In Vitro and in Vivo Models of Non-Alcoholic Fatty Liver Disease (NAFLD)

Abstract
:1. Introduction
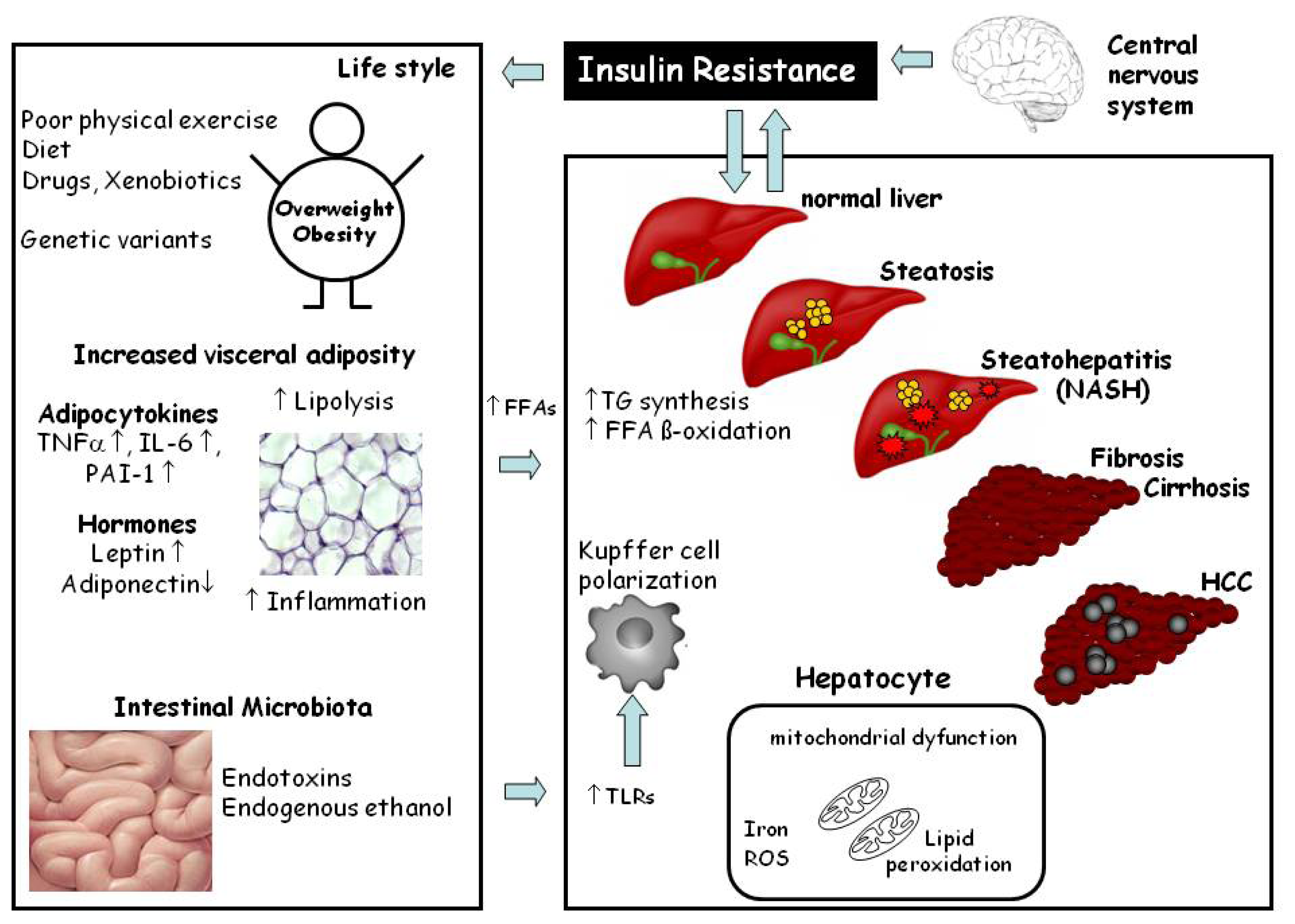
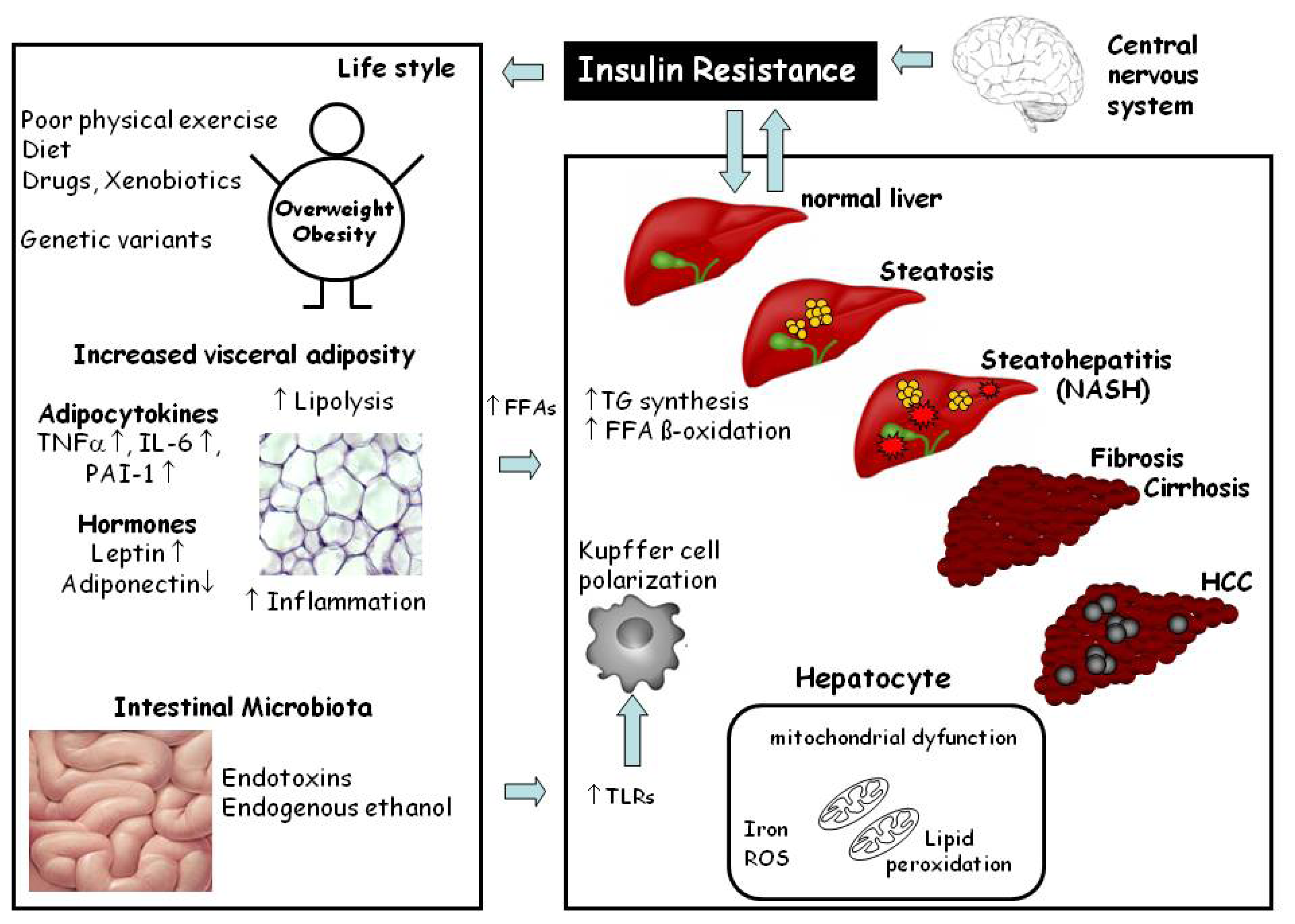
2. Histopathology and Pathogenesis of NAFLD
3. Animal Models of NAFLD
4. Genetic Rodent Models of NAFLD
4.1. Ob/ob, db/db and Obese (fa/fa) Zucker Rat
4.2. Agouti Gene [KK-Ay/a]
4.3. Melanocortin 4 Receptor (MC4R)
4.4. Sterol Regulator Element-Binding Protein 1c (SREBP)
5. Dietary Rodent Models of NAFLD
5.1. Methionine- and Choline-Deficient (MCD) Diet
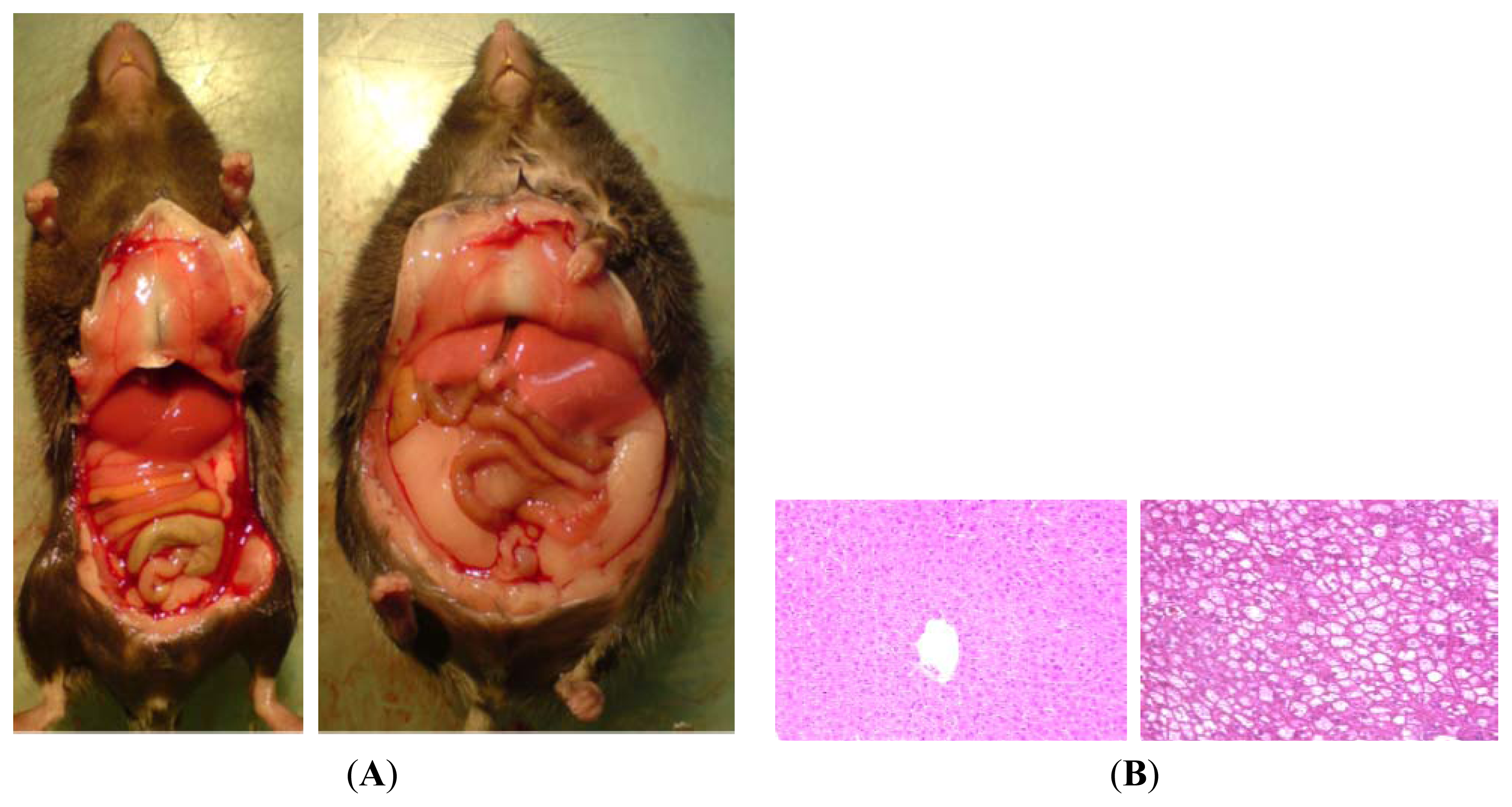
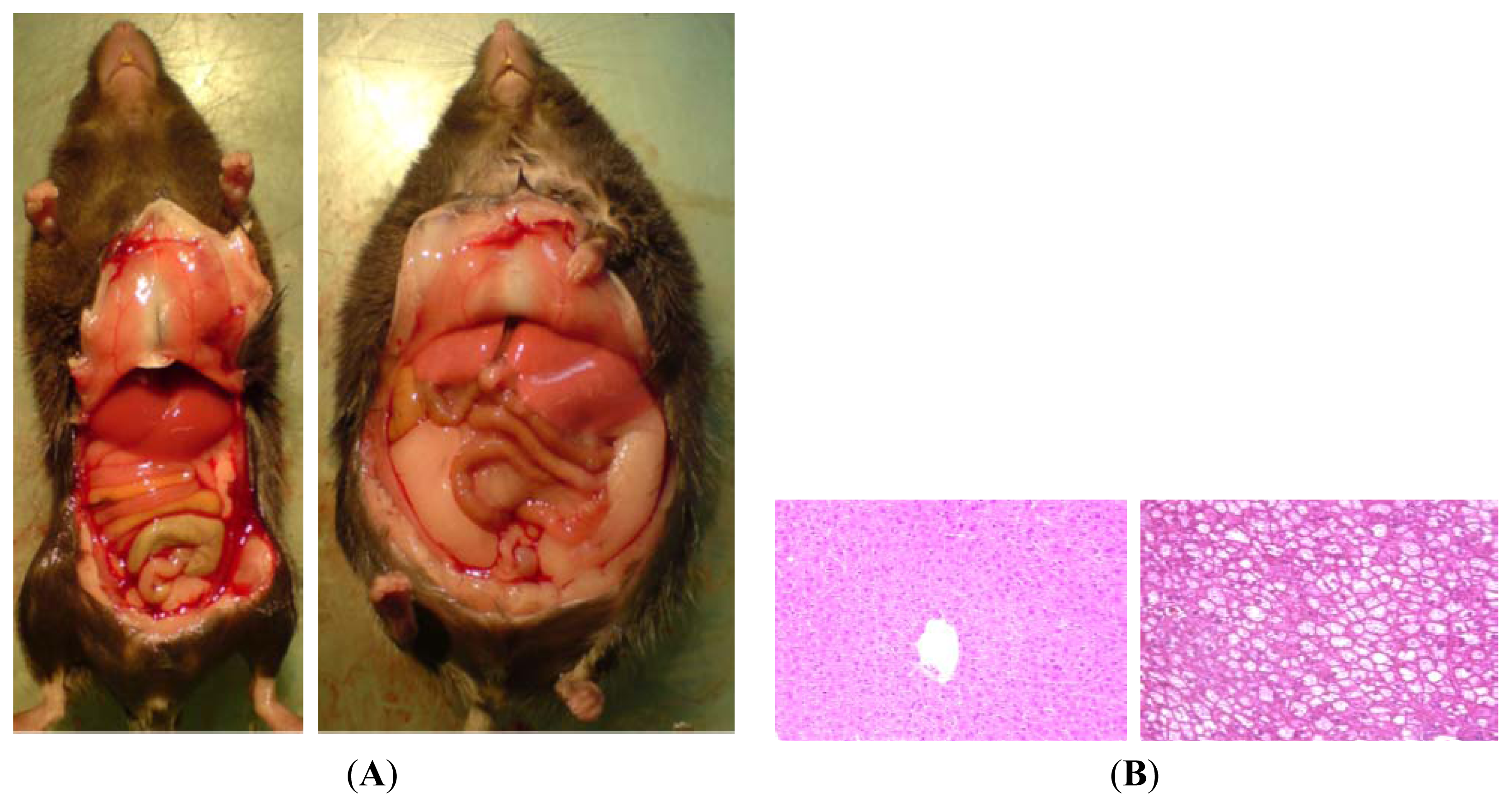
5.2. High Fat Diet (HFD)
5.3. Fructose-Rich Diets
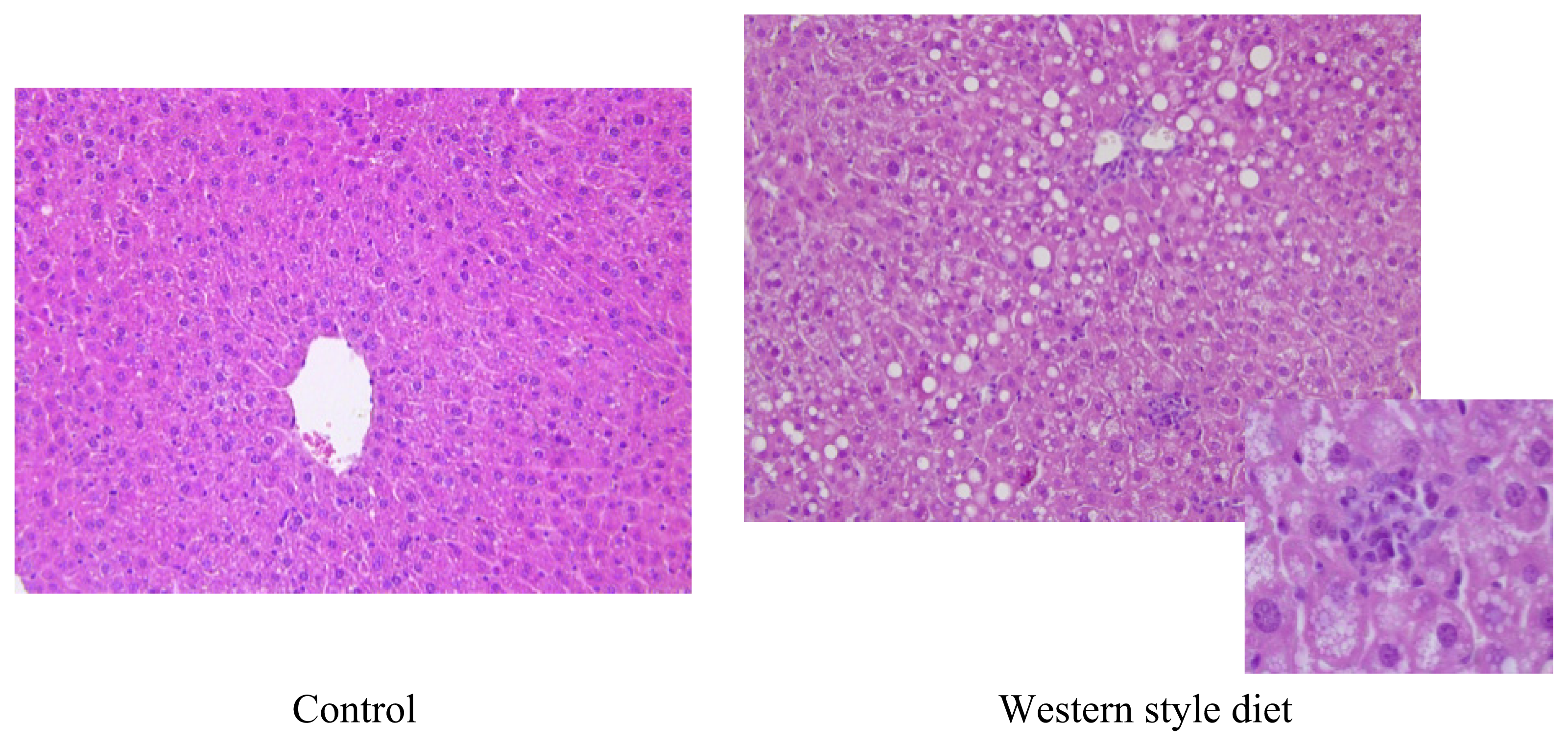
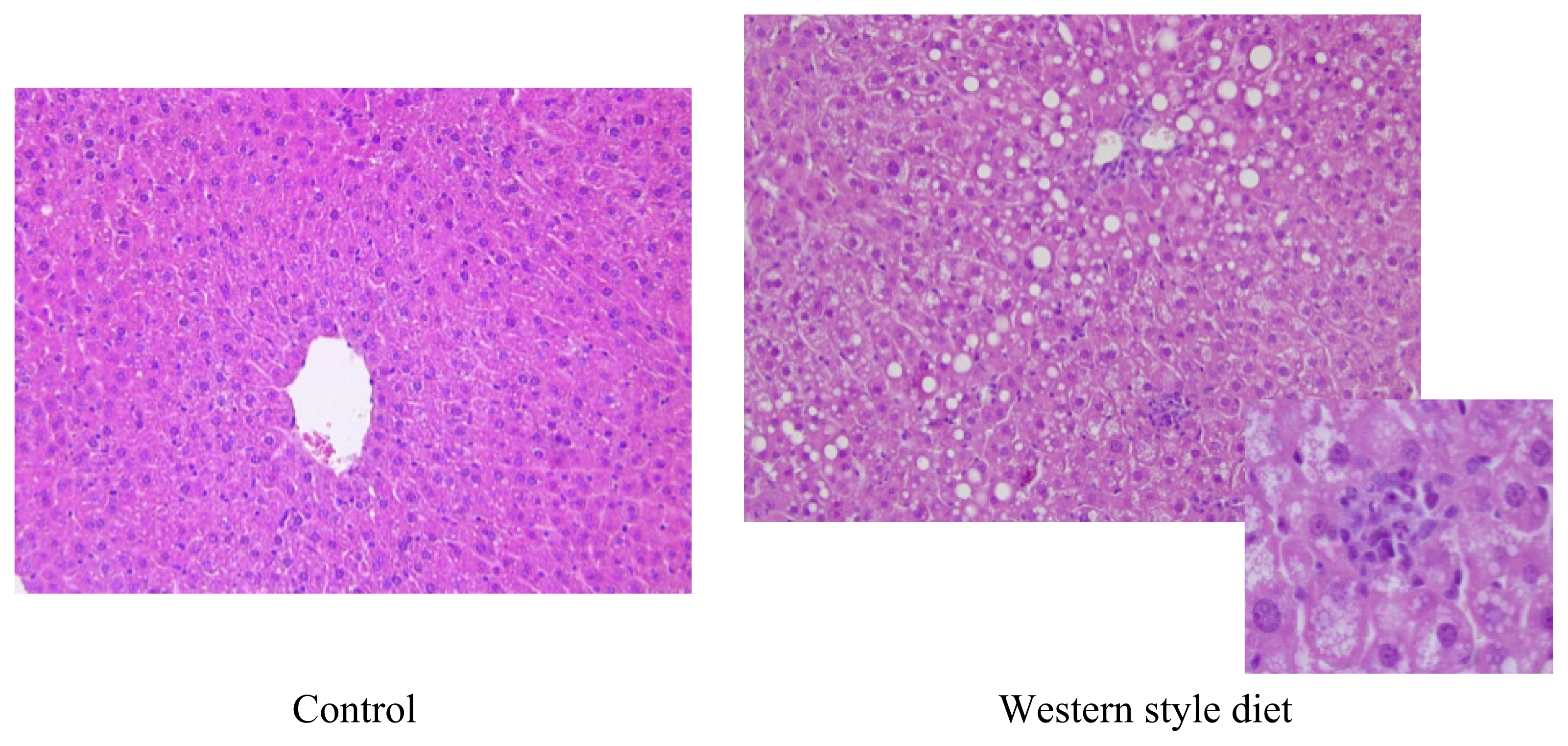
5.4. Western-Style or Fast Food Diet: Combination of Fat and Sugar
5.5. “Non-rodent” Animal Models of NAFLD
5.6. In Vitro Models for NAFLD
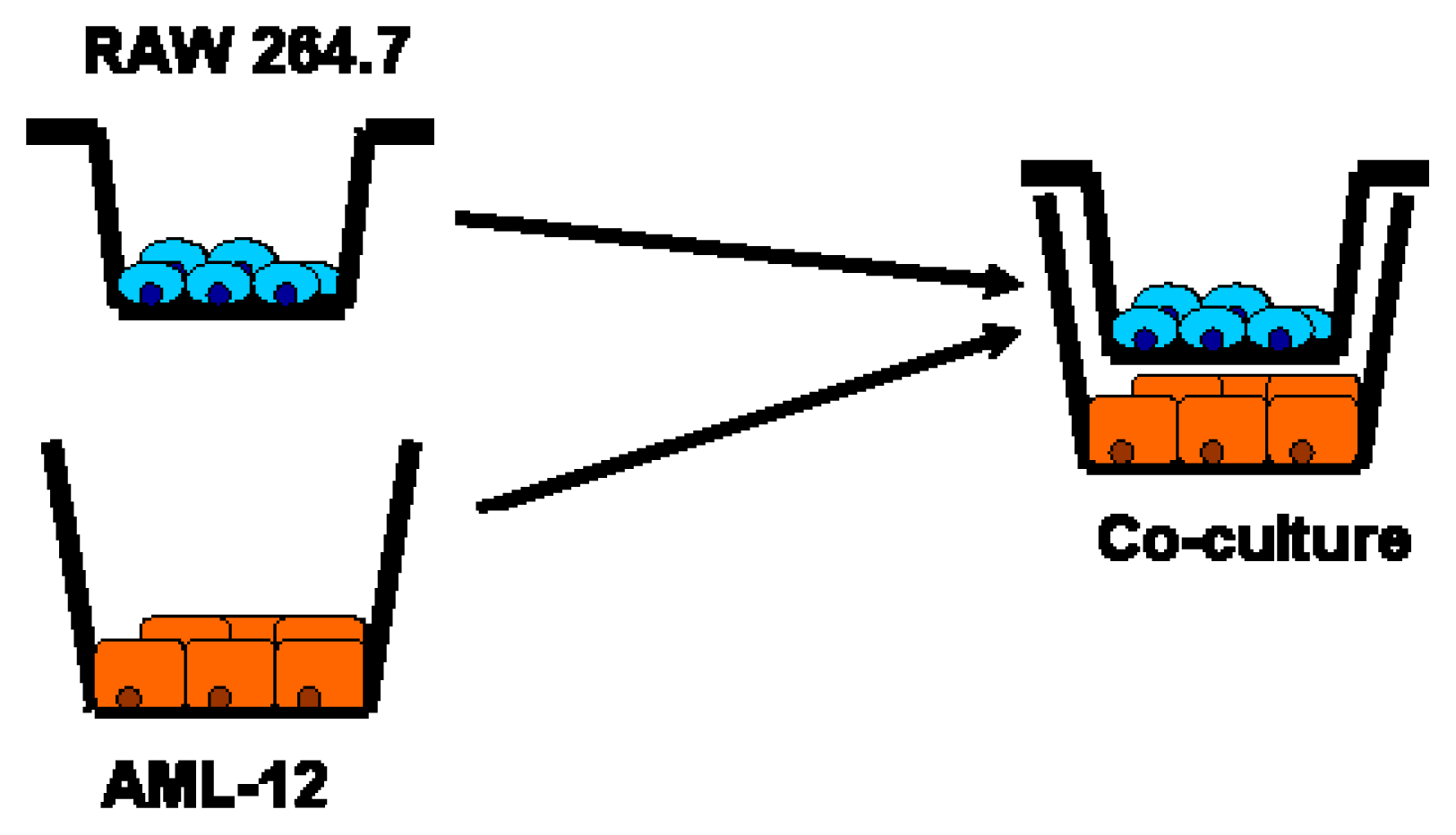
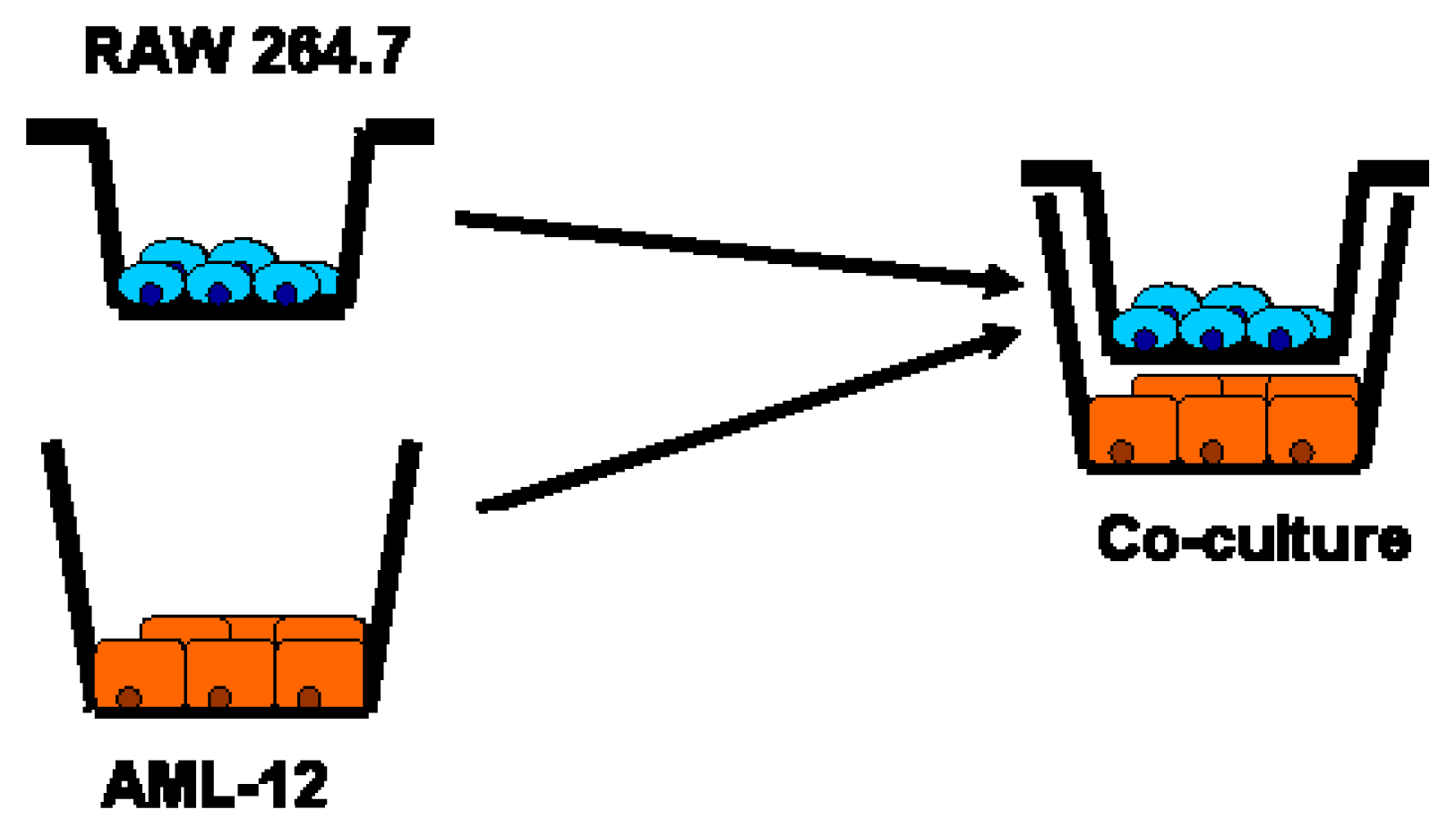
5.7. Co-Culture Model: Interaction of RAW 264.7 Macrophages and AML-12 Cells
5.8. Three-Dimensional Cell Culture Model
6. Conclusions
Acknowledgments
Abbreviations
| NAFLD | non-alcoholic fatty liver disease |
| NFκB | nuclear factor kappa B |
| ROS | reactive oxygen species |
| TNFα | tumor necrosis factor α |
| TG | triglycerides |
| IL-6 | interleukin 6 |
| MCD | methionine- and choline-deficient |
| MC4R | melanocortin 4 receptor |
| HFD | high-fat diet |
| SREBP1 | sterol regulator element-binding protein 1c |
| HFCS | high fructose corn syrup |
Conflict of Interest
References
- Marchesini, G.; Babini, M. Nonalcoholic fatty liver disease and the metabolic syndrome. Minerva Cardioangiol 2006, 54, 229–239. [Google Scholar]
- Adams, L.A.; Lymp, J.F.; St Sauver, J. The natural history of nonalcoholic fatty liver disease: A population-based cohort study. Gastroenterology 2005, 129, 113–121. [Google Scholar]
- Neuschwander-Tetri, B.A. Nonalcoholic steatohepatitis and the metabolic syndrome. Am. J. Med. Sci 2005, 330, 326–335. [Google Scholar]
- Krawczyk, M.; Bonfrate, L.; Portincasa, P. Nonalcoholic fatty liver disease. Best Pract. Res. Clin. Gastroenterol 2010, 24, 695–708. [Google Scholar]
- Gross, L.S.; Li, L.; Ford, E.S.; Liu, S. Increased consumption of refined carbohydrates and the epidemic of type 2 diabetes in the United States: An ecologic assessment. Am. J. Clin. Nutr 2004, 79, 774–779. [Google Scholar]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar]
- Cohen, J.C.; Horton, J.D.; Hobbs, H.H. Human fatty liver disease: Old questions and new insights. Science 2011, 332, 1519–1523. [Google Scholar]
- Takahashi, H.; Ono, N.; Eguchi, Y. Evaluation of acoustic radiation force impulse elastography for fibrosis staging of chronic liver disease: A pilot study. Liver Int 2010, 30, 538–545. [Google Scholar]
- Brunt, E.M.; Kleiner, D.E.; Wilson, L.A. Nonalcoholic fatty liver disease [NAFLD] activity score and the histopathologic diagnosis in NAFLD: Distinct clinicopathologic meanings. Hepatology 2011, 53, 810–820. [Google Scholar]
- Mayer, J.; Bates, M.W.; Dickie, M.M. Hereditary diabetes in genetically obese mice. Science 1951, 113, 746–747. [Google Scholar]
- Mayer, J.; Dickie, M.M.; Bates, M.W. Free selection of nutrients by hereditarily obese mice. Science 1951, 113, 745–746. [Google Scholar]
- Chalasani, N.; Crabb, D.W.; Cummings, O.W. Does leptin play a role in the pathogenesis of human nonalcoholic steatohepatitis? Am. J. Gastroenterol 2003, 98, 2771–2776. [Google Scholar]
- Machado, M.V.; Cortez-Pinto, H. Gut microbiota and nonalcoholic fatty liver disease. Ann. Hepatol 2012, 11, 440–449. [Google Scholar]
- Zelber-Sagi, S.; Ratziu, V.; Zvibel, I. The association between adipocytokines and biomarkers for nonalcoholic fatty liver disease-induced liver injury: A study in the general population. Eur. J. Gastroenterol. Hepatol 2012, 24, 262–269. [Google Scholar]
- Swellam, M.; Hamdy, N. Association of nonalcoholic fatty liver disease with a single nucleotide polymorphism on the gene encoding leptin receptor. IUBMB Life 2012, 64, 180–186. [Google Scholar]
- Aller, R.; de Luis, D.A.; Izaola, O. Lys656Asn polymorphism of leptin receptor, leptin levels and insulin resistance in patients with non alcoholic fatty liver disease. Eur. Rev. Med. Pharmacol. Sci 2012, 16, 335–341. [Google Scholar]
- Faggioni, R.; Fantuzzi, G.; Gabay, C. Leptin deficiency enhances sensitivity to endotoxin-induced lethality. Am. J. Physiol 1999, 276, R136–R142. [Google Scholar]
- Brix, A.E.; Elgavish, A.; Nagy, T.R. Evaluation of liver fatty acid oxidation in the leptin-deficient obese mouse. Mol. Genet. Metab 2002, 75, 219–226. [Google Scholar]
- Wortham, M.; He, L.; Gyamfi, M. The transition from fatty liver to NASH associates with SAMe depletion in db/db mice fed a methionine choline-deficient diet. Dig. Dis. Sci 2008, 53, 2761–2774. [Google Scholar]
- Godbole, V.; York, D.A. Lipogenesis in situ in the genetically obese Zucker fatty rat [fa/fa]: Role of hyperphagia and hyperinsulinaemia. Diabetologia 1978, 14, 191–197. [Google Scholar]
- Oana, F.; Takeda, H.; Hayakawa, K. Physiological difference between obese [fa/fa] Zucker rats and lean Zucker rats concerning adiponectin. Metabolism 2005, 54, 995–1001. [Google Scholar]
- Masaki, T.; Chiba, S.; Tatsukawa, H. Adiponectin protects LPS-induced liver injury through modulation of TNF-alpha in KK-Ay obese mice. Hepatology 2004, 40, 177–184. [Google Scholar]
- Balthasar, N.; Dalgaard, L.T.; Lee, C.E. Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell 2005, 123, 493–505. [Google Scholar]
- Vaisse, C.; Clement, K.; Durand, E. Melanocortin-4 receptor mutations are a frequent and heterogeneous cause of morbid obesity. J. Clin. Invest 2000, 106, 253–262. [Google Scholar]
- Marsh, D.J.; Hollopeter, G.; Huszar, D. Response of melanocortin-4 receptor-deficient mice to anorectic and orexigenic peptides. Nat. Genet 1999, 21, 119–122. [Google Scholar]
- Huszar, D.; Lynch, C.A.; Fairchild-Huntress, V. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell 1997, 88, 131–141. [Google Scholar]
- Albarado, D.C.; McClaine, J.; Stephens, J.M. Impaired coordination of nutrient intake and substrate oxidation in melanocortin-4 receptor knockout mice. Endocrinology 2004, 145, 243–252. [Google Scholar]
- Itoh, M.; Suganami, T.; Nakagawa, N. Melanocortin 4 receptor-deficient mice as a novel mouse model of nonalcoholic steatohepatitis. Am. J. Pathol 2011, 179, 2454–2463. [Google Scholar]
- Shimomura, I.; Hammer, R.E.; Richardson, J.A. Insulin resistance and diabetes mellitus in transgenic mice expressing nuclear SREBP-1c in adipose tissue: Model for congenital generalized lipodystrophy. Genes Dev 1998, 12, 3182–3194. [Google Scholar]
- Nakayama, H.; Otabe, S.; Ueno, T. Transgenic mice expressing nuclear sterol regulatory element-binding protein 1c in adipose tissue exhibit liver histology similar to nonalcoholic steatohepatitis. Metabolism 2007, 56, 470–475. [Google Scholar]
- Anstee, Q.M.; Goldin, R.D. Mouse models in non-alcoholic fatty liver disease and steatohepatitis research. Int. J. Exp. Pathol 2006, 87, 1–16. [Google Scholar]
- Leclercq, I.A.; Farrell, G.C.; Field, J. CYP2E1 and CYP4A as microsomal catalysts of lipid peroxides in murine nonalcoholic steatohepatitis. J. Clin. Invest 2000, 105, 1067–1075. [Google Scholar]
- Larter, C.Z.; Yeh, M.M.; Williams, J. MCD-induced steatohepatitis is associated with hepatic adiponectin resistance and adipogenic transformation of hepatocytes. J. Hepatol 2008, 49, 407–416. [Google Scholar]
- Rinella, M.E.; Elias, M.S.; Smolak, R.R. Mechanisms of hepatic steatosis in mice fed a lipogenic methionine choline-deficient diet. J. Lipid Res 2008, 49, 1068–1076. [Google Scholar]
- Rangnekar, A.S.; Lammert, F.; Igolnikov, A.; Green, R.M. Quantitative trait loci analysis of mice administered the methionine-choline deficient dietary model of experimental steatohepatitis. Liver Int 2006, 26, 1000–1005. [Google Scholar]
- Yamazaki, Y.; Kakizaki, S.; Takizawa, D. Interstrain differences in susceptibility to non-alcoholic steatohepatitis. J. Gastroenterol. Hepatol 2008, 23, 276–282. [Google Scholar]
- Pogribny, I.P.; Tryndyak, V.P.; Bagnyukova, T.V. Hepatic epigenetic phenotype predetermines individual susceptibility to hepatic steatosis in mice fed a lipogenic methyl-deficient diet. J. Hepatol 2009, 51, 176–186. [Google Scholar]
- Bray, G.A.; Paeratakul, S.; Popkin, B.M. Dietary fat and obesity: A review of animal, clinical and epidemiological studies. Physiol. Behav 2004, 83, 549–555. [Google Scholar]
- Takahashi, Y.; Soejima, Y.; Fukusato, T. Animal models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J. Gastroenterol 2012, 18, 2300–2308. [Google Scholar]
- Buettner, R.; Scholmerich, J.; Bollheimer, L.C. High-fat diets: Modeling the metabolic disorders of human obesity in rodents. Obesity [Silver Spring] 2007, 15, 798–808. [Google Scholar]
- Lanthier, N.; Molendi-Coste, O.; Cani, P.D. Kupffer cell depletion prevents but has no therapeutic effect on metabolic and inflammatory changes induced by a high-fat diet. FASEB J 2011, 25, 4301–4311. [Google Scholar]
- Omagari, K.; Kato, S.; Tsuneyama, K. Effects of a long-term high-fat diet and switching from a high-fat to low-fat, standard diet on hepatic fat accumulation in Sprague-Dawley rats. Dig. Dis. Sci 2008, 53, 3206–3212. [Google Scholar]
- Varela-Rey, M.; Embade, N.; Ariz, U. Non-alcoholic steatohepatitis and animal models: Understanding the human disease. Int. J. Biochem. Cell Biol 2009, 41, 969–976. [Google Scholar]
- Li, Z.; Soloski, M.J.; Diehl, A.M. Dietary factors alter hepatic innate immune system in mice with nonalcoholic fatty liver disease. Hepatology 2005, 42, 880–885. [Google Scholar]
- Cong, W.N.; Tao, R.Y.; Tian, J.Y.; Liu, G.T.; Ye, F. The establishment of a novel non-alcoholic steatohepatitis model accompanied with obesity and insulin resistance in mice. Life Sci 2008, 82, 983–990. [Google Scholar]
- Deng, Q.G.; She, H.; Cheng, J.H. Steatohepatitis induced by intragastric overfeeding in mice. Hepatology 2005, 42, 905–914. [Google Scholar]
- Tsukamoto, H.; French, S.W.; Benson, N. Severe and progressive steatosis and focal necrosis in rat liver induced by continuous intragastric infusion of ethanol and low fat diet. Hepatology 1985, 5, 224–232. [Google Scholar]
- Nanji, A.A. Role of different dietary fatty acids in the pathogenesis of experimental alcoholic liver disease. Alcohol 2004, 34, 21–25. [Google Scholar]
- Tipoe, G.L.; Ho, C.T.; Liong, E.C. Voluntary oral feeding of rats not requiring a very high fat diet is a clinically relevant animal model of non-alcoholic fatty liver disease [NAFLD]. Histol. Histopathol 2009, 24, 1161–1169. [Google Scholar]
- Morimoto, M.; Zern, M.A.; Hagbjork, A.L.; Ingelman-Sundberg, M.; French, S.W. Fish oil, alcohol, and liver pathology: Role of cytochrome P450 2E1. Proc. Soc. Exp. Biol. Med 1994, 207, 197–205. [Google Scholar]
- Nanji, A.A.; Zhao, S.; Sadrzadeh, S.M. Markedly enhanced cytochrome P450 2E1 induction and lipid peroxidation is associated with severe liver injury in fish oil-ethanol-fed rats. Alcohol Clin. Exp. Res 1994, 18, 1280–1285. [Google Scholar]
- Chen, H.W.; Tsai, C.W.; Yang, J.J. The combined effects of garlic oil and fish oil on the hepatic antioxidant and drug-metabolizing enzymes of rats. Br. J. Nutr 2003, 89, 189–200. [Google Scholar]
- Zong, H.; Armoni, M.; Harel, C. Cytochrome P-450 CYP2E1 knockout mice are protected against high-fat diet-induced obesity and insulin resistance. Am. J. Physiol. Endocrinol. Metab 2012, 302, E532–E539. [Google Scholar]
- Van den Berg, S.A.; Guigas, B.; Bijland, S. High levels of dietary stearate promote adiposity and deteriorate hepatic insulin sensitivity. Nutr. Metab. [Lond. ] 2010, 7, 24. [Google Scholar]
- Syn, W.K.; Yang, L.; Chiang, D.J. Genetic differences in oxidative stress and inflammatory responses to diet-induced obesity do not alter liver fibrosis in mice. Liver Int 2009, 29, 1262–1272. [Google Scholar]
- Hill-Baskin, A.E.; Markiewski, M.M.; Buchner, D.A. Diet-induced hepatocellular carcinoma in genetically predisposed mice. Hum. Mol. Genet 2009, 18, 2975–2988. [Google Scholar]
- Jeong, W.I.; Jeong, D.H.; Do, S.H. Mild hepatic fibrosis in cholesterol and sodium cholate diet-fed rats. J. Vet. Med. Sci 2005, 67, 235–242. [Google Scholar]
- Kumar, S.A.; Sudhahar, V.; Varalakshmi, P. Protective role of eicosapentaenoate-lipoate [EPA-LA] derivative in combating oxidative hepatocellular injury in hypercholesterolemic atherogenesis. Atherosclerosis 2006, 189, 115–122. [Google Scholar]
- Shockley, K.R.; Witmer, D.; Burgess-Herbert, S.L. Effects of atherogenic diet on hepatic gene expression across mouse strains. Physiol. Genomics 2009, 39, 172–182. [Google Scholar]
- Spruss, A.; Bergheim, I. Dietary fructose and intestinal barrier: Potential risk factor in the pathogenesis of nonalcoholic fatty liver disease. J. Nutr. Biochem 2009, 20, 657–662. [Google Scholar]
- Tappy, L.; Le, K.A.; Tran, C. Fructose and metabolic diseases: New findings, new questions. Nutrition 2010, 26, 1044–1049. [Google Scholar]
- Bergheim, I.; Weber, S.; Vos, M. Antibiotics protect against fructose-induced hepatic lipid accumulation in mice: Role of endotoxin. J. Hepatol 2008, 48, 983–992. [Google Scholar]
- Spruss, A.; Kanuri, G.; Wagnerberger, S. Toll-like receptor 4 is involved in the development of fructose-induced hepatic steatosis in mice. Hepatology 2009, 50, 1094–1104. [Google Scholar]
- Ackerman, Z.; Oron-Herman, M.; Grozovski, M. Fructose-induced fatty liver disease: Hepatic effects of blood pressure and plasma triglyceride reduction. Hypertension 2005, 45, 1012–1018. [Google Scholar]
- Kanuri, G.; Spruss, A.; Wagnerberger, S. Role of tumor necrosis factor alpha [TNFalpha] in the onset of fructose-induced nonalcoholic fatty liver disease in mice. J. Nutr. Biochem 2011, 22, 527–534. [Google Scholar]
- Anderson, B.; Rafferty, A.P.; Lyon-Callo, S. Fast-food consumption and obesity among Michigan adults. Prev. Chronic. Dis 2011, 8, A71. [Google Scholar]
- Bezerra, I.N.; Curioni, C.; Sichieri, R. Association between eating out of home and body weight. Nutr. Rev 2012, 70, 65–79. [Google Scholar]
- Charlton, M.; Krishnan, A.; Viker, K. Fast food diet mouse: Novel small animal model of NASH with ballooning, progressive fibrosis, and high physiological fidelity to the human condition. Am. J. Physiol. Gastrointest. Liver Physiol 2011, 301, G825–G834. [Google Scholar]
- Tsuchiya, H.; Ebata, Y.; Sakabe, T. High-fat, high-fructose diet induces hepatic iron overload via a hepcidin-independent mechanism prior to the onset of liver steatosis and insulin resistance in mice. Metabolism 2013, 62, 62–69. [Google Scholar]
- Hashmi, S.; Wang, Y.; Parhar, R.S. A C. elegans model to study human metabolic regulation. Nutr. Metab. [Lond. ] 2013, 10, 31. [Google Scholar]
- Farrell, G.C.; van, R.D. Liver cholesterol: Is it playing possum in NASH? Am. J. Physiol. Gastrointest. Liver Physiol 2012, 303, G9–G11. [Google Scholar]
- Lee, L.; Alloosh, M.; Saxena, R. Nutritional model of steatohepatitis and metabolic syndrome in the Ossabaw miniature swine. Hepatology 2009, 50, 56–67. [Google Scholar]
- Lant, B.; Storey, K.B. An overview of stress response and hypometabolic strategies in Caenorhabditis elegans: Conserved and contrasting signals with the mammalian system. Int. J. Biol. Sci 2010, 6, 9–50. [Google Scholar]
- Chan, J.; Sharkey, F.E.; Kushwaha, R.S. Steatohepatitis in laboratory opossums exhibiting a high lipemic response to dietary cholesterol and fat. Am. J. Physiol. Gastrointest. Liver Physiol 2012, 303, G12–G19. [Google Scholar]
- Puri, P.; Baillie, R.A.; Wiest, M.M. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology 2007, 46, 1081–1090. [Google Scholar]
- Dyson, M.C.; Alloosh, M.; Vuchetich, J.P. Components of metabolic syndrome and coronary artery disease in female Ossabaw swine fed excess atherogenic diet. Comp. Med 2006, 56, 35–45. [Google Scholar]
- Schattenberg, J.M.; Galle, P.R. Animal models of non-alcoholic steatohepatitis: Of mice and man. Dig. Dis 2010, 28, 247–254. [Google Scholar]
- Gomez-Lechon, M.J.; Donato, M.T.; Castell, J.V. Human hepatocytes in primary culture: The choice to investigate drug metabolism in man. Curr. Drug Metab 2004, 5, 443–462. [Google Scholar]
- Dambach, D.M.; Andrews, B.A.; Moulin, F. New technologies and screening strategies for hepatotoxicity: Use of in vitro models. Toxicol. Pathol 2005, 33, 17–26. [Google Scholar]
- Chavez-Tapia, N.C.; Rosso, N.; Tiribelli, C. In vitro models for the study of non-alcoholic fatty liver disease. Curr. Med. Chem 2011, 18, 1079–1084. [Google Scholar]
- Thakur, V.; Pritchard, M.T.; McMullen, M.R. Adiponectin normalizes LPS-stimulated TNF-alpha production by rat Kupffer cells after chronic ethanol feeding. Am. J. Physiol. Gastrointest. Liver Physiol 2006, 290, G998–G1007. [Google Scholar]
- Mandal, P.; Roychowdhury, S.; Park, P.H. Adiponectin and heme oxygenase-1 suppress TLR4/MyD88-independent signaling in rat Kupffer cells and in mice after chronic ethanol exposure. J. Immunol 2010, 185, 4928–4937. [Google Scholar]
- Shi, L.; Kishore, R.; McMullen, M.R. Chronic ethanol increases lipopolysaccharide-stimulated Egr-1 expression in RAW 264.7 macrophages: Contribution to enhanced tumor necrosis factor alpha production. J. Biol. Chem 2002, 277, 14777–14785. [Google Scholar]
- Kishore, R.; McMullen, M.R.; Cocuzzi, E. Lipopolysaccharide-mediated signal transduction: Stabilization of TNF-alpha mRNA contributes to increased lipopolysaccharide-stimulated TNF-alpha production by Kupffer cells after chronic ethanol feeding. Comp. Hepatol 2004, 3, S31. [Google Scholar]
- Thuy, S.; Ladurner, R.; Volynets, V. Nonalcoholic fatty liver disease in humans is associated with increased plasma endotoxin and plasminogen activator inhibitor 1 concentrations and with fructose intake. J. Nutr 2008, 138, 1452–1455. [Google Scholar]
- Volynets, V.; Kuper, M.A.; Strahl, S. Nutrition, intestinal permeability, and blood ethanol levels are altered in patients with nonalcoholic fatty liver disease [NAFLD]. Dig. Dis. Sci 2012, 57, 1932–1941. [Google Scholar]
- Harte, A.L.; da Silva, N.F.; Creely, S.J. Elevated endotoxin levels in non-alcoholic fatty liver disease. J. Inflamm 2010, 7, 15. [Google Scholar]
- Spruss, A.; Kanuri, G.; Uebel, K. Role of the inducible nitric oxide synthase in the onset of fructose-induced steatosis in mice. Antioxid. Redox Signal 2011, 14, 2121–2135. [Google Scholar]
- Fiegel, H.C.; Kneser, U.; Kluth, D. Hepatic tissue engineering. Handchir. Mikrochir. Plast Chir 2010, 42, 337–341. [Google Scholar]
- Janorkar, A.V.; Harris, L.M.; Murphey, B.S. Use of three-dimensional spheroids of hepatocyte-derived reporter cells to study the effects of intracellular fat accumulation and subsequent cytokine exposure. Biotechnol. Bioeng 2011, 108, 1171–1180. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ingredients | g/100 g diet | Ingredients | g/100 g diet |
|---|---|---|---|
| Casein | 20.0 | Vitamin mixture | 1 |
| DL methionine | 0.3 | Choline bitartrate | 0.2 |
| Corn strach | 15.0 | Corn oil | 5 |
| Sucrose | 48.7 | Sodium cholate | 0.3 |
| Cellulose powder | 5 | Cholesterol | 1 |
| Mineral mixture | 3.5 |
| Control diet | % of energy from Nutrients | Western style diet | % of energy from Nutrients |
|---|---|---|---|
| Total sugars | 23.3 | Fructose | 50 |
| Starch | 39 | Glucose | 5 |
| Protein | 11 | Starch | 5 |
| Fat | 24 | Protein | 15 |
| Cholesterol | 0.2 | ||
| Fat | 25 |
| In vitro models | Cell lines | Pros | Cons |
|---|---|---|---|
| Primary cell cultures | Hepatocytes from NAFLD patients/rodents Kupffer cells/stellate cells/iNKT cells from human patients/rodents | Mimic in vivo settings | Isolation problems Ethical issues Varying reproducibility in experiments Limited culture time |
| Immortalized cell lines | RAW 264.7, AML-12, J774A, HepG2, HuH7, H4IIE, H4IIEC3, PAV-1, LX2 | Continuous growth Easy to culture Stable phenotype | Expression of several enzymes and nuclear factors alter according to the immortalization method |
| Co-culture models | RAW 264.7 and AML-12 Human hepatocytes and adipocytes | Mimic in vivo liver architecture Important tools in cellular cross talk studies | Difficult to cultivate |
| 3D cultures | H35 rat hepatoma cell line | Mimic in vivo liver architecture Liver specific differentiation and function Tools for transcriptional regulation studies | Difficult to cultivate |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kanuri, G.; Bergheim, I. In Vitro and in Vivo Models of Non-Alcoholic Fatty Liver Disease (NAFLD). Int. J. Mol. Sci. 2013, 14, 11963-11980. https://doi.org/10.3390/ijms140611963
Kanuri G, Bergheim I. In Vitro and in Vivo Models of Non-Alcoholic Fatty Liver Disease (NAFLD). International Journal of Molecular Sciences. 2013; 14(6):11963-11980. https://doi.org/10.3390/ijms140611963
Chicago/Turabian StyleKanuri, Giridhar, and Ina Bergheim. 2013. "In Vitro and in Vivo Models of Non-Alcoholic Fatty Liver Disease (NAFLD)" International Journal of Molecular Sciences 14, no. 6: 11963-11980. https://doi.org/10.3390/ijms140611963