The Uneven Rate of the Molecular Evolution of Gene Sequences of DNA-Dependent RNA Polymerase I of the Genus Lamium L.


Abstract
:1. Introduction
2. Results and Discussion
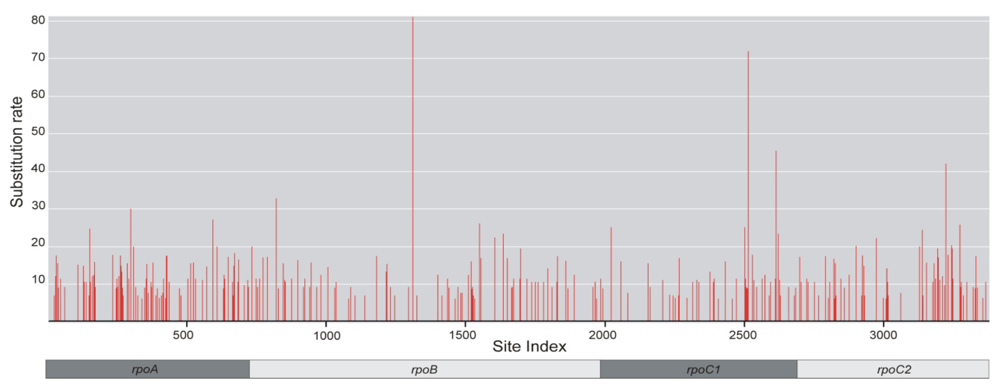
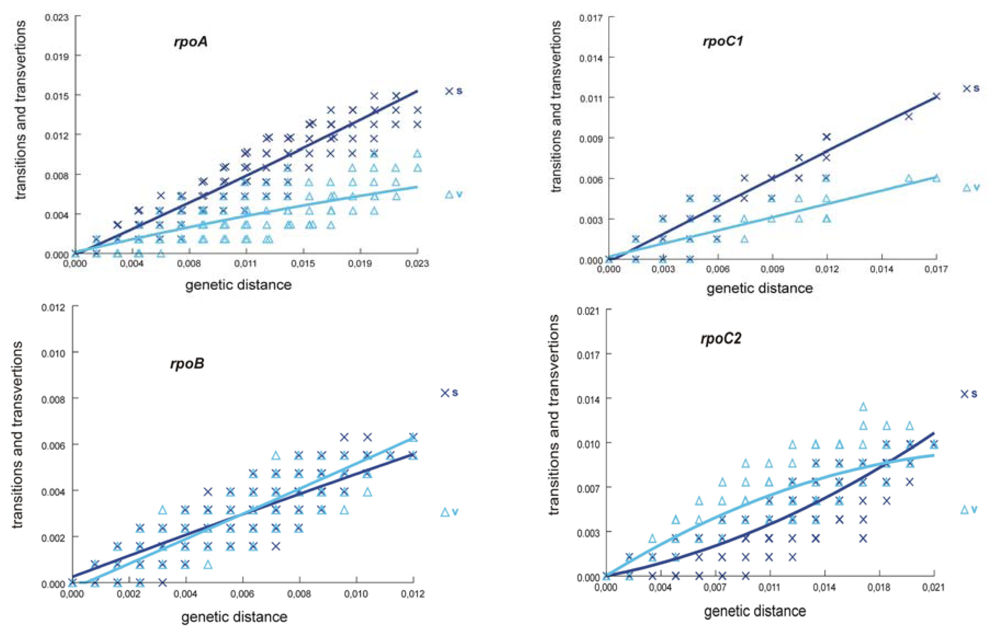
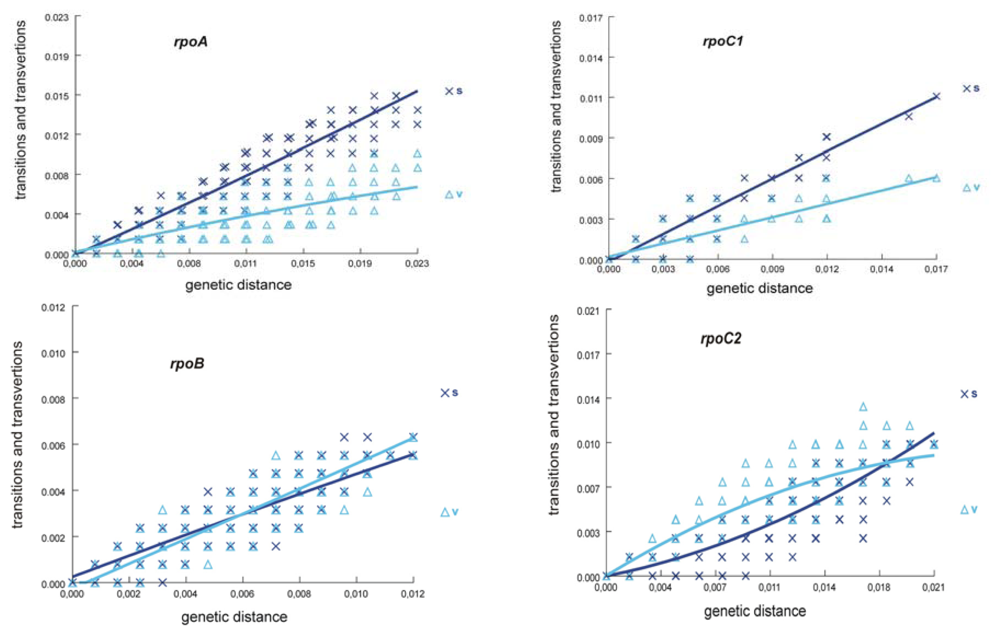
2.1. Sequence Characteristics
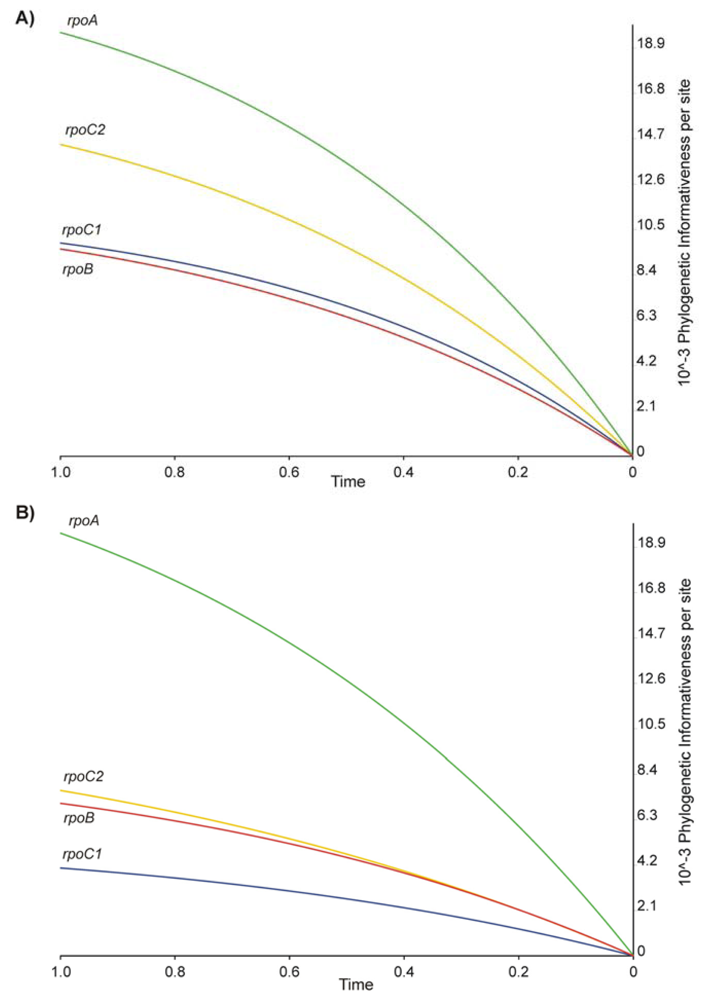
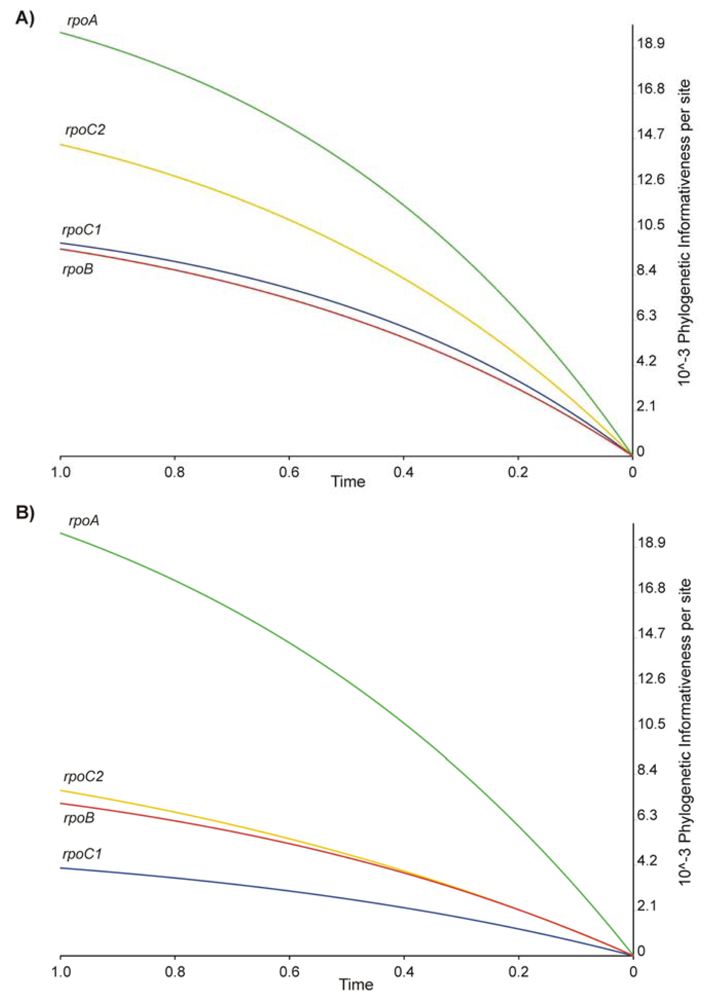
2.2. Phylogenetic Signal and Informativeness
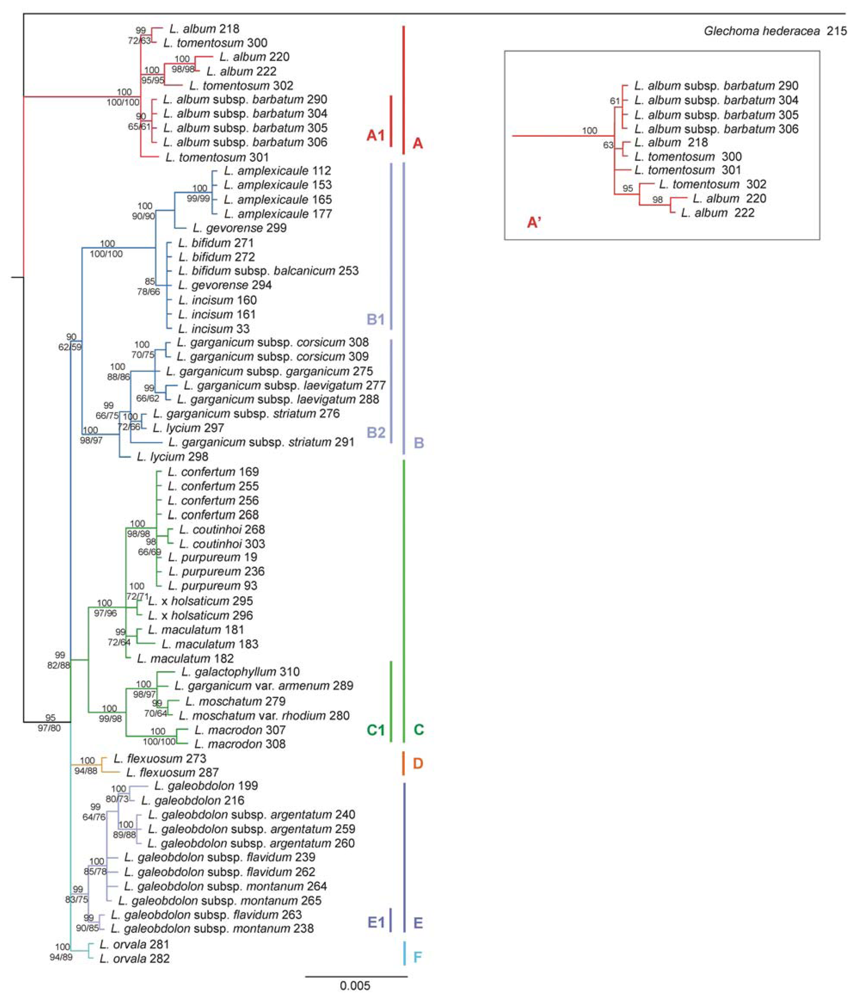
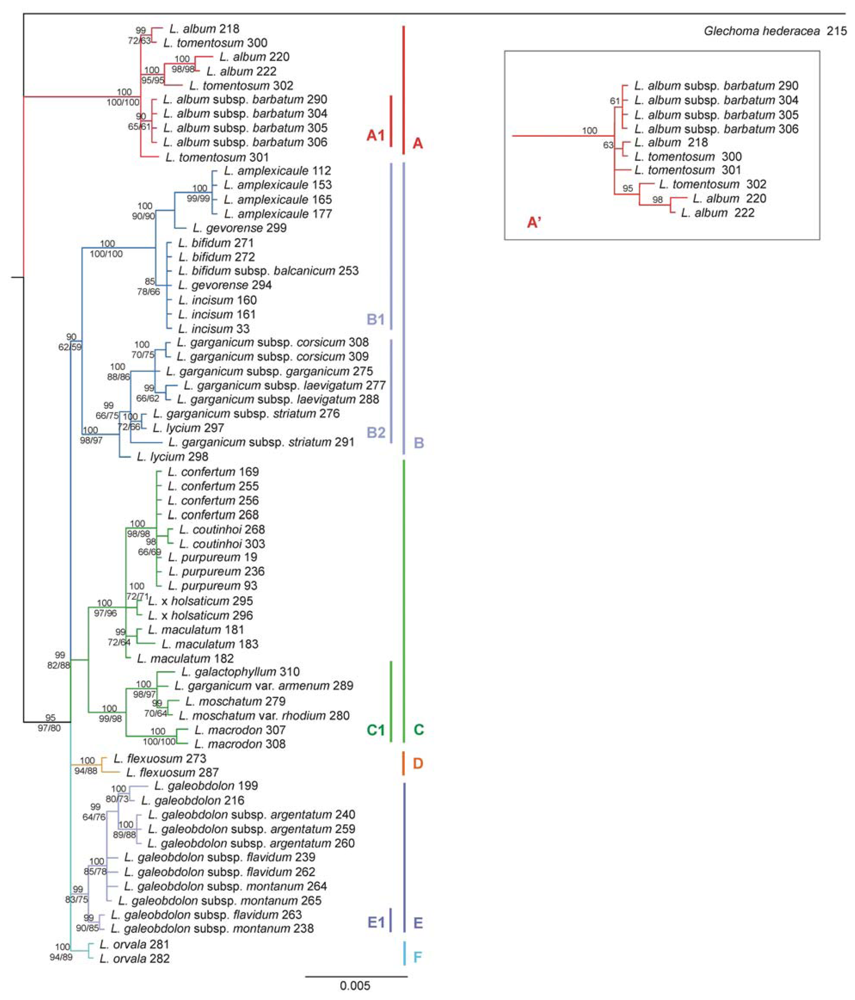
2.3. Phylogenetic Tree
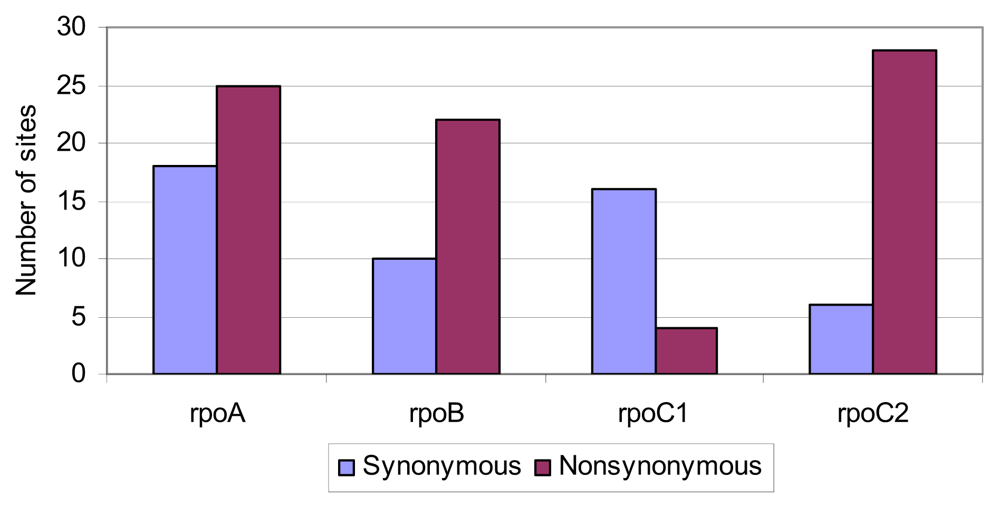
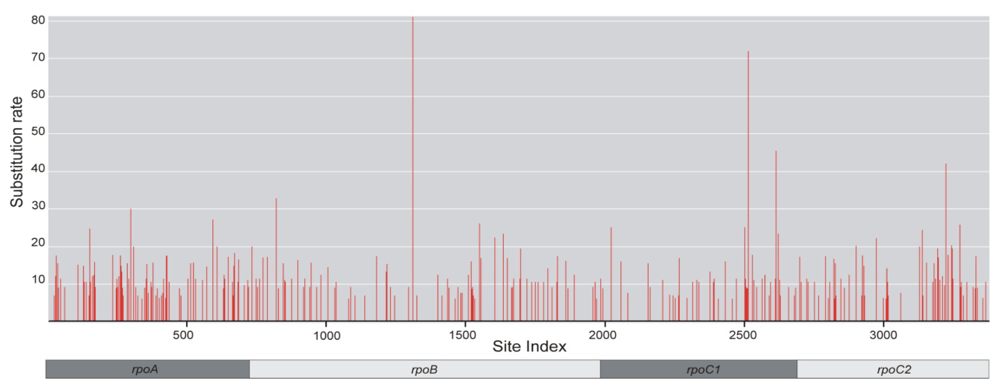
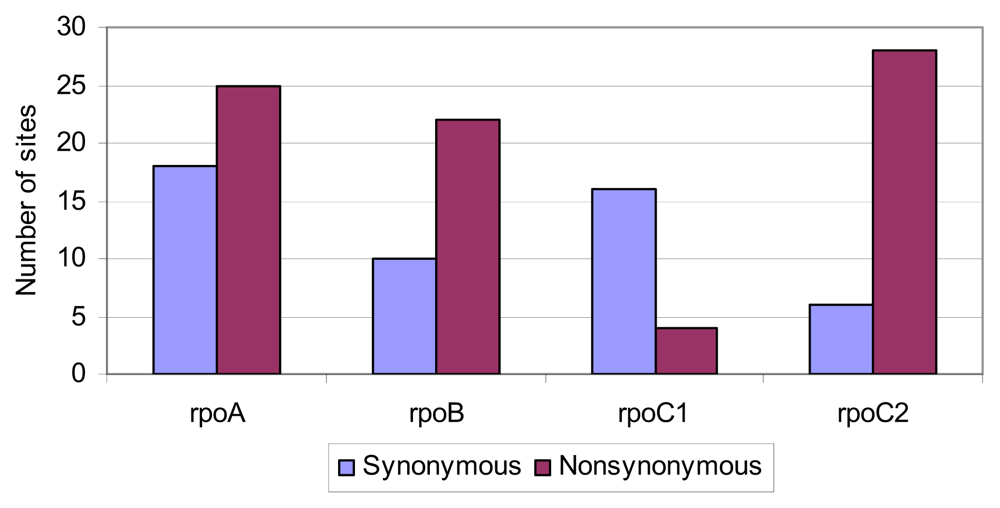
2.4. Positive Selective Pressure
3. Experimental Section
3.1. Plant Material
3.2. Experimental Procedures and Sequence Alignment
3.3. Patterns of Substitutions and Phylogenetic Informativeness
3.4. Phylogenetic Analysis
3.5. Detection of Positive Selection
4. Conclusions
Acknowledgements
Abbreviations
| PEP | Plastid-encoded plastid RNA polymerase |
| NEP | Nucleus-encoded plastid RNA polymerase |
| PI | Phylogenetic informativeness |
| Pi | Parsimony informative |
| MP | Maximum Parsimony |
| ML | Maximum likelihood |
| BI | Bayesian inference |
Conflict of Interest
References
- Little, M.C.; Hallick, R.B. Chloroplast rpoA, rpoB, and rpoC genes specify at least three components of a chloroplast DNA-dependent RNA polymerase active in tRNA and mRNA transcription. J. Biol. Chem 1988, 263, 14302–14307. [Google Scholar]
- Gabor, L.I.; Meinke, A.; Döry, I.; Kössel, H. Nucleotide sequence of the maize chloroplast rpoB/CI/C2 operon: Comparison between the derived protein primary structures from various organisms with respect to functional domains. Mol. Gen. Genet 1990, 221, 379–394. [Google Scholar]
- Serino, G.; Maliga, P. RNA polymerase subunits encoded by the plastid rpo genes are not shared with the nucleus-encoded plastid enzyme. Plant Physiol 1998, 117, 1165–1170. [Google Scholar]
- Allison, L.A.; Simon, L.D.; Maliga, P. Deletion of rpoB reveals a second distinct transcription system in plastids of higher plants. EMBO J 1996, 15, 2802–2809. [Google Scholar]
- Hajdukiewicz, P.T.J.; Allison, L.A.; Maliga, P. The two RNA polymerases encoded by the nuclear and the plastid compartments transcribe distinct groups of genes in tobacco plastids. EMBO J 1997, 16, 4041–4048. [Google Scholar]
- Krause, K.; Falk, J.; Humbeck, K.; Krupinska, K. Responses of the transcriptional apparatus of barley chloroplasts to a prolonged dark period and to subsequent reillumination. Physiol. Plant 1998, 104, 143–152. [Google Scholar]
- Logacheva, M.D.; Penin, A.A.; Samigullin, T.H.; Vallejo-Roman, C.M.; Antonov, A.S. Phylogeny of flowering plants by the chloroplast genome sequences: In search of a “Lucky Gene”. Biochemistry 2007, 72, 1324–1330. [Google Scholar]
- Korczak, B.; Christensen, H.; Emler, S.; Frey, J.; Kuhnert, P. Phylogeny of the family Pasteurellaceae based on rpoB sequences. Int. J. Syst. Evol. Microbiol 2004, 54, 1393–1399. [Google Scholar]
- Chelo, I.M.; Zé-Zé, L.; Tenreiro, R. Congruence of evolutionary relationships inside the Leuconostoc–Oenococcus–Weissella clade assessed by phylogenetic analysis of the 16S rRNA gene, dnaA, gyrB, rpoC and dnaK. Int. J. Syst. Evol. Microbiol 2007, 57, 276–286. [Google Scholar]
- Petersen, G.; Seberg, O. Phylogenetic analysis of the Triticaceae (Poaceae) based on rpoA sequence data. Mol. Phyl. Evol 1997, 7, 217–230. [Google Scholar]
- Liu, Y.; Yan, H.F.; Cao, T.; Ge, X.J. Evaluation of 10 plant barcodes in Bryophyta (Mosses). J. Syst. Evol 2010, 48, 36–46. [Google Scholar]
- Newmaster, S.G.; Fazekas, A.J.; Steeves, R.A.D.; Janovec, J. Testing candidate plant barcode regions in the Myristicaceae. Mol. Ecol. Res 2008, 8, 480–490. [Google Scholar]
- Chase, M.W.; Cowan, R.S.; Hollingsworth, P.M.; van den Berg, C.; Madrińán, S.; Petersen, G.; Seberg, O.; Jřrgsensen, T.; Cameron, K.M.; Carine, M.; et al. A proposal for a standardised protocol to barcode all land plants. Taxon 2007, 56, 295–299. [Google Scholar]
- Jeffroy, O.; Brinkmann, H.; Delsuc, F.; Philippe, H. Phylogenomics: The beginning of incongruence? Trends. Genet 2006, 22, 225–231. [Google Scholar]
- Bendiksby, M.; Brysting, A.K.; Thorbek, L.; Gussarova, G.; Ryding, O. Molecular phylogeny and taxonomy of the genus Lamium L. (Lamiaceae): Disentangling origins of presumed allotetraploids. Taxon 2011, 60, 986–1000. [Google Scholar]
- Nei, M.; Kumar, S. Molecular Evolution and Phylogenetics; Oxford University Press: New York, NY, USA, 2000. [Google Scholar]
- Yang, Z. Among site rate variation and its impact on phylogenetic analyses. Trends. Ecol. Evol 1996, 11, 367–372. [Google Scholar]
- Lió, P.; Goldman, N. Models of molecular evolution and phylogeny. Genome Res 1998, 8, 1233–1244. [Google Scholar]
- Christelová, P.; Valárik, M.; Hřibová, E.; de Langhe, E.; Doležel, J. A multi gene sequence-based phylogeny of the Musaceae (banana) family. BMC Evol. Biol. 2011. [Google Scholar] [CrossRef]
- Townsend, J.P. Profiling phylogenetic informativeness. Syst. Biol 2007, 56, 222–231. [Google Scholar]
- Townsend, J.P.; Leuenberger, C. Taxon sampling and the optimal rates of evolution for phylogenetic inference. Syst. Biol 2011, 60, 358–365. [Google Scholar]
- Townsend, J.P.; López-Giráldez, F.; Friedman, R. The phylogenetic informativeness of nucleotide and amino acid sequences for reconstructing the vertebrate tree. J. Mol. Syst. Evol. 2008, 67, 437–447. [Google Scholar]
- Pamilo, P.; Nei, M. Relationships between gene trees and species trees. Mol. Biol. Evol 1988, 5, 568–583. [Google Scholar]
- Maddison, W.P.; Knowles, L.L. Inferring phylogeny despite incomplete lineage sorting. Syst. Biol 2006, 55, 21–30. [Google Scholar]
- Jörgensen, C.A. Cytological and experimental studies on the genus Lamium. Hereditas 1927, 9, 126–136. [Google Scholar]
- Jones, S.B., Jr; Jones, C.A. Status of Lamium hybridum Vill. (Labiatae). Am. Midl. Nat. 1965, 74, 503–505. [Google Scholar]
- Bernström, P. Cytogenetic studies on relationships between annual species of Lamium. Hereditas 1955, 41, 1–122. [Google Scholar]
- Taylor, R. The origin of Lamium hybridum, a case study in the search for the parents of hybrid species. Northwest Sci 1991, 65, 116–124. [Google Scholar]
- Dvorakova, M. Lamium L.—hluchavka. In KvětenaČeskěRepubliky; Slavík, B., Ed.; Academia: Praha, Czech Republic, 2000; Volume 6, pp. 596–603. [Google Scholar]
- Govaerts, R.; Paton, A.; Harvey, Y.; Navarro, T. World checklist of Lamiaceae and Verbenaceae. 2010. Available online: http://www.kew.org/wcsp/lamiaceae (accessed on 15 November 2012).
- Harley, R.M.; Atkins, S.; Budantsev, A.L.; Cantino, P.D.; Conn, B.J.; Grayer, R.; Harley, M.M.; de Kok, R.; Krestovskaya, T.; Morales, R.; et al. The Families and Genera of Vascular Plants; Springer: Heidelberg, Germany, 2004. [Google Scholar]
- Botanical Garden in Kew website. Available online: http://www.kew.org/barcoding/protocols/ (accessed on 24 March 2012).
- Benson, D.A.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Wheeler, D.L. GenBank. Nucleic Acids Res 2005, 40, 34–38. [Google Scholar]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl. Acids. Symp. Ser 1999, 41, 95–98. [Google Scholar]
- Pond, S.L.K.; Muse, S.V. HyPhy: Hypothesis testing using phylogenies. Bioinformatics 2005, 21, 676–679. [Google Scholar]
- Xia, X.; Xie, Z. DAMBE: Software package for data analysis in molecular biology and evolution. J. Hered 2001, 92, 371–373. [Google Scholar]
- Lopez-Giraldez, F.; Townsend, J.P. PhyDesign: an online application for profiling phylogenetic informativeness. BMC Evol. Biol. 2011. [Google Scholar] [CrossRef]
- Britton, T.; Anderson, C.L.; Jacquet, D.; Lundqvist, S.; Bremer, K. Estimating divergence times in large phylogenetic trees. Syst. Biol 2007, 56, 741–752. [Google Scholar]
- Mayrose, I.; Graur, D.; Ben-Tal, N.; Pupko, T. Comparison of site-specific rate-inference methods for protein sequences: Empirical Bayesian methods are superior. Mol. Biol. Evol 2004, 21, 1781–1791. [Google Scholar]
- Hulsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar]
- Posada, D. jModelTest: Phylogenetic Model Averaging. Mol. Biol. Evol 2008, 25, 1253–1256. [Google Scholar]
- Fig Tree, version 1.4.0. Software for molecular evolution, phylogenetics and epidemiology. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 05 December 2012).
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular Evolutionary Genetics Analysis using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Mol. Biol. Evol. 2011. [Google Scholar] [CrossRef]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar]
- Woolley, S.; Johnson, J.; Smith, M.J.; Crandall, K.A.; McClellan, D.A. TreeSAAP: Selection on amino acid properties using phylogenetic trees. Bioinformatics 2003, 19, 671–672. [Google Scholar]
- McClellan, D.A.; McCracken, K.G. Estimating the influence of selection on the variable amino acid sites of the cytochrome b protein functional domains. Mol. Biol. Evol 2001, 18, 917–925. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| rpoA | rpoB | rpoC1 | rpoC2 | |
|---|---|---|---|---|
| Alignment length | 728 | 1269 | 701 | 676 |
| Sequence length | 713–728 | 1269 | 701 | 676 |
| Site rates | 1.423 | 0.773 | 0.825 | 1.152 |
| Variable characters (V) | 43 (5.91%) | 32 (2.52%) | 20 (2.85%) | 34 (5.03%) |
| Parsimony-informative sites (Pi) | 35 (4.81%) | 25 (1.97%) | 18 (2.57%) | 27 (3.99%) |
| Purines % | 53.375 | 53.381 | 53.458 | 50.727 |
| Pyrimidynes % | 46.625 | 46.619 | 46.542 | 49.273 |
| GC % | 37.564 | 36.311 | 41.821 | 43.221 |
| Transition/Transversion bias (R) | 1.84 | 1.33 | 1.73 | 0.48 |
| Gene | Codon | Branch | Amino acid change | Property | Category of change | Statistical error |
|---|---|---|---|---|---|---|
| rpoA | 79 | clade C | C into S | Ns | 7 | 0.010 |
| 128 | clade B2 | R into I | Ns | 6 | 0.050 | |
| 138 | clade E | H into Y | am | 7 | 0.001 | |
| 155 | clade B2 | R into Q | pHi | 7 | 0.050 | |
| 84 | L. tomentosum 302 | C into R | C | 7 | 0.001 | |
| 84 | L. tomentosum 302 | C into R | Ns | 7 | 0.010 | |
| 110 | L. tomentosum 301 | H into Y | am | 7 | 0.001 | |
| rpoB | 90 | clade B2 | I into L | pK′ | 8 | 0.010 |
| 160 | clade A | S into L | αn | 6 | 0.010 | |
| 192 | clade F | I into M | pK′ | 8 | 0.010 | |
| 192 | cladeA1 | I into M | pK′ | 8 | 0.010 | |
| 192 | L. maculatum 183 | I into M | pK′ | 8 | 0.010 | |
| rpoC1 | 146 | clade E1 | T into A | Pα | 6 | 0.050 |
| 207 | L. garganicum subsp. laevigatum 277 | I into V | pK′ | 8 | 0.050 | |
| 207 | L. garganicum subsp. striatum 291 | I into V | pK′ | 8 | 0.050 | |
| rpoC2 | 77 | clade A | A into T | Pα | 6 | 0.050 |
| 43 | L. gevorense 299 | A into V | Pα | 6 | 0.050 | |
| Region | Forward primer’s sequence (5′-3′) | Reverse primer’s sequence (5′-3′) |
|---|---|---|
| rpoA | CTACTCGGACACTACAGTGG | GGGAGACAATTCGGATTGATC |
| rpoB-1 | CCGTCTCTACCCAAGATCCC | GTCGACCAATCCCTTCCTAA |
| rpoB-2 | GCGGGGATCCGGTATTTTC | GGCTTTCTAGAGATCCCCAA |
| rpoC1 | TATGAAACCAGAATGGATGG | GAAAACATAAGTARRCGWGC |
| rpoC2 | CAAAGCAATTTACGCGAAGG | GCAATCACTTGTTCCGATTC |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Krawczyk, K.; Sawicki, J. The Uneven Rate of the Molecular Evolution of Gene Sequences of DNA-Dependent RNA Polymerase I of the Genus Lamium L. Int. J. Mol. Sci. 2013, 14, 11376-11391. https://doi.org/10.3390/ijms140611376
Krawczyk K, Sawicki J. The Uneven Rate of the Molecular Evolution of Gene Sequences of DNA-Dependent RNA Polymerase I of the Genus Lamium L. International Journal of Molecular Sciences. 2013; 14(6):11376-11391. https://doi.org/10.3390/ijms140611376
Chicago/Turabian StyleKrawczyk, Katarzyna, and Jakub Sawicki. 2013. "The Uneven Rate of the Molecular Evolution of Gene Sequences of DNA-Dependent RNA Polymerase I of the Genus Lamium L." International Journal of Molecular Sciences 14, no. 6: 11376-11391. https://doi.org/10.3390/ijms140611376