Phospholipids at the Interface: Current Trends and Challenges

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
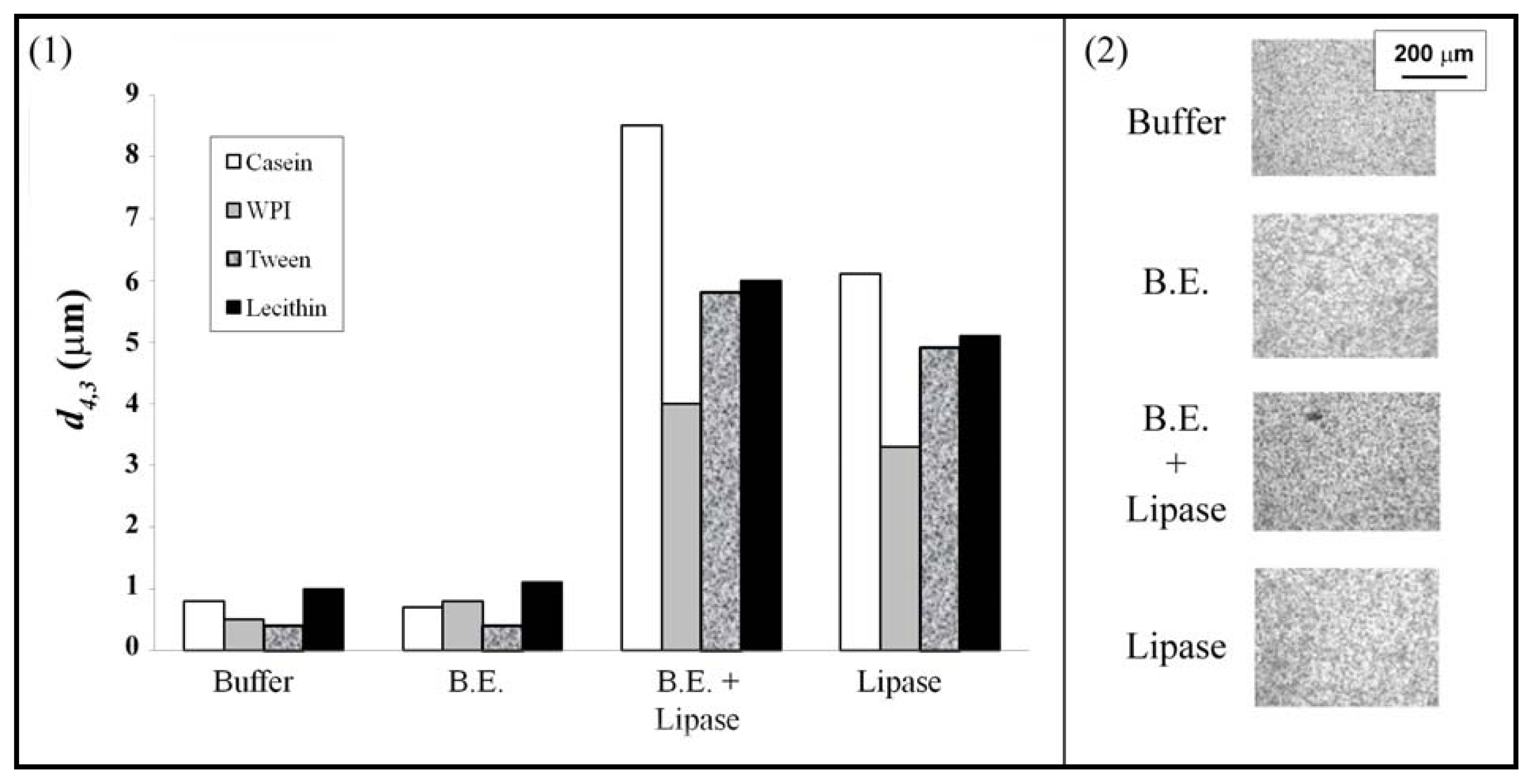{kind=link}
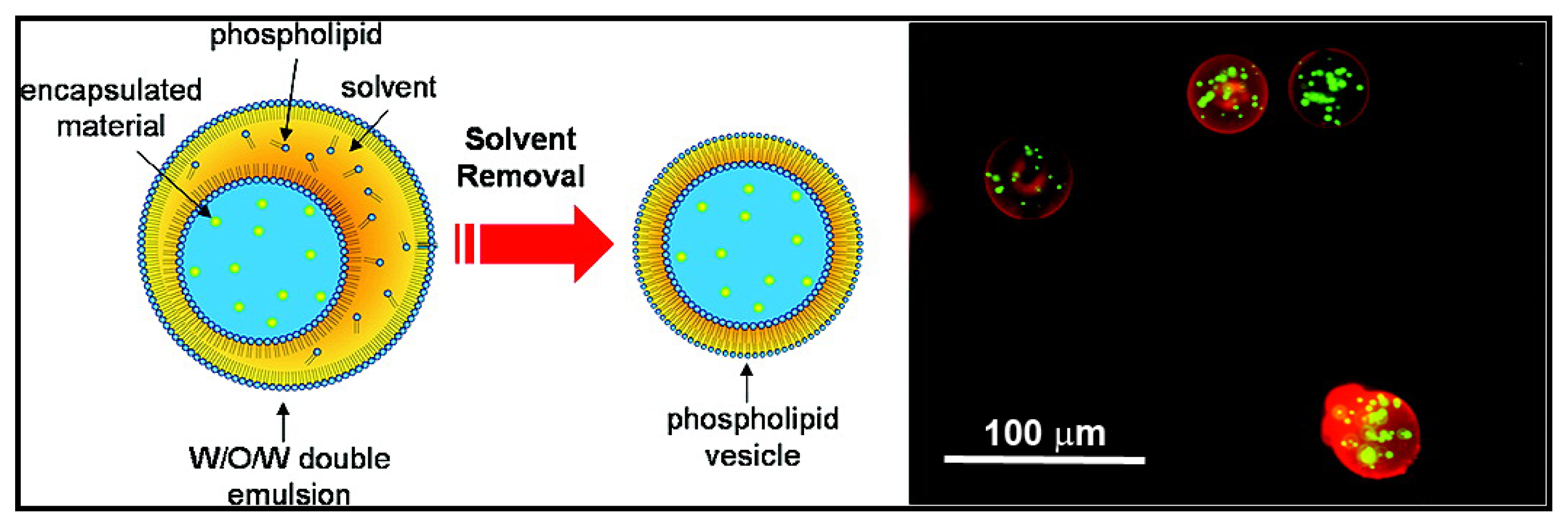{kind=link}
Abstract
:1. Introduction
2. Interfacial Properties of Phospholipids
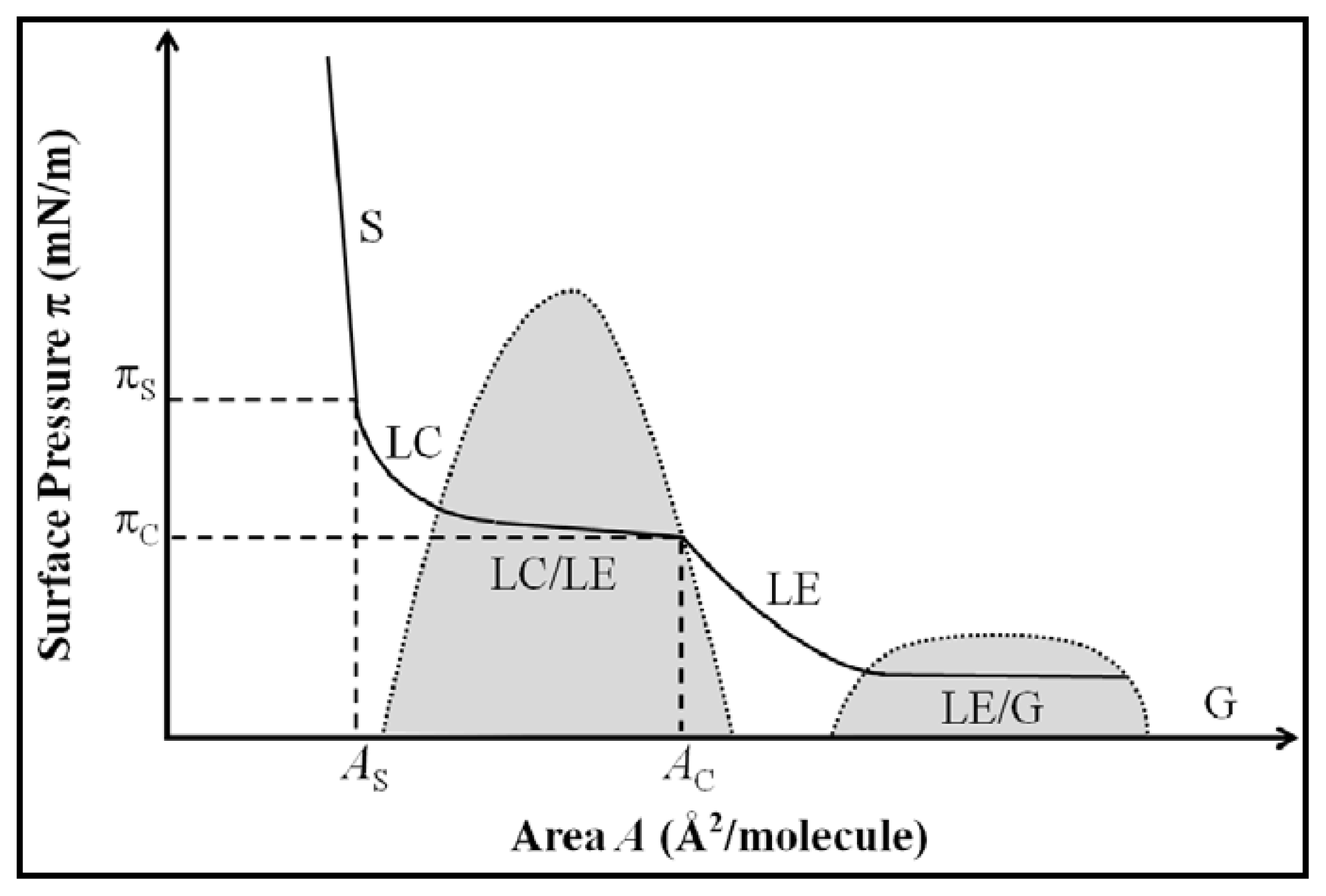
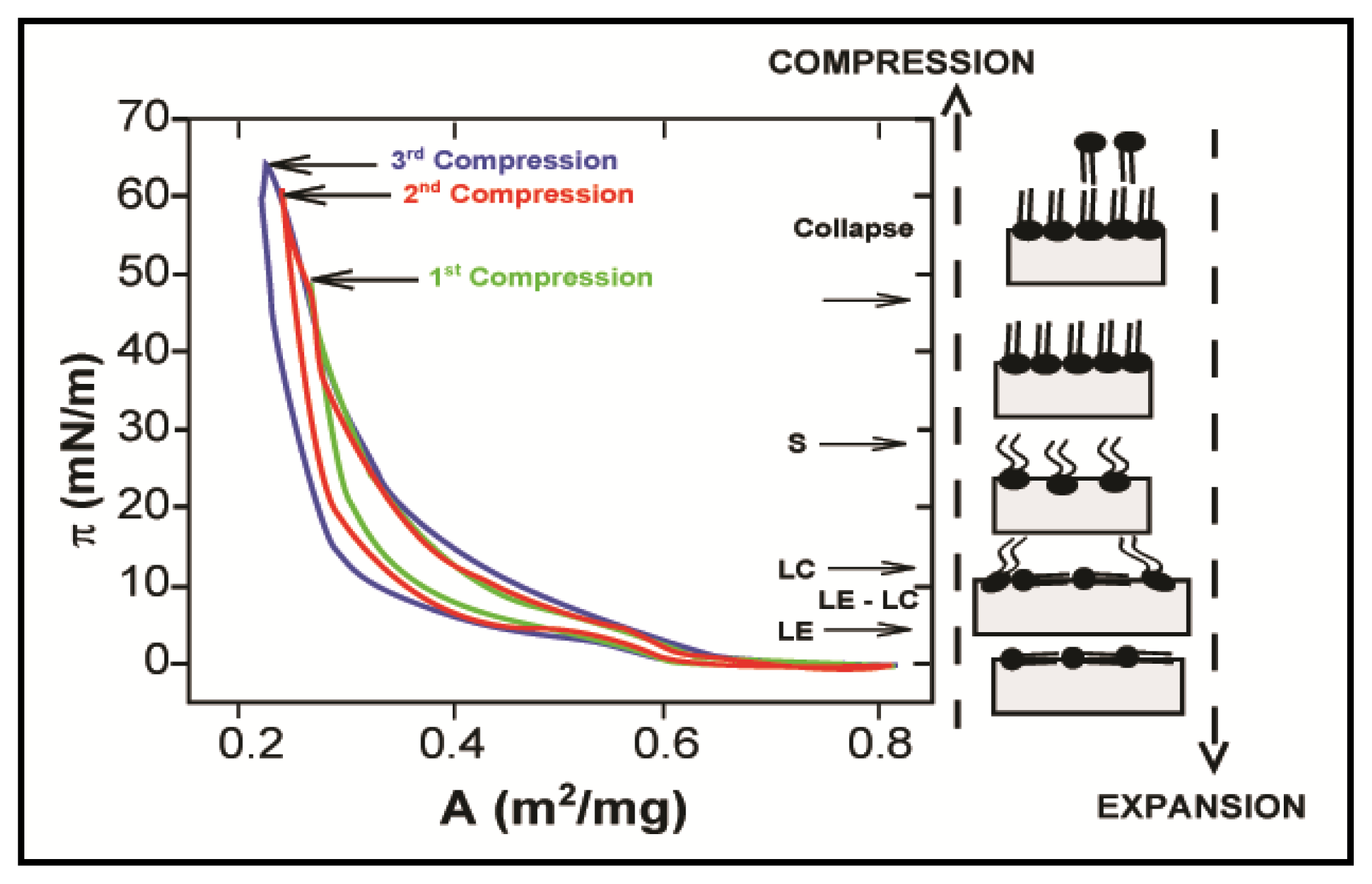
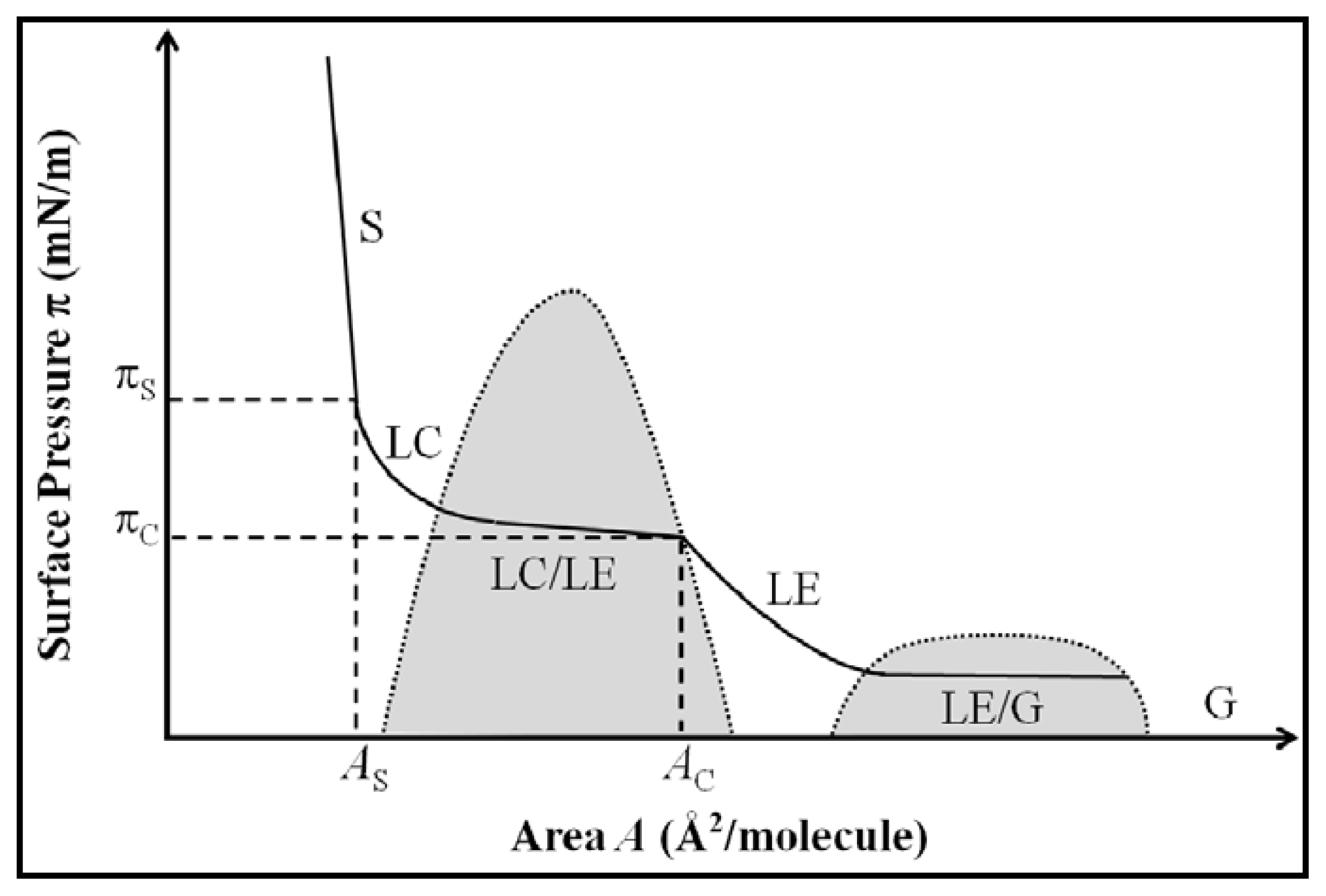
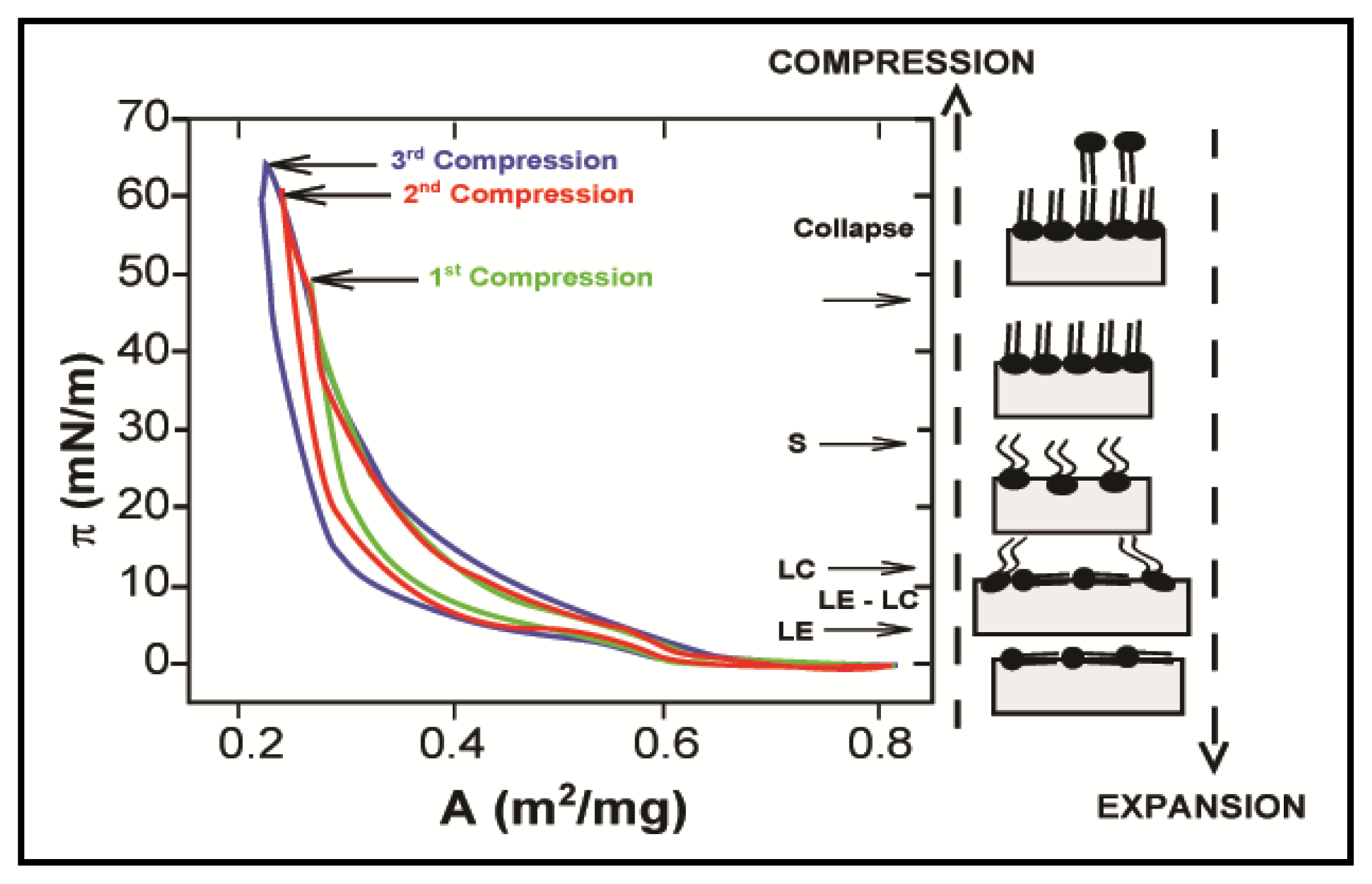
2.1. Properties of Phospholipids at the Air/Water Interface
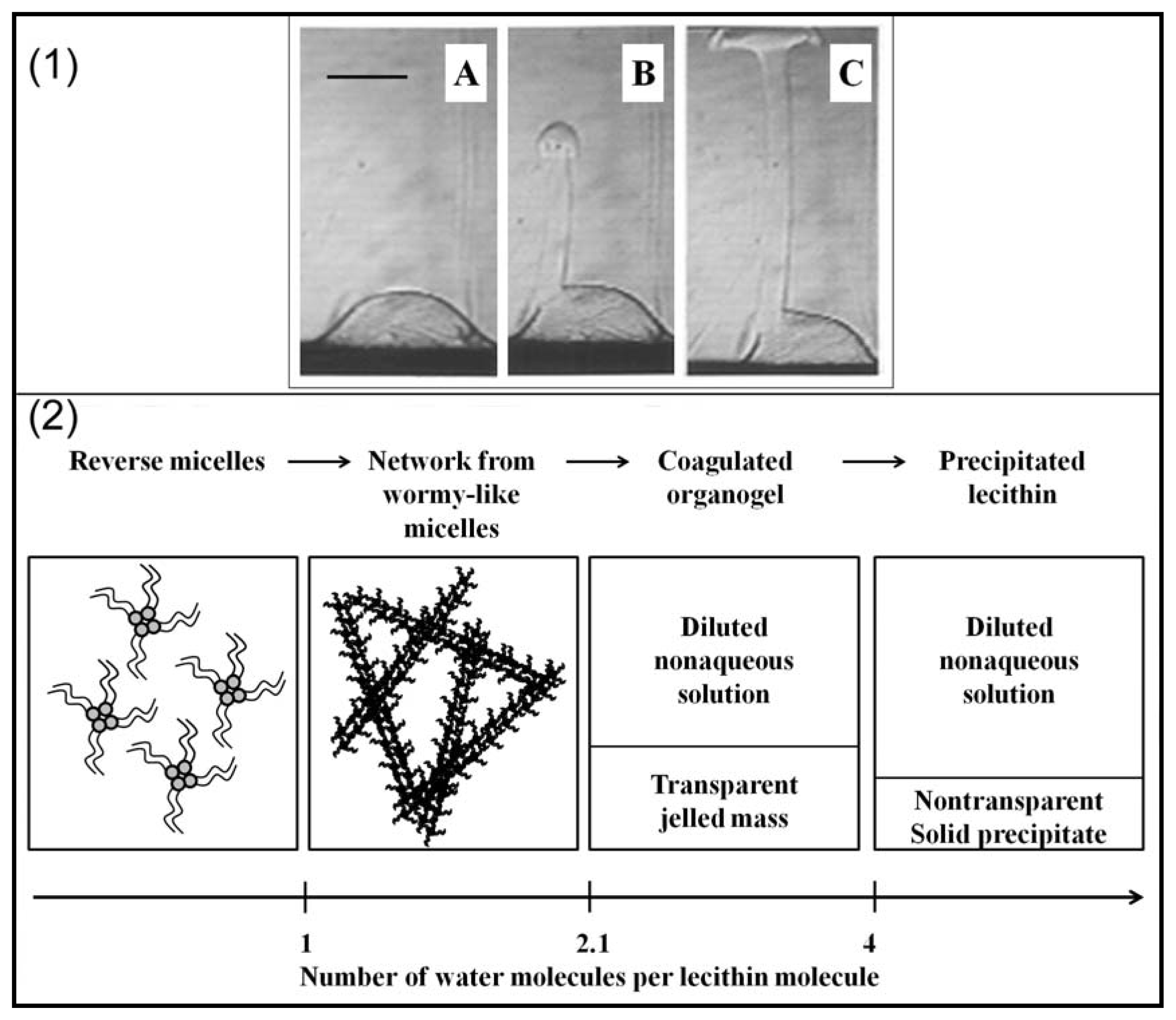
2.2. Properties of Phospholipids at the Oil/Water Interface
3. Applications
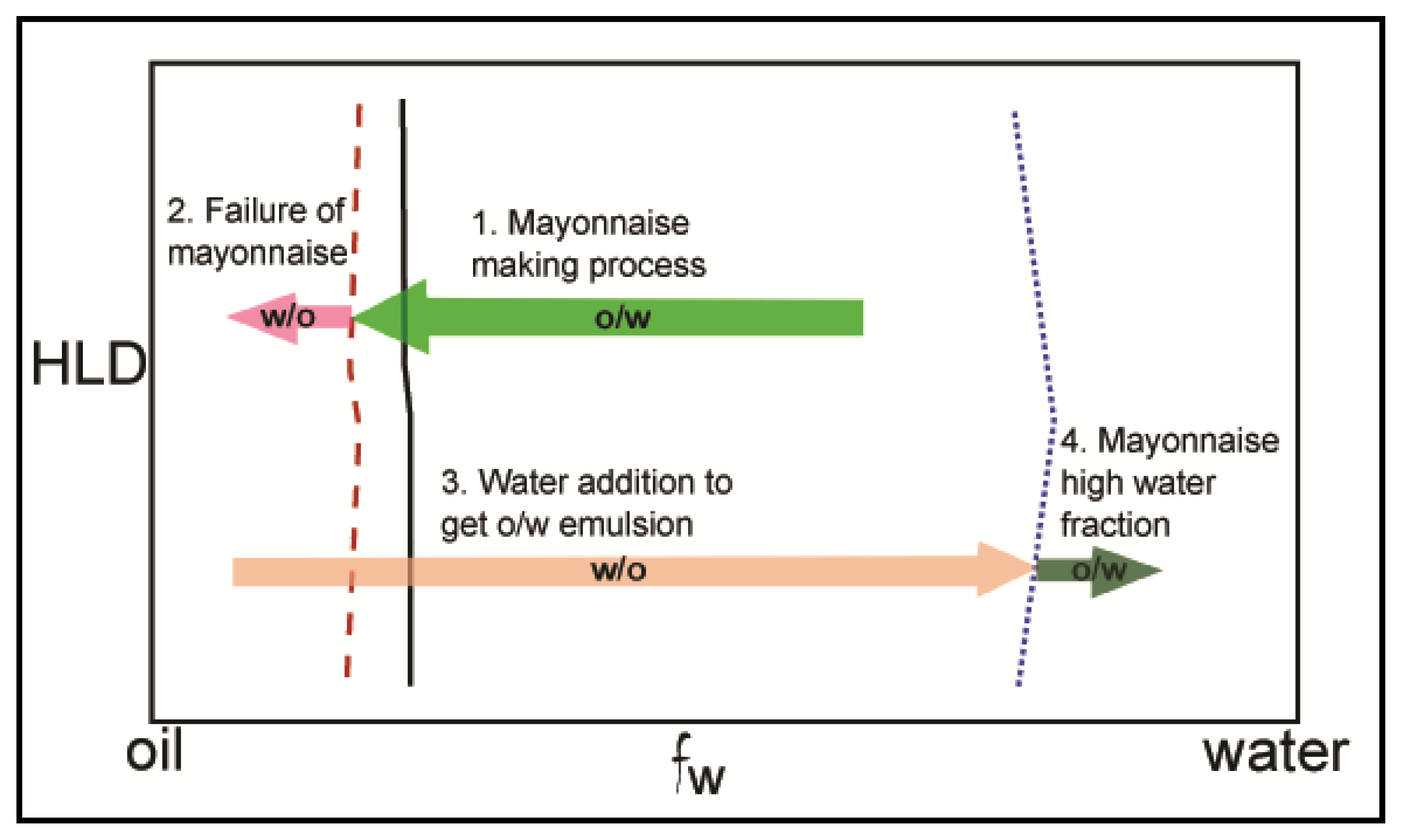
3.1. Emulsions
3.2. Other Applications
4. Conclusion and Challenges
Abbreviations
| α-DML | α-Dimyristoyllecithin |
| α-DPL | α-Dipalmitoyllecithin |
| β-DPL | β-Dipalmitoyllecithin |
| DHPC | Dihexadecylphosphatidylcholine |
| DLPC | l-α-dilauroylphosphatidylcholine |
| DMPA | Dimyristoylphosphatidic acid |
| DMPE | l-α-Dimyristoyl phosphatidylethanolamine |
| DOPC | 1,2-dioleoyl-3-sn-phosphatidylcholine |
| DSPC | distearoylphosphatidylcholine |
| DPPC | Dipalmitoyl phosphatidylcholine |
| LC | Liquid-Condensed phase |
| LE | Liquid-Expanded phase |
| PA | Phosphatidic Acid |
| PC | Phosphatidylcholine |
| PE | Phosphatidylethanolamine |
| PG | Phosphatidylglyerol |
| PI | Phosphatidylinositol |
| PL | Phospholipid |
| PS | Phosphatidylserine |
References
- Cecv, G.; Marsh, D. Phospholipid Bilayers: Physical Principles and Models; John Wiley & Sons, Ltd: Toronto, Canada, 1987. [Google Scholar]
- Cecv, G. Phospholipids Handbook; Marcel Dekker, Inc: New York, NY, USA, 1993. [Google Scholar]
- Verheij, H.M. Organic Chemistry of Phospholipids. In Fundamentals of Lipid Chemistry; Burton, R.M., Guerra, F.C., Eds.; BI-Science Publications: Webster Groves, MO, USA, 1972; pp. 225–249. [Google Scholar]
- Lundberg, B.; Svens, E.; Ekman, S. The hydration of phospholipids and phospholipid-cholesterol complexes. Chem. Phys. Lipids 1978, 22, 285–292. [Google Scholar]
- McIntosh, T.J.; Magid, A.D. Phospholipid Hydration. In Phospholipids Handbook; Cecv, G., Ed.; Marcel Dekker, Inc: New York, NY, USA, 1993; pp. 553–577. [Google Scholar]
- Seddon, J.M.; Cecv, G. Lipid Polymorphism: Structure and Stability of Lyotropic Mesophases of Phospholipids. In Phospholipids Handbook; Cecv, G., Ed.; Marcel Dekker, Inc: New York, NY, USA, 1993; pp. 403–454. [Google Scholar]
- Seddon, J.M. Structure of the inverted hexagonal (HII) phase, and non-lamellar phase transitions of lipids. Biochim. Biophys. Acta 1990, 1031, 1–69. [Google Scholar]
- Taylor, D.J.F.; Thomas, R.K.; Penfold, J. Polymer/surfactant interactions at the air/water interface. Adv. Colloid Interface Sci 2007, 132, 69–110. [Google Scholar]
- Mackie, A.; Wilde, P. The role of interactions in defining the structure of mixed protein-surfactant interfaces. Adv. Colloid Interface Sci 2005, 117, 3–13. [Google Scholar]
- Möhwald, H. Phospholipid Monolayers. In Handbook of Biological Physics—Structure and Dynamics of Membranes; Lipowsky, R., Sackman, E., Eds.; Elsevier: Amsterdam, The Netherlands, 1995; pp. 161–211. [Google Scholar]
- Möhwald, H. Phospholipid and phospholipid-protein monolayers at the air/water interface. Annu. Rev. Phys. Chem 1990, 41, 441–476. [Google Scholar]
- Phillips, M.C.; Hauser, H. Spreading of solid glycerides and phospholipids at the air-water interface. J. Colloid Interface Sci 1974, 49, 31–39. [Google Scholar]
- Albrecht, O.; Gruler, H.; Sackmann, E. Polymorphism of phospholipid monolayers. J. Phys 1978, 39, 301–313. [Google Scholar]
- Albrecht, O.; Gruler, H.; Sackmann, E. Pressure-composition phase diagrams of cholesterol/lecithin, cholesterol/phosphatidic acid, and lecithin/phosphatidic acid mixed monolayers: A Langmuir film balance study. J. Colloid Interface Sci 1981, 79, 319–338. [Google Scholar]
- Miller, A.; Möhwald, H. Diffusion limited growth of crystalline domains in phospholipid monolayers. J. Chem. Phys 1987, 86, 4258–4265. [Google Scholar]
- Pallas, N.R.; Pethica, B.A. Liquid-expanded to liquid-condensed transition in lipid monolayers at the air/water interface. Langmuir 1985, 1, 509–513. [Google Scholar]
- McConnell, H.M. Structures and transitions in lipid monolayers at the air-water interface. Annu. Rev. Phys. Chem 1991, 42, 171–195. [Google Scholar]
- Vogel, V.; Möbius, D. Hydrated polar groups in lipid monolayers: Effective local dipole moments and dielectric properties. Thin Solid Films 1988, 159, 73–81. [Google Scholar]
- Blaudez, D.; Buffeteau, T.; Desbat, B.; Marie Turlet, J. Infrared and Raman spectroscopies of monolayers at the air-water interface. Curr. Opin. Colloid Interface Sci 1999, 4, 265–272. [Google Scholar]
- Mellier, A.; Auge, O.; Crouigneau, P. Molecular interactions at the phospholipid-water interface. Infrared spectrum of dimyristoyl-Lα-lecithin adsorbed on hydrated potassium bromide. Colloids Surf 1983, 7, 325–337. [Google Scholar]
- Lösche, M.; Rabe, J.; Fischer, A.; Rucha, B.U.; Knoll, W.; Möhwald, H. Microscopically observed preparation of Langmuir-Blodgett films. Thin Solid Films 1984, 117, 269–280. [Google Scholar]
- Kirstein, S.; Möhwald, H.; Shimomura, M. Crystalline two-dimensional domains of cyanine dyes at interfaces. Chem. Phys. Lett 1989, 154, 303–308. [Google Scholar]
- Flörsheimer, M.; Möhwald, H. Growth of large liquid crystalline domains of phospholipids at air-water interfaces. Thin Solid Films 1990, 189, 379–387. [Google Scholar]
- Lösche, M.; Duwe, H.P.; Möhwald, H. Quantitative analysis of surface textures in phospholipid monolayer phase transitions. J. Colloid Interface Sci 1988, 126, 432–444. [Google Scholar]
- Makino, M.; Yoshikawa, K. Dynamic properties of a phospholipid thin film at an air/water interface with a periodic change in surface area. Langmuir 1997, 13, 7125–7134. [Google Scholar]
- Rodríguez Niño, R.; Lucero, A.; Rodríguez Patino, J.M. Relaxation phenomena in phospholipid monolayers at the air-water interface. Colloids Surf. A 2008, 320, 260–270. [Google Scholar]
- MacDonald, R.C.; Simon, S.A. Lipid monolayer states and their relationships to bilayers. Proc. Natl. Acad. Sci. USA 1987, 84, 4089–4093. [Google Scholar]
- Blume, A. A comparative study of the phase transitions of phospholipid bilayers and monolayers. Biochim. Biophy. Acta 1979, 557, 32–44. [Google Scholar]
- Pattus, F.; Desnuelle, P.; Verger, R. Spreading of liposomes at the air/water interface. Biochim. Biophy. Acta 1978, 507, 62–70. [Google Scholar]
- Mansour, H.; Wang, D.S.; Chen, C.S.; Zografi, G. Comparison of bilayer and monolayer properties of phospholipid systems containing dipalmitoylphosphatidylglycerol and dipalmitoylphosphatidylinositol. Langmuir 2001, 17, 6622–6632. [Google Scholar]
- Mansour, H.M.; Zografi, G. Relationships between equilibrium spreading pressure and phase equilibria of phospholipid bilayers and monolayers at the air-water interface. Langmuir 2007, 23, 3809–3819. [Google Scholar]
- Evans, R.W. Aggregates of saturated phospholipids at the air-water interface. Chem. Phys. Lipids 1995, 78, 163–175. [Google Scholar]
- Hayashi, M.; Muramatsu, T.; Hara, I.; Seimiya, T. Phase transitions of phospholipids in monolayers and surface viscosity. Chem. Phys. Lipids 1975, 15, 209–215. [Google Scholar]
- Krägel, J.; Kretzschmar, G.; Li, J.B.; Loglio, G.; Miller, R.; Möhwald, H. Surface rheology of monolayers. Thin Solid Films 1996, 284–285, 361–364. [Google Scholar]
- Vrânceanu, M.; Winkler, K.; Nirschl, H.; Leneweit, G. Surface rheology and phase transitions of monolayers of phospholipid/cholesterol mixtures. Biophys. J 2008, 94, 3924–3934. [Google Scholar]
- Anton, M.; Gandemer, G. Effect of pH on interface composition and on quality of oil-in-water emulsions made with hen egg yolk. Colloids Surf. B 1999, 12, 351–358. [Google Scholar]
- Brzozowska, I.; Figaszewski, Z.A. The influence of pH on phosphatidylcholine monolayer at the air/aqueous solution interface. Colloids Surf. B 2003, 27, 303–309. [Google Scholar]
- Martínez-Landeira, P.; López-Fontán, J.L.; Ruso, J.M.; Prieto, G.; Sarmiento, F. Surface behaviour of C5, C6, C7 and C8 lecithins at the aqueous solution/air interface. Colloids Surf. A 2003, 216, 91–96. [Google Scholar]
- Petelska, A.D.; Figaszewski, Z.A. Interfacial tension of phosphatidylcholine-phosphatidylserine system in bilayer lipid membrane. Biophys. Chem 2006, 120, 199–206. [Google Scholar]
- Hayashi, M.; Muramatsu, T.; Hara, I. Surface properties of synthetic phospholipids II. Thermal phase transitions in monolayers. Biochim. Biophy. Acta 1973, 291, 335–343. [Google Scholar]
- Hayashi, M.; Kobayashi, T.; Seimiya, T.; Muramatsu, T.; Hara, I. Ionic properties of phospholipids at the oil/water interface. Chem. Phys. Lipids 1980, 27, 1–8. [Google Scholar]
- Joos, P. Cholesterol as liquifier in phospholipid membranes studied by surface viscosity measurements of mixed monolayers. Chem. Phys. Lipids 1970, 4, 162–168. [Google Scholar]
- Henry, J.V.L.; Fryer, P.J.; Frith, W.J.; Norton, I.T. The influence of phospholipids and food proteins on the size and stability of model sub-micron emulsions. Food Hydrocoll 2010, 24, 66–71. [Google Scholar]
- Pichot, R.; Spyropoulos, F.; Norton, I.T. Competitive adsorption of surfactants and hydrophilic silica particles at the oil-water interface: Interfacial tension and contact angle studies. J. Colloid Interface Sci 2012, 377, 396–405. [Google Scholar]
- Brzozowska, I.; Figaszewski, Z.A. Interfacial tension of phosphatidylcholine-cholesterol system in monolayers at the air/water interface. Biophys. Chem 2002, 95, 173–179. [Google Scholar]
- Petelska, A.D.; Figaszewski, Z.A. Effect of pH on the interfacial tension of bilayer lipid membrane formed from phosphatidylcholine or phosphatidylserine. Biochim. Biophy. Acta 2002, 1561, 135–146. [Google Scholar]
- Petelska, A.D.; Figaszewski, Z.A. The equilibria of phosphatidylcholine-fatty acid and phosphatidylcholine-amine in monolayers at the air/water interface. Colloids Surf. B 2011, 82, 340–344. [Google Scholar]
- Brooks, J.H.; Pethica, B.A. Properties of ionized monolayers. Part 6.—Film pressures for ionized spread monolayers at the heptane/water interface. Trans. Faraday Soc 1964, 60, 208–215. [Google Scholar]
- Girault, H.H.J.; Schiffrin, D.J. Adsorption of phosphatidylcholine and phosphatidyl-ethanolamine at the polarised water/1,2-dichloroethane interface. J. Electroanal. Chem. Interfacial Electrochem 1984, 179, 277–284. [Google Scholar]
- Girault, H.H.J.; Schiffrin, D.J. Charge effects on phospholipid monolayers in relation to cell motility. Biochim. Biophy. Acta 1986, 857, 251–258. [Google Scholar]
- Wandlowski, T.; Marecc̆ek, V.; Samec, Z. Adsorption of phospholipids at the interface between two immiscible electrolyte solutions: Part I. Equilibrium adsorption of phosphatidylcholines at the water/nitrobenzene interface. J. Electroanal. Chem. Interfacial Electrochem 1988, 242, 277–290. [Google Scholar]
- Wandlowski, T.; Račinský, S.; Mareček, V.; Samec, Z. Adsorption of phospholipids at the interface between two immiscible electrolyte solutions. J. Electroanal. Chem. Interfacial Electrochem 1987, 227, 281–285. [Google Scholar]
- Kontturi, A.K.; Kontturi, K.; Murtomäki, L.; Quinn, B.; Cunnane, V.J. Study of ion transfer across phospholipid monolayers adsorbed at micropipette ITIES. J. Electroanal. Chem 1997, 424, 69–74. [Google Scholar]
- Manzanares, J.A.; Allen, R.M.; Kontturi, K. Enhanced ion transfer rate due to the presence of zwitterionic phospholipid monolayers at the ITIES. J. Electroanal. Chem 2000, 483, 188–196. [Google Scholar]
- Quinn, B.; Kontturi, K. Aspects of electron transfer at ITIES. J. Electroanal. Chem 2000, 483, 124–134. [Google Scholar]
- Kakiuchi, T.; Nakanishi, M.; Senda, M. The electrocapillary curves of the phosphatidylcholine monolayer at the polarized oil-water interface. II. Double layer structure of dilauroylphosphatidylcholine monolayer at the nitrobenzene-water interface. Bull. Chem. Soc. Jpn 1989, 62, 403–409. [Google Scholar]
- Kakiuchi, T.; Nakanishi, M.; Senda, M. The electrocapillary curves of the phosphatidylcholine monolayer at the polarized oil–water interface. I. Measurement of interfacial tension using a computer-aided pendant-drop method. Bull. Chem. Soc. Jpn 1988, 61, 1845–1851. [Google Scholar]
- Samec, Z.; Trojanek, A.; Krtil, P. Dynamics of phospholipid monolayers on polarised liquid-liquid interfaces. Faraday Discuss 2005, 129, 301–313. [Google Scholar]
- Santos, H.A.; García-Morales, V.; Pereira, C.M. Electrochemical properties of phospholipid monolayers at liquid-liquid interfaces. ChemPhysChem 2010, 11, 28–41. [Google Scholar]
- Shchipunov, Y.A.; Kolpakov, A.F. Phospholipids at the oil/water interface: Adsorption and interfacial phenomena in an electric field. Adv. Colloid Interface Sci 1991, 35, 31–138. [Google Scholar]
- Yue, B.Y.; Jackson, C.M.; Taylor, J.A.G.; Mingins, J.; Pethica, B.A. Phospholipid monolayers at non-polar oil/water interfaces. Part 1.—Phase transitions in distearoly-lecithin films at the n-heptane aqueous sodium chloride interface. J. Chem. Soc. Faraday Trans. 1 1976, 72, 2685–2693. [Google Scholar]
- Taylor, J.A.G.; Mingins, J.; Pethica, B.A. Phospholipid monolayers at the n-heptane/water interface. Part 2.—Dilute monolayers of saturated 1,2-diacyl-lecithins and -cephalins. J. Chem. Soc. Faraday Trans. 1 1976, 72, 2694–2702. [Google Scholar]
- Alexander, A.E.; Teorell, T.; Rideal, E.K. A study of films at the liquid/liquid interface. Trans. Faraday Soc 1939, 35, 727–737. [Google Scholar]
- Ogino, K.; Onishi, M. Interfacial action of natural surfactants in oil/water systems. J. Colloid Interface Sci 1981, 83, 18–25. [Google Scholar]
- Friberg, S.; Jansson, P.O.; Cederberg, E. Surfactant association structure and emulsion stability. J. Colloid Interface Sci 1976, 55, 614–623. [Google Scholar]
- Li, J.; Miller, R.; Möhwald, H. Characterisation of phospholipid layers at liquid interfaces 2. Comparison of isotherms of insoluble and soluble films of phospholipids at different fluid/water interfaces. Colloids Surf. A 1996, 114, 123–130. [Google Scholar]
- Grandell, D.; Murtomäki, L. Surface pressure control of phospholipid monolayers at the water/1,2-dichloroethane interface. Langmuir 1998, 14, 556–559. [Google Scholar]
- Shchipunov, Y.; Schmiedel, P. Phase behavior of lecithin at the oil/water interface. Langmuir 1996, 12, 6443–6445. [Google Scholar]
- Ma, Z.; Friberg, S.E.; Neogi, P. Observation of temporary liquid crystals in water-in-oil microemulsion systems. Colloids Surf 1988, 33, 249–258. [Google Scholar]
- Adalsteinsson, T.; Yu, H. Lipid lateral diffusion in multi-bilayers, and in monolayers at the air/water and heptane/water interfaces. Langmuir 2000, 16, 9410–9413. [Google Scholar]
- Negishi, M.; Seto, H.; Hase, M.; Yoshikawa, K. How does the mobility of phospholipid molecules at a water/oil interface reflect the viscosity of the surrounding oil? Langmuir 2008, 24, 8431–8434. [Google Scholar]
- Walder, R.B.; Honciuc, A.; Schwartz, D.K. Phospholipid diffusion at the oil-water interface. J. Phys. Chem. B 2010, 114, 11484–11488. [Google Scholar]
- Barenholz, Y. Liposome application: Problems and prospects. Curr. Opin. Colloid Interface Sci 2001, 6, 66–77. [Google Scholar]
- Van Swaay, D.; deMello, A. Microfluidic methods for forming liposomes. Lab Chip 2013, 13, 752–767. [Google Scholar]
- Wagner, A.; Vorauer-Uhl, K. Liposome technology for industrial purposes. J. Drug Deliv 2010, 2011, 591325. [Google Scholar]
- Yang, B.; Matsumura, H.; Furusawa, K. Adsorption behavior of phospholipid vesicles at oil/water interfaces. Colloids Surf. B 1999, 14, 161–168. [Google Scholar]
- Hase, M.; Yamada, A.; Hamada, T.; Yoshikawa, K. Transport of a cell-sized phospholipid micro-container across water/oil interface. Chem. Phys. Lett 2006, 426, 441–444. [Google Scholar] [Green Version]
- Mitsche, M.A.; Wang, L.; Small, D.M. Adsorption of egg phosphatidylcholine to an air/water and triolein/water bubble interface: Use of the 2-dimensional phase rule to estimate the surface composition of a phospholipid/triolein/water surface as a function of surface pressure. J. Phys. Chem. B 2010, 114, 3276–3284. [Google Scholar]
- Friberg, S.; Mandell, L.; Larsson, M. Mesomorphous phases, a factor of importance for the properties of emulsions. J. Colloid Interface Sci 1969, 29, 155–156. [Google Scholar]
- Friberg, S.; Mandell, L. Phase equilibria and their influence on the properties of emulsions. J. Am. Oil Chem. Soc 1970, 47, 149–152. [Google Scholar]
- Friberg, S. Liquid crystalline phases in emulsions. J. Colloid Interface Sci 1971, 37, 291–295. [Google Scholar]
- Mirtallo, J.M.; Dasta, J.F.; Kleinschmidt, K.C.; Varon, J. State of the art review: Intravenous fat emulsions: Current applications, safety profile, and clinical implications. Ann. Pharmacother 2010, 44, 688–700. [Google Scholar]
- Rydhag, L.; Wilton, I. The function of phospholipids of soybean lecithin in emulsions. J. Am. Oil Chem. Soc 1981, 58, 830–837. [Google Scholar]
- Handa, T.; Saito, H.; Miyajima, K. Phospholipid monolayers at the triolein-saline interface: Production of microemulsion particles and conversion of monolayers to bilayers. Biochemistry 1990, 29, 2884–2890. [Google Scholar]
- Cornelus, C.; Giulieri, F.; Krafft, M.-P.; Riess, J.G. Impact of the structure of phospholipid dispersions on the stability of fluorocarbon/phospholipid emulsions for biomedical uses. Colloids Surf. A 1993, 70, 233–238. [Google Scholar]
- Thakur, R.K.; Villette, C.; Aubry, J.M.; Delaplace, G. Dynamic emulsification and catastrophic phase inversion of lecithin-based emulsions. Colloids Surf. A 2008, 315, 285–293. [Google Scholar]
- Evstigneeva, R. Chemical Stability. In Phospholipid Handbook; Cecv, G., Ed.; Marcel Dekker, Inc: New York, NY, USA, 1993; pp. 323–334. [Google Scholar]
- Thakur, R.K.; Villette, C.; Aubry, J.M.; Delaplace, G. Formulation-composition map of a lecithin-based emulsion. Colloids Surf. A 2007, 310, 55–61. [Google Scholar]
- Knoth, A.; Scherze, I.; Muschiolik, G. Stability of water-in-oil-emulsions containing phosphatidylcholine-depleted lecithin. Food Hydrocoll 2005, 19, 635–640. [Google Scholar]
- Scherze, I.; Knoth, A.; Muschiolik, G. Effect of emulsification method on the properties of lecithin- and pgpr-stabilized water–in–oil–emulsions. J. Dispers. Sci. Technol 2006, 27, 427–434. [Google Scholar]
- Watrobska-Swietlikowska, D.; Sznitowska, M. Partitioning of parabens between phases of submicron emulsions stabilized with egg lecithin. Int. J. Pharm 2006, 312, 174–178. [Google Scholar]
- Lucks, J.S.; Müller, B.; Klütsch, K. Parental Fat Emulsions: Structure, Stability and Applications. In Pharmaceutical Emulsions and Suspensions; Nielloud, F., Marti-Mestres, G., Eds.; Marcel Dekker, Inc: New York, NY, USA, 2000; pp. 229–245. [Google Scholar]
- Férézou, J.; Gulik, A.; Domingo, N.; Milliat, F.; Dedieu, J.C.; Dunel-Erb, S.; Chevalier, C.; Bach, A.C. Intralipid 10%: Physicochemical characterization. Nutrition 2001, 17, 930–933. [Google Scholar]
- Han, J.; Washington, C. Partition of antimicrobial additives in an intravenous emulsion and their effect on emulsion physical stability. Int. J. Pharm 2005, 288, 263–271. [Google Scholar]
- Han, J.; Davis, S.S.; Washington, C. Physical properties and stability of two emulsion formulations of propofol. Int. J. Pharm 2001, 215, 207–220. [Google Scholar]
- Sznitowska, M.; Janicki, S.; Dabrowska, E.A.; Gajewska, M. Physicochemical screening of antimicrobial agents as potential preservatives for submicron emulsions. Eur. J. Pharm. Sci 2002, 15, 489–495. [Google Scholar]
- Burnham, W.R.; Hansrani, P.K.; Knott, C.E.; Cook, J.A.; Davis, S.S. Stability of a fat emulsion based intravenous feeding mixture. Int. J. Pharm 1982, 13, 9–22. [Google Scholar]
- Washington, C.; Athersuch, A.; Kynoch, D.J. The electrokinetic properties of phospholipid stabilized fat emulsions. IV. The effect of glucose and of pH. Int. J. Pharm 1990, 64, 217–222. [Google Scholar]
- Jumaa, M.; Müller, B.W. The effect of oil components and homogenization conditions on the physicochemical properties and stability of parenteral fat emulsions. Int. J. Pharm 1998, 163, 81–89. [Google Scholar]
- Jumaa, M.; Müller, B.W. Parenteral emulsions stabilized with a mixture of phospholipids and PEG-660-12-hydroxy-stearate: Evaluation of accelerated and long-term stability. Eur. J. Pharm. Biopharm 2002, 54, 207–212. [Google Scholar]
- Klang, S.H.; Baszkin, A.; Benita, S. The stability of piroxicam incorporated in a positively-charged submicron emulsion for ocular administration. Int. J. Pharm 1996, 132, 33–44. [Google Scholar]
- Yu, Y.L.; Lu, Y.; Tang, X.; Cui, F.D. Formulation, preparation and evaluation of an intravenous emulsion containing brucea javanica oil and coix seed oil for anti-tumor application. Biol. Pharm. Bull 2008, 31, 673–680. [Google Scholar]
- Zurowska-Pryczkowska, K.; Sznitowska, M.; Janicki, S. Studies on the effect of pilocarpine incorporation into a submicron emulsion on the stability of the drug and the vehicle. Eur. J. Pharm. Biopharm 1999, 47, 255–260. [Google Scholar]
- Klinkesorn, U.; Sophanodora, P.; Chinachoti, P.; McClements, D.J.; Decker, E.A. Increasing the oxidative stability of liquid and dried tuna oil-in-water emulsions with electrostatic layer-by-layer deposition technology. J. Agric. Food Chem 2005, 53, 4561–4566. [Google Scholar]
- Pokorný, J. Production, Separation and Modification of Phospholipids for Use in Food. In Modifying Lipids for Use in Food; Gunstone, F.D., Ed.; Woodhead Publishing: Cambridge, UK, 2006; pp. 383–386. [Google Scholar]
- Garti, N. What can nature offer from an emulsifier point of view: Trends and progress? Colloids Surf. A 1999, 152, 125–146. [Google Scholar]
- Fillery-Travis, A.J.; Foster, L.H.; Robins, M.M. Stability of emulsions stabilised by two physiological surfactants: l-α-phosphatidylcholine and sodium taurocholate. Biophys. Chem 1995, 54, 253–260. [Google Scholar]
- Wickham, M.; Garrood, M.; Leney, J.; Wilson, P.D.G.; Fillery-Travis, A. Modification of a phospholipid stabilized emulsion interface by bile salt: Effect on pancreatic lipase activity. J. Lipid Res 1998, 39, 623–632. [Google Scholar]
- Mun, S.; Decker, E.A.; McClements, D.J. Influence of emulsifier type on in vitro digestibility of lipid droplets by pancreatic lipase. Food Res. Int 2007, 40, 770–781. [Google Scholar]
- Li, Y.; Le Maux, S.; Xiao, H.; McClements, D.J. Emulsion-based delivery systems for tributyrin, a potential colon cancer preventative agent. J. Agric. Food Chem 2009, 57, 9243–9249. [Google Scholar]
- Benita, S.; Friedman, D.; Weinstock, M. Physostigmine emulsion: A new injectable controlled release delivery system. Int. J. Pharm 1986, 30, 47–55. [Google Scholar]
- Benita, S.; Levy, M.Y. Submicron emulsions as colloidal drug carriers for intravenous administration: Comprehensive physicochemical characterization. J. Pharm. Sci 1993, 82, 1069–1079. [Google Scholar]
- Bylaite, E.; Nylander, T.; Venskutonis, R.; Jönsson, B. Emulsification of caraway essential oil in water by lecithin and β-lactoglobulin: Emulsion stability and properties of the formed oil-aqueous interface. Colloids Surf. B 2001, 20, 327–340. [Google Scholar]
- Donsì, F.; Wang, Y.; Huang, Q. Freeze-thaw stability of lecithin and modified starch-based nanoemulsions. Food Hydrocoll 2011, 25, 1327–1336. [Google Scholar]
- Pichot, R.; Spyropoulos, F.; Norton, I.T. O/W emulsions stabilised by both low molecular weight surfactants and colloidal particles: The effect of surfactant type and concentration. J. Colloid Interface Sci 2010, 352, 128–135. [Google Scholar]
- Ghouchi Eskandar, N.; Simovic, S.; Prestidge, C.A. Synergistic effect of silica nanoparticles and charged surfactants in the formation and stability of submicron oil-in-water emulsions. Phys. Chem. Chem. Phys 2007, 9, 6426–6434. [Google Scholar]
- Westesen, K.; Bunjes, H. Do nanoparticles prepared from lipids solid at room temperature always possess a solid lipid matrix? Int. J. Pharm 1995, 115, 129–131. [Google Scholar]
- Westesen, K.; Siekmann, B. Investigation of the gel formation of phospholipid-stabilized solid lipid nanoparticles. Int. J. Pharm 1997, 151, 35–45. [Google Scholar]
- Sjöström, B.; Bergenståhl, B. Preparation of submicron drug particles in lecithin-stabilized ow emulsions: I. Model studies of the precipitation of cholesteryl acetate. Int. J. Pharm 1992, 84, 107–116. [Google Scholar]
- Shum, H.C.; Lee, D.; Yoon, I.; Kodger, T.; Weitz, D.A. Double emulsion templated monodisperse phospholipid vesicles. Langmuir 2008, 24, 7651–7653. [Google Scholar]
- Funakoshi, K.; Suzuki, H.; Takeuchi, S. Formation of giant lipid vesiclelike compartments from a planar lipid membrane by a pulsed jet flow. J. Am. Chem. Soc 2007, 129, 12608–12609. [Google Scholar]
- Campbell, A.; Taylor, P.; Cayre, O.J.; Paunov, V.N. Preparation of aqueous gel beads coated by lipid bilayers. Chem. Commun. 2004, 2378–2379. [Google Scholar]
- Yamanaka, T.; Hayashi, M.; Matuura, R. Studies on the soap films stabilized by phospholipids: I. Effects of metal ions on the free energy of black soap films. J. Colloid Interface Sci 1982, 88, 458–466. [Google Scholar]
- Sarker, D.K.; Wilde, P.J.; Clark, D.C. Competitive adsorption of l-α-lysophosphatidylcholine/β-lactoglobulin mixtures at the interfaces of foams and foam lamellae. Colloids Surf. B 1995, 3, 349–356. [Google Scholar]

© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pichot, R.; Watson, R.L.; Norton, I.T. Phospholipids at the Interface: Current Trends and Challenges. Int. J. Mol. Sci. 2013, 14, 11767-11794. https://doi.org/10.3390/ijms140611767
Pichot R, Watson RL, Norton IT. Phospholipids at the Interface: Current Trends and Challenges. International Journal of Molecular Sciences. 2013; 14(6):11767-11794. https://doi.org/10.3390/ijms140611767
Chicago/Turabian StylePichot, Roman, Richard L. Watson, and Ian T. Norton. 2013. "Phospholipids at the Interface: Current Trends and Challenges" International Journal of Molecular Sciences 14, no. 6: 11767-11794. https://doi.org/10.3390/ijms140611767