mack, M.; Grone, H.J.; et al. A chemokine receptor CCR-1 antagonist reduces renal fibrosis after unilateral ureter ligation. J. Clin. Invest 2002, 109, 251–259. [Google Scholar]
- Anders, H.J.; Belemezova, E.; Eis, V.; Segerer, S.; Vielhauer, V.; Perez de Lema, G.; Kretzler, M.; Cohen, C.D.; Frink, M.; Horuk, R.; et al. Late onset of treatment with a chemokine receptor CCR1 antagonist prevents progression of lupus nephritis in MRL-Fas(lpr) mice. J. Am. Soc. Nephrol 2004, 15, 1504–1513. [Google Scholar]
- Vielhauer, V.; Berning, E.; Eis, V.; Kretzler, M.; Segerer, S.; Strutz, F.; Horuk, R.; Grone, H.J.; Schlondorff, D.; Anders, H.J. CCR1 blockade reduces interstitial inflammation and fibrosis in mice with glomerulosclerosis and nephrotic syndrome. Kidney Int 2004, 66, 2264–2278. [Google Scholar]
- Ninichuk, V.; Gross, O.; Reichel, C.; Khandoga, A.; Pawar, R.D.; Ciubar, R.; Segerer, S.; Belemezova, E.; Radomska, E.; Luckow, B.; et al. Delayed chemokine receptor 1 blockade prolongs survival in collagen 4A3-deficient mice with Alport disease. J. Am. Soc. Nephrol 2005, 16, 977–985. [Google Scholar]
- Ninichuk, V.; Khandoga, A.G.; Segerer, S.; Loetscher, P.; Schlapbach, A.; Revesz, L.; Feifel, R.; Khandoga, A.; Krombach, F.; Nelson, P.J.; et al. The role of interstitial macrophages in nephropathy of type 2 diabetic db/db mice. Am. J. Pathol 2007, 170, 1267–1276. [Google Scholar]
- Sakai, N.; Furuichi, K.; Shinozaki, Y.; Yamauchi, H.; Toyama, T.; Kitajima, S.; Okumura, T.; Kokubo, S.; Kobayashi, M.; Takasawa, K.; et al. Fibrocytes are involved in the pathogenesis of human chronic kidney disease. Hum. Pathol 2010, 41, 672–678. [Google Scholar]
- Wada, T.; Sakai, N.; Matsushima, K.; Kaneko, S. Fibrocytes: A new insight into kidney fibrosis. Kidney Int 2007, 72, 269–273. [Google Scholar]
- Reich, B.; Schmidbauer, K.; Rodriguez Gomez, M.; Johannes Hermann, F.; Gobel, N.; Bruhl, H.; Ketelsen, I.; Talke, Y.; Mack, M. Fibrocytes develop outside the kidney but contribute to renal fibrosis in a mouse model. Kidney Int. 2013. [Google Scholar] [CrossRef]
- Sakai, N.; Wada, T.; Yokoyama, H.; Lipp, M.; Ueha, S.; Matsushima, K.; Kaneko, S. Secondary lymphoid tissue chemokine (SLC/CCL21)/CCR7 signaling regulates fibrocytes in renal fibrosis. Proc. Natl. Acad. Sci. USA 2006, 103, 14098–14103. [Google Scholar]
- Duffield, J.S.; Lupher, M.; Thannickal, V.J.; Wynn, T.A. Host responses in tissue repair and fibrosis. Annu. Rev. Pathol 2013, 8, 241–276. [Google Scholar]
- Schrimpf, C.; Xin, C.; Campanholle, G.; Gill, S.E.; Stallcup, W.; Lin, S.L.; Davis, G.E.; Gharib, S.A.; Humphreys, B.D.; Duffield, J.S. Pericyte TIMP3 and ADAMTS1 modulate vascular stability after kidney injury. J. Am. Soc. Nephrol 2012, 23, 868–883. [Google Scholar]
- Fligny, C.; Duffield, J.S. Activation of pericytes: Recent insights into kidney fibrosis and microvascular rarefaction. Curr. Opin. Rheumatol 2013, 25, 78–86. [Google Scholar]

© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hagemann, J.H.; Haegele, H.; Müller, S.; Anders, H.-J. Danger Control Programs Cause Tissue Injury and Remodeling. Int. J. Mol. Sci. 2013, 14, 11319-11346. https://doi.org/10.3390/ijms140611319
Hagemann JH, Haegele H, Müller S, Anders H-J. Danger Control Programs Cause Tissue Injury and Remodeling. International Journal of Molecular Sciences. 2013; 14(6):11319-11346. https://doi.org/10.3390/ijms140611319
Chicago/Turabian StyleHagemann, Jan H., Holger Haegele, Susanna Müller, and Hans-Joachim Anders. 2013. "Danger Control Programs Cause Tissue Injury and Remodeling" International Journal of Molecular Sciences 14, no. 6: 11319-11346. https://doi.org/10.3390/ijms140611319