Design, Synthesis, Biological Activity and Molecular Dynamics Studies of Specific Protein Tyrosine Phosphatase 1B Inhibitors over SHP-2

Abstract
:1. Introduction
2. Results and Discussion
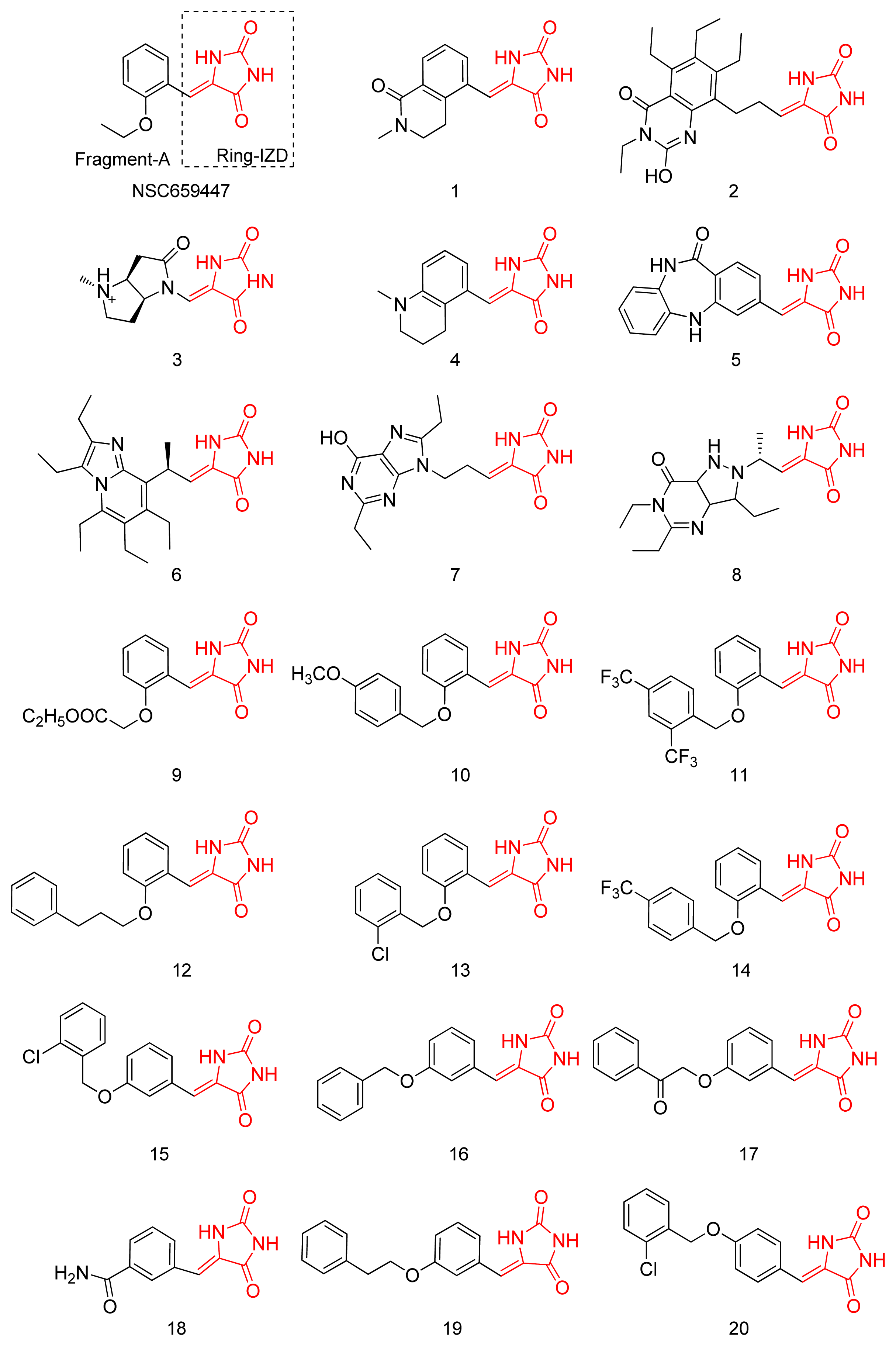
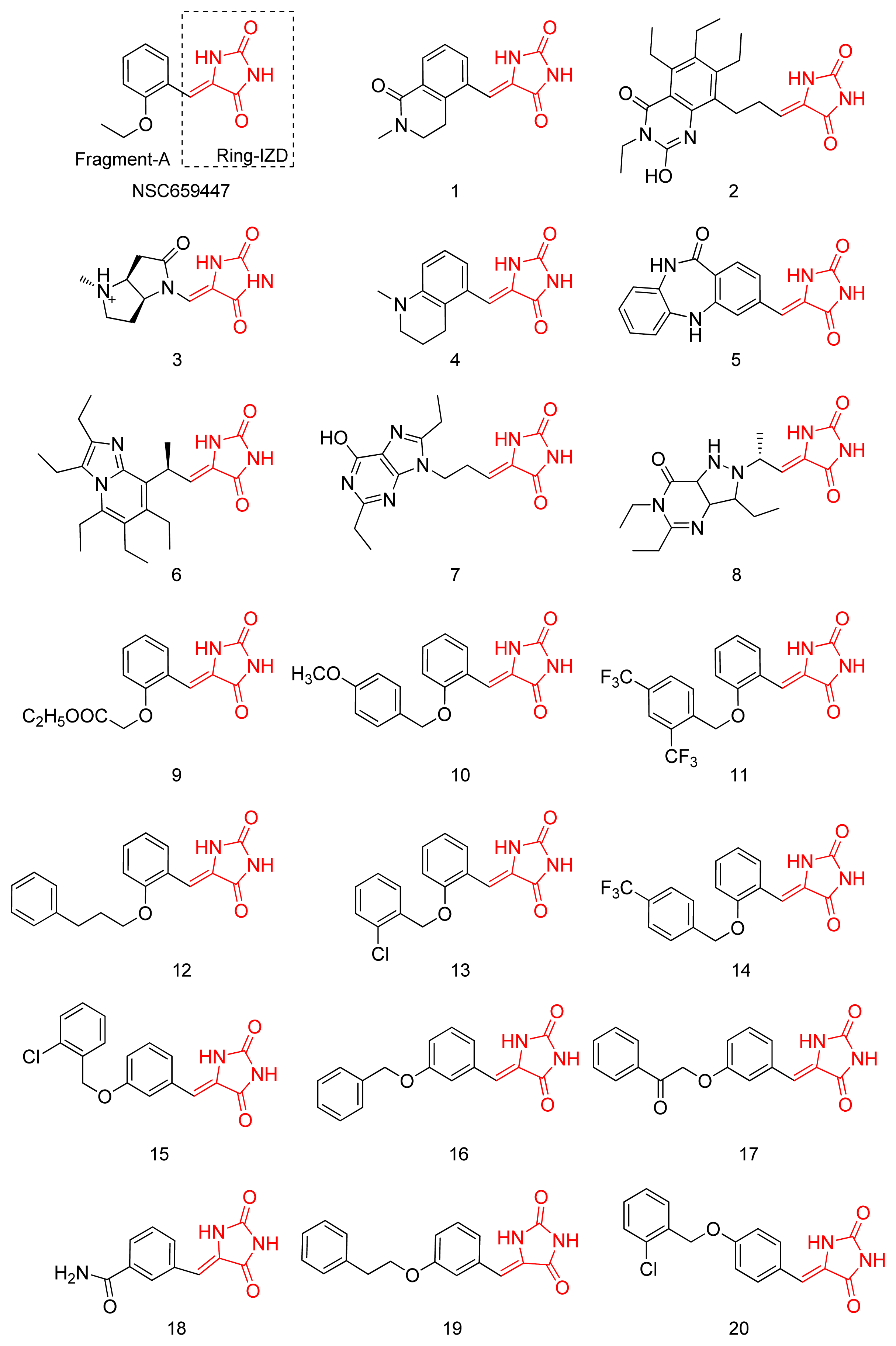
2.1. Virtual Screening and Core-Hopping
2.2. Synthesis
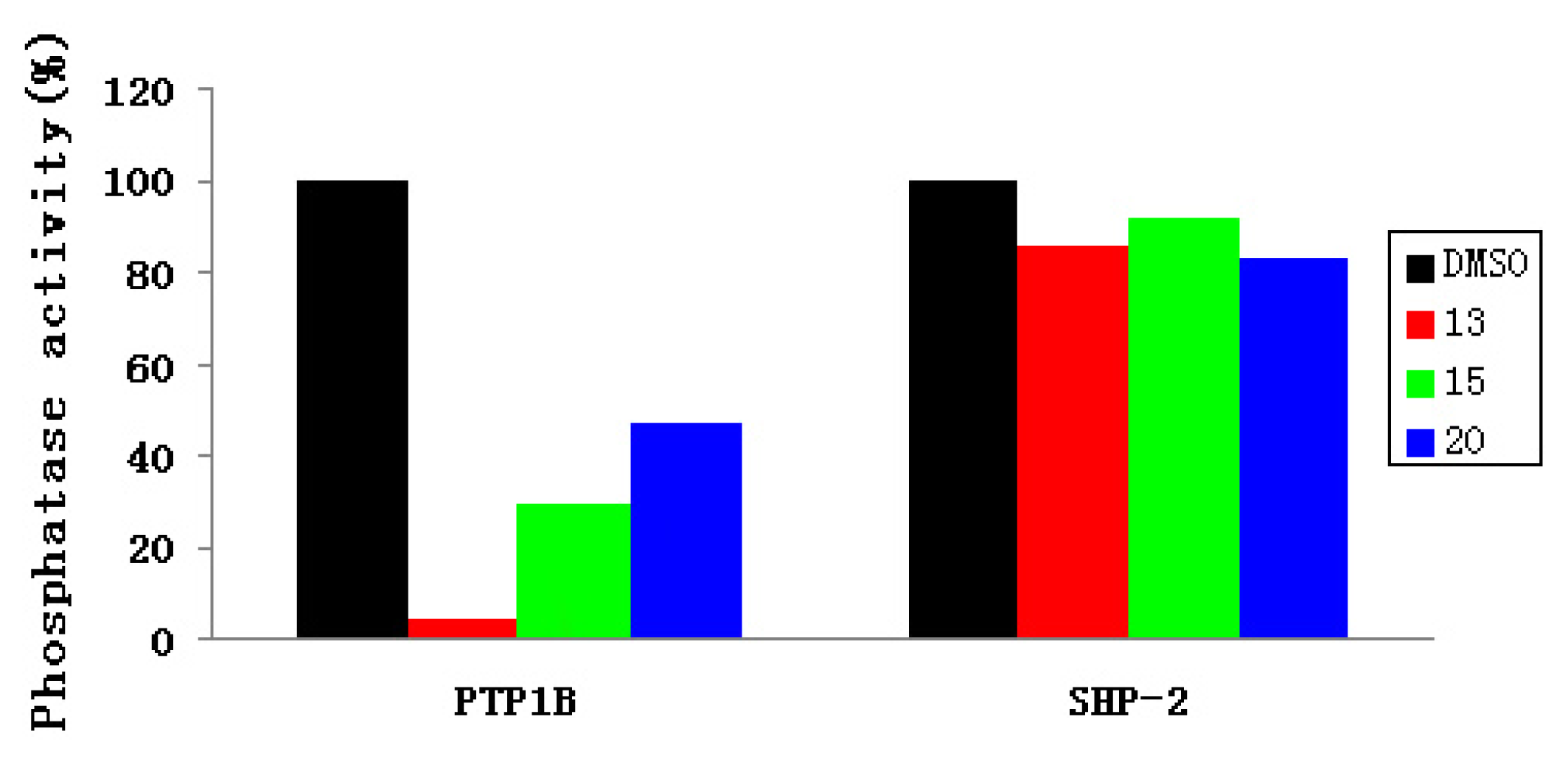
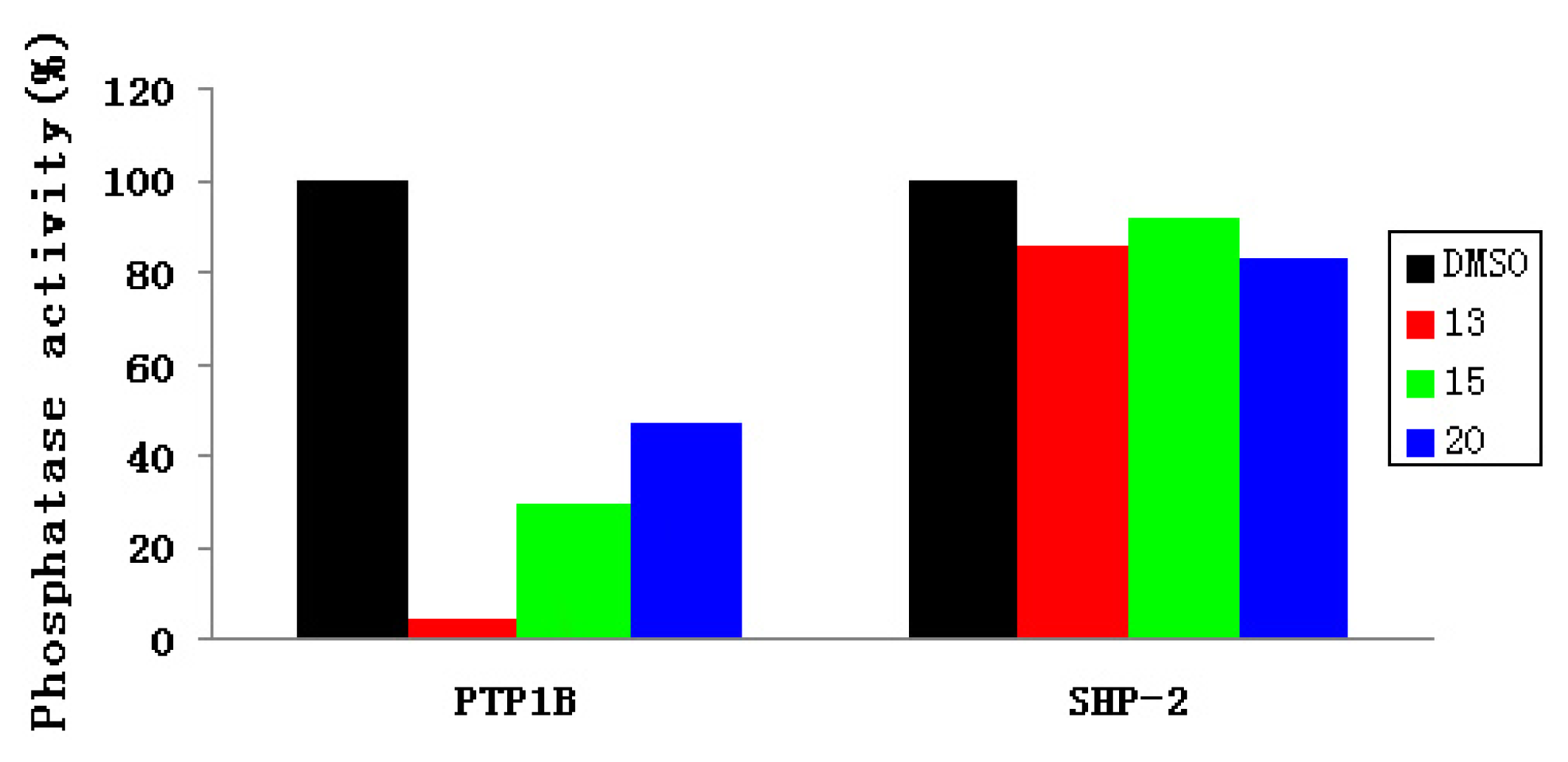
2.3. Experimental Assay
3. Molecular Dynamics Simulations
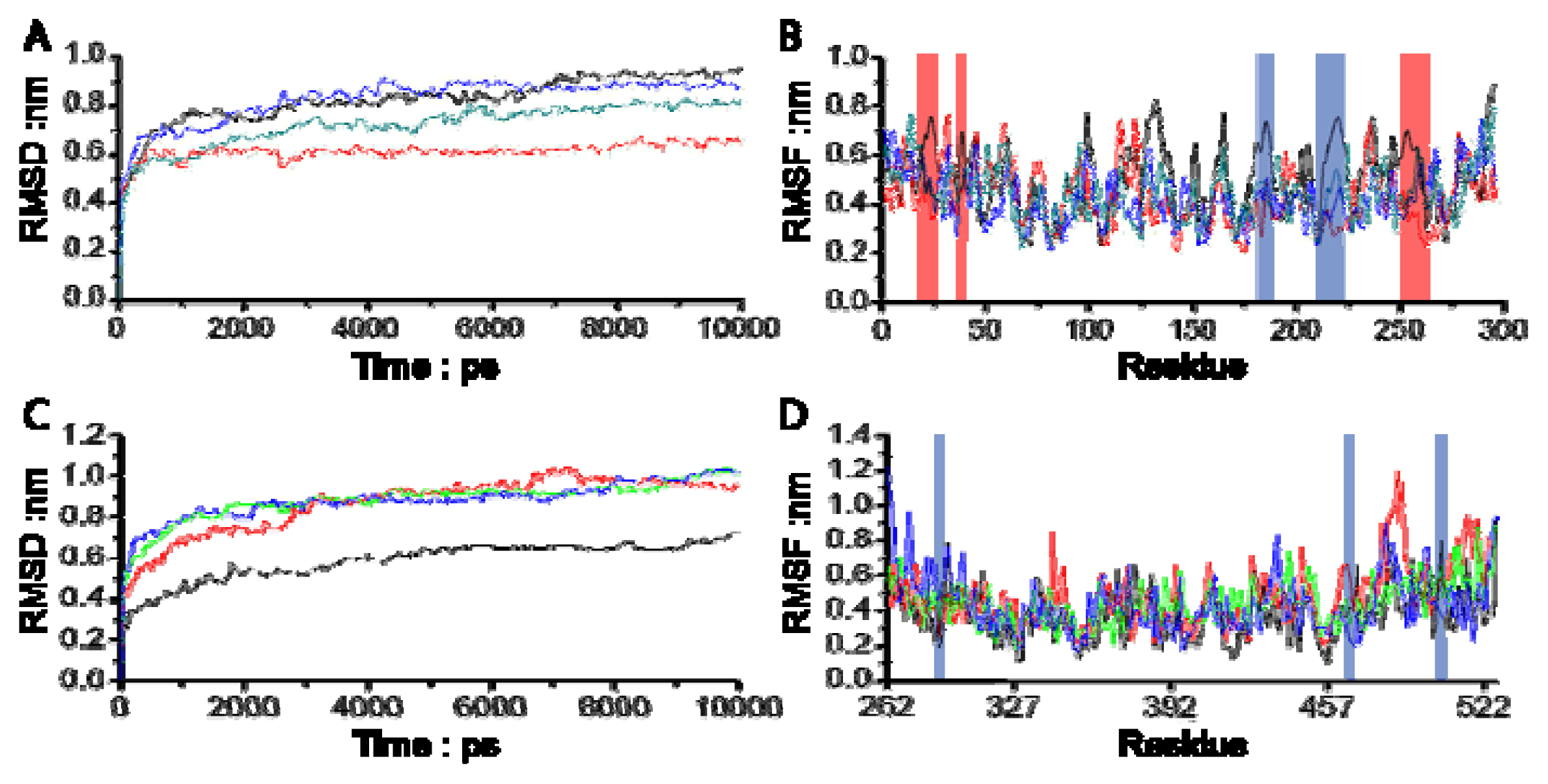
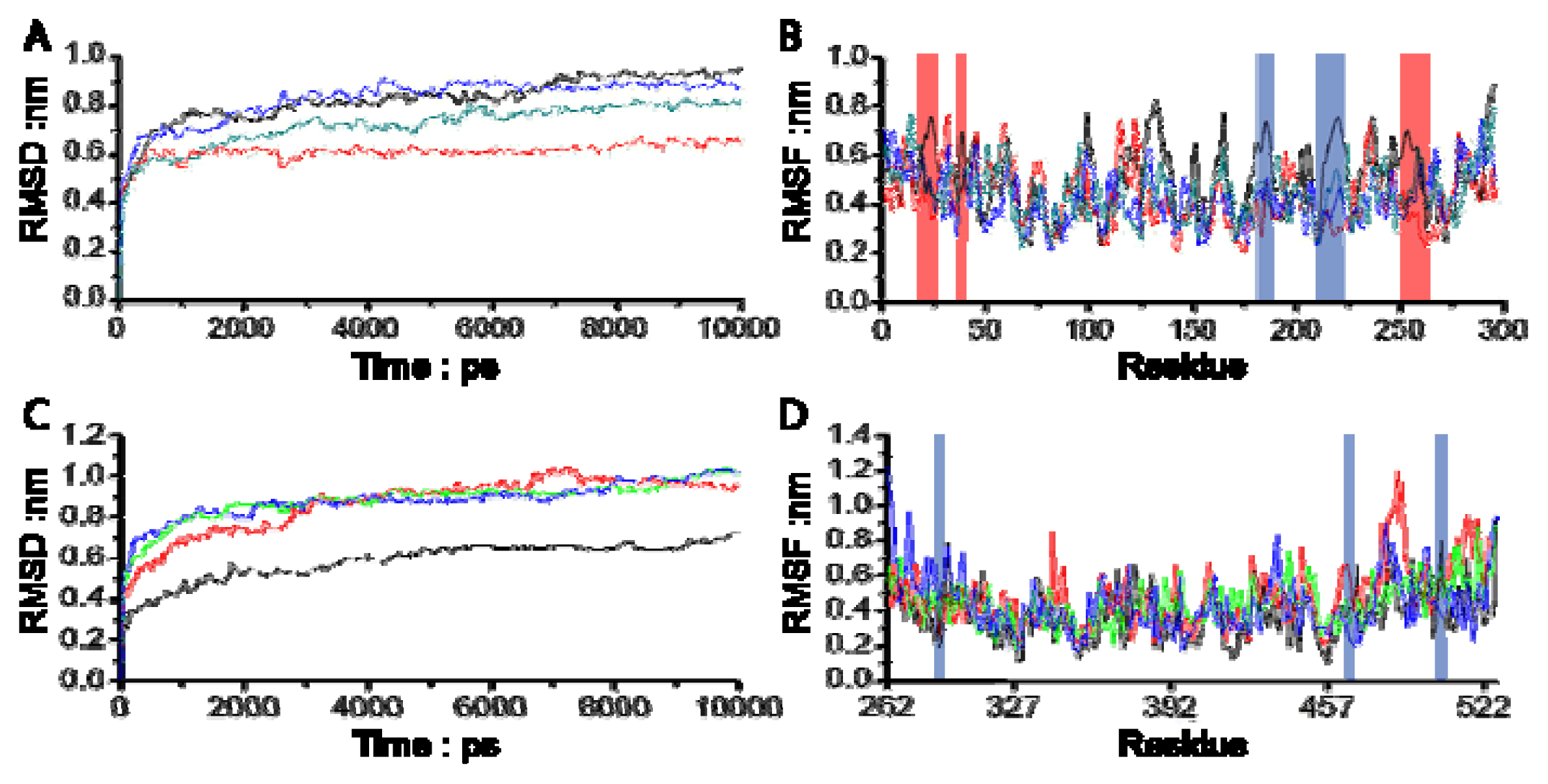
3.1. Molecular Dynamics Trajectory Analysis
3.2. Combinational Ways and Energy Analysis during the Simulations
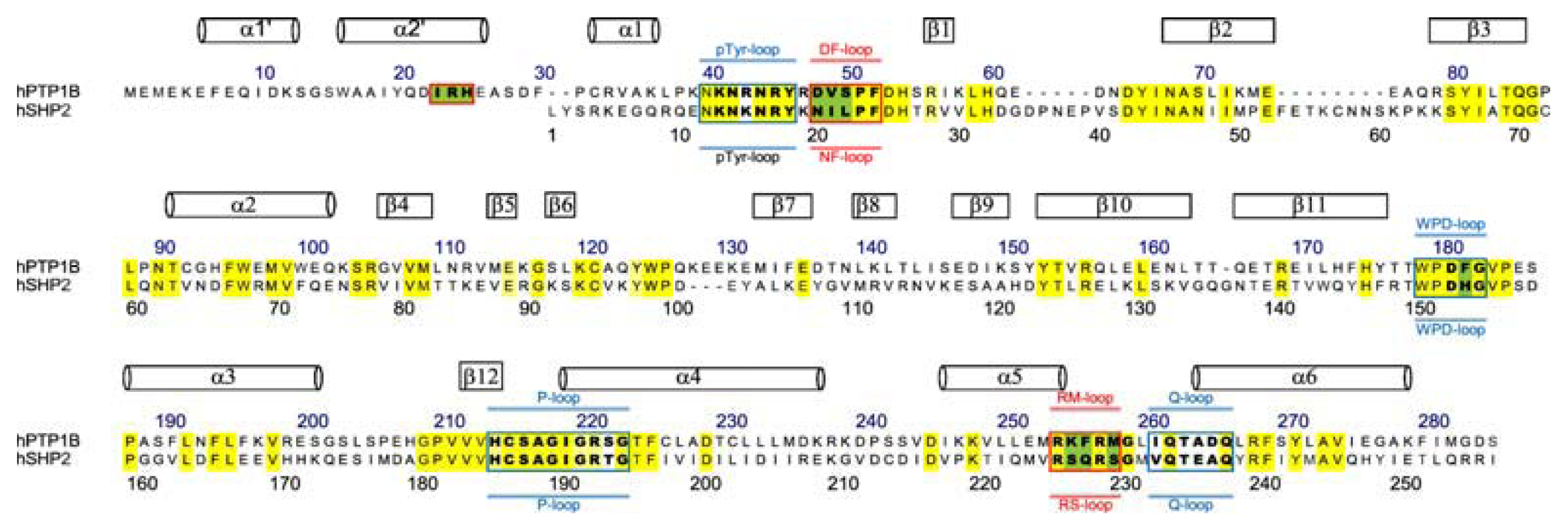
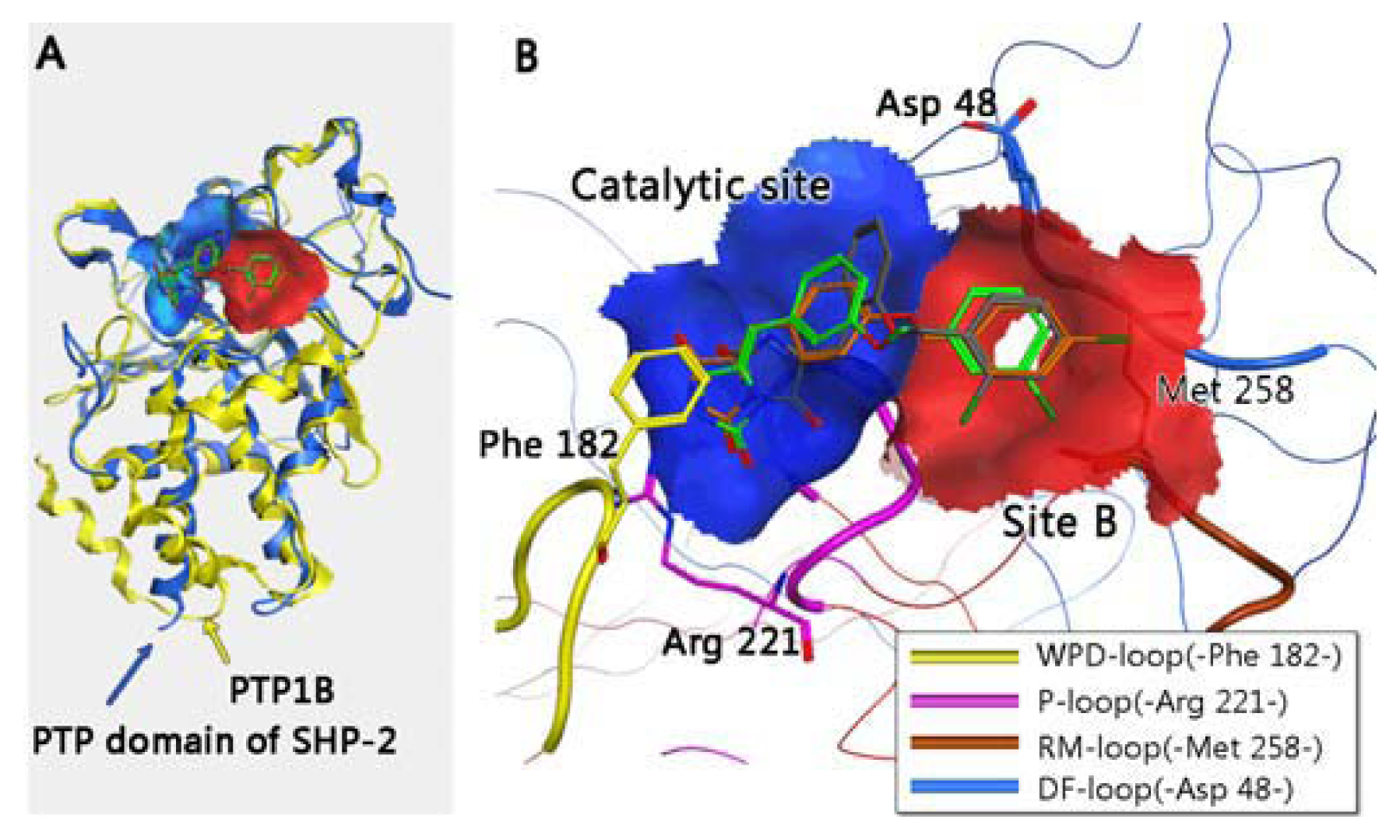
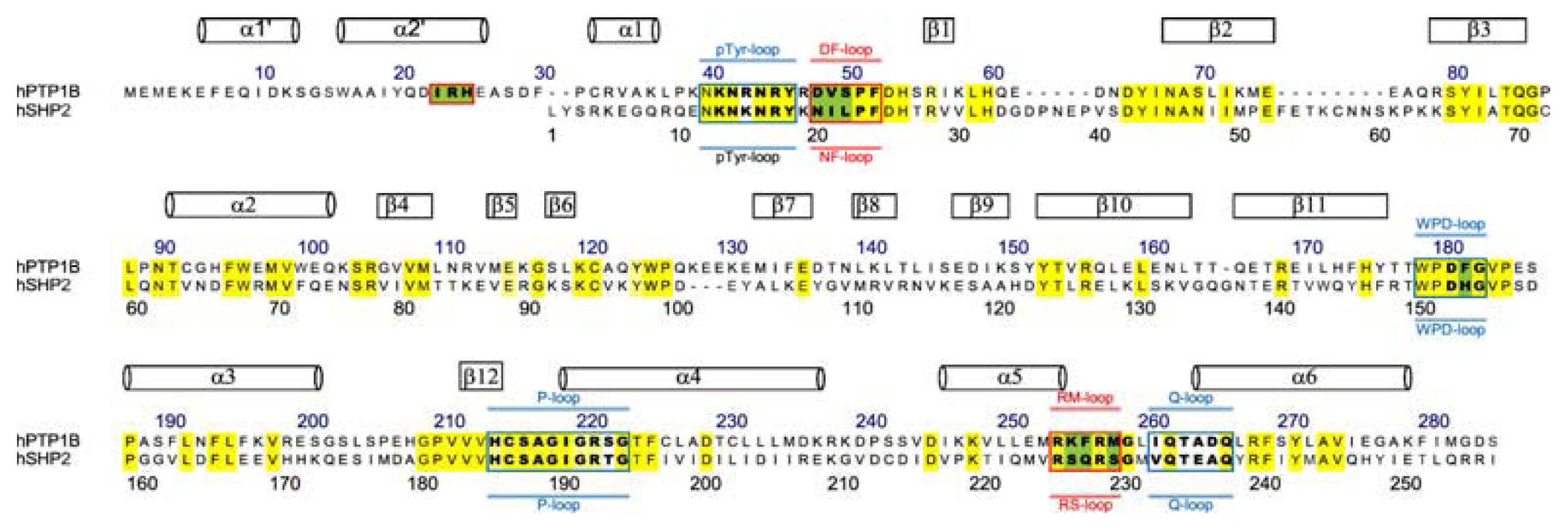
3.3. Residues Involved in the Interaction between Ligand and Key Residues
4. Material and Methods
4.1. Starting Structures
4.2. Virtual Screening and Core-Hopping Procedure on Silicon
5. Conclusions
Supplementary Information
ijms-14-12661-s001.pdfAcknowledgments
Conflict of Interest
References
- Zhang, S.; Zhang, Z.Y. PTP1B as a drug target: Recent developments in PTP1B inhibitor discovery. Drug Discov. Today 2007, 12, 373–381. [Google Scholar]
- Scapin, G.; Patel, S.B.; Becker, J.W.; Wang, Q.; Desponts, C.; Waddleton, D.; Skorey, K.; Cromlish, W.; Bayly, C.; Therien, M.; et al. The structural basis for the selectivity of benzotriazole inhibitors of PTP1B. Biochemistry 2003, 42, 11451–11459. [Google Scholar]
- Bentires-Alj, M.; Paez, J.G.; David, F.S.; Keilhack, H.; Halmos, B.; Naoki, K.; Maris, J.M.; Richardson, A.; Bardelli, A.; Sugarbaker, D.J.; et al. Activating mutations of the noonan syndrome-associated SHP2/PTPN11 gene in human solid tumors and adult acute myelogenous leukemia. Cancer Res 2004, 64, 8816–8820. [Google Scholar]
- Malamas, M.S.; Sredy, J.; Gunawan, I.; Mihan, B.; Sawicki, D.R.; Seestaller, L.; Sullivan, D.; Flam, B.R. New azolidinediones as inhibitors of protein tyrosine phosphatase 1B with antihyperglycemic properties. J. Med. Chem 2000, 43, 995–1010. [Google Scholar]
- Zhou, M.; Ji, M. Molecular docking and 3D-QSAR on 2-(oxalylamino) benzoic acid and its analogues as protein tyrosine phosphatase 1B inhibitors. Bioorganic Med. Chem. Lett 2005, 15, 5521–5525. [Google Scholar]
- Zhang, W.; Hong, D.; Zhou, Y.; Zhang, Y.; Shen, Q.; Li, J.Y.; Hu, L.H.; Li, J. Ursolic acid and its derivative inhibit protein tyrosine phosphatase 1B, enhancing insulin receptor phosphorylation and stimulating glucose uptake. Biochim. Biophys. Acta 2006, 1760, 1505–1512. [Google Scholar]
- Maccari, R.; Paoli, P.; Ottana, R.; Jacomelli, M.; Ciurleo, R.; Manao, G.; Steindl, T.; Langer, T.; Vigorita, M.G.; Camici, G. 5-Arylidene-2,4-thiazolidinediones as inhibitors of protein tyrosine phosphatases. Bioorganic Med. Chem 2007, 15, 5137–5149. [Google Scholar]
- Zinker, B.A.; Rondinone, C.M.; Trevillyan, J.M.; Gum, R.J.; Clampit, J.E.; Waring, J.F.; Xie, N.; Wilcox, D.; Jacobson, P.; Frost, L.; et al. PTP1B antisense oligonucleotide lowers PTP1B protein, normalizes blood glucose, and improves insulin sensitivity in diabetic mice. Proc. Natl. Acad. Sci. USA 2002, 99, 11357–11362. [Google Scholar]
- Loh, M.L.; Vattikuti, S.; Schubbert, S.; Reynolds, M.G.; Carlson, E.; Lieuw, K.H.; Cheng, J.W.; Lee, C.M.; Stokoe, D.; Bonifas, J.M.; et al. Mutations in PTPN11 implicate the SHP-2 phosphatase in leukemogenesis. Blood 2004, 103, 2325–2331. [Google Scholar]
- Tartaglia, M.; Mehler, E.L.; Goldberg, R.; Zampino, G.; Brunner, H.G.; Kremer, H.; van der Burgt, I.; Crosby, A.H.; Ion, A.; Jeffery, S.; et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat. Genet 2001, 29, 465–468. [Google Scholar]
- Hellmuth, K.; Grosskopf, S.; Lum, C.T.; Wurtele, M.; Roder, N.; von Kries, J.P.; Rosario, M.; Rademann, J.; Birchmeier, W. Specific inhibitors of the protein tyrosine phosphatase Shp2 identified by high-throughput docking. Proc. Natl. Acad. Sci. USA 2008, 105, 7275–7280. [Google Scholar]
- Andersen, J.N.; Mortensen, O.H.; Peters, G.H.; Drake, P.G.; Iversen, L.F.; Olsen, O.H.; Jansen, P.G.; Andersen, H.S.; Tonks, N.K.; Moller, N.P. Structural and evolutionary relationships among protein tyrosine phosphatase domains. Mol. Cell. Biol 2001, 21, 7117–7136. [Google Scholar]
- Bhattarai, B.R.; Kafle, B.; Hwang, J.S.; Khadka, D.; Lee, S.M.; Kang, J.S.; Ham, S.W.; Han, I.O.; Park, H.; Cho, H. Thiazolidinedione derivatives as PTP1B inhibitors with antihyperglycemic and antiobesity effects. Bioorganic Med. Chem. Lett 2009, 19, 6161–6165. [Google Scholar]
- Vella, A.; de Groen, P.C.; Dinneen, S.F. Fatal hepatotoxicity associated with troglitazone. Ann. Intern. Med 1998, 129, 1080. [Google Scholar]
- Schwartz, S.; Raskin, P.; Fonseca, V.; Graveline, J.F. Effect of troglitazone in insulin-treated patients with type II diabetes mellitus. Troglitazone and Exogenous Insulin Study Group. N. Engl. J. Med 1998, 338, 861–866. [Google Scholar]
- Pei, Z.; Li, X.; Liu, G.; Abad-Zapatero, C.; Lubben, T.; Zhang, T.; Ballaron, S.J.; Hutchins, C.W.; Trevillyan, J.M.; Jirousek, M.R. Discovery and SAR of novel, potent and selective protein tyrosine phosphatase 1B inhibitors. Bioorganic Med. Chem. Lett 2003, 13, 3129–3132. [Google Scholar]
- Puius, Y.A.; Zhao, Y.; Sullivan, M.; Lawrence, D.S.; Almo, S.C.; Zhang, Z.Y. Identification of a second aryl phosphate-binding site in protein-tyrosine phosphatase 1B: A paradigm for inhibitor design. Proc. Natl. Acad. Sci. USA 1997, 94, 13420–13425. [Google Scholar]
- Voigt, J.H.; Bienfait, B.; Wang, S.; Nicklaus, M.C. Comparison of the NCI open database with seven large chemical structural databases. J. Chem. Inf. Comput. Sci 2001, 41, 702–712. [Google Scholar]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1.Method and assessment of docking accuracy. J. Med. Chem 2004, 47, 177–182. [Google Scholar]
- Billek, G. P-hydroxyphenylpyruvic Acid. Org. Synth 1973, 5, 627–629. [Google Scholar]
- Wang, R.L.; Li, W.M.; Liu, M.Y.; Xu, W.R. Synthesis and biological activity evaluation of novel imidazolidinedione derivatives, as potent antidiabetic agent. J. Chin. Chem. Soc 2009, 56, 34–39. [Google Scholar]
- Yu, W.M.; Guvench, O.; Mackerell, A.D.; Qu, C.K. Identification of small molecular weight inhibitors of Src homology 2 domain-containing tyrosine phosphatase 2 (SHP-2) via in silico database screening combined with experimental assay. J. Med. Chem 2008, 51, 7396–7404. [Google Scholar]
- Oostenbrink, C.; Soares, T.A.; van der Vegt, N.F.; van Gunsteren, W.F. Validation of the 53A6 GROMOS forcefield. Eur. Biophys. J 2005, 34, 273–284. [Google Scholar]
- Schuttelkopf, A.W.; van Aalten, D. PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr 2004, 60, 1355–1363. [Google Scholar]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et al. The protein data bank. Acta Crystallogr. D Biol. Crystallogr 2002, 58, 899–907. [Google Scholar]
- Wang, J.F.; Gong, K.; Wei, D.Q.; Li, Y.X.; Chou, K.C. Molecular dynamics studies on the interactions of PTP1B with inhibitors: From the first phosphate-binding site to the second one. Protein Eng. Des. Sel 2009, 22, 349–355. [Google Scholar]
- Barford, D.; Flint, A.J.; Tonks, N.K. Crystal structure of human protein tyrosine phosphatase 1B. Science 1994, 263, 1397–1404. [Google Scholar]
- Qu, C.K. Role of the SHP-2 tyrosine phosphatase in cytokine-induced signaling and cellular response. Biochim. Biophys. Acta 2002, 1592, 297–301. [Google Scholar]
- Barr, A.J.; Ugochukwu, E.; Lee, W.H.; King, O.N.; Filippakopoulos, P.; Alfano, I.; Savitsky, P.; Burgess-Brown, N.A.; Muller, S.; Knapp, S. Large-scale structural analysis of the classical human protein tyrosine phosphatome. Cell 2009, 136, 352–363. [Google Scholar]
- Zhang, X.; He, Y.; Liu, S.; Yu, Z.; Jiang, Z.X.; Yang, Z.; Dong, Y.; Nabinger, S.C.; Wu, L.; Gunawan, A.M.; et al. Salicylic acid based small molecule inhibitor for the oncogenic Src homology-2 domain containing protein tyrosine phosphatase-2 (SHP2). J. Med. Chem 2010, 53, 2482–2493. [Google Scholar]
- Irwin, J.J.; Shoichet, B.K. ZINC—A free database of commercially available compounds for virtual screening. J. Chem. Inf. Model 2005, 45, 177–182. [Google Scholar]
- Li, X.B.; Wang, S.Q.; Xu, W.R.; Wang, R.L.; Chou, K.C. Novel inhibitor design for hemagglutinin against H1N1 influenza virus by core hopping method. PLoS One 2011, 6, e28111. [Google Scholar]
- Banks, J.L.; Beard, H.S.; Cao, Y.; Cho, A.E.; Damm, W.; Farid, R.; Felts, A.K.; Halgren, T.A.; Mainz, D.T.; Maple, J.R.; et al. Integrated modeling program, applied chemical theory (IMPACT). J. Comput. Chem 2005, 26, 1752–1780. [Google Scholar]
- Eldridge, M.D.; Murray, C.W.; Auton, T.R.; Paolini, G.V.; Mee, R.P. Empirical scoring functions: I the development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J. Comput. Aided Mol. Des 1997, 11, 425–445. [Google Scholar]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach fo rrapid, accurate docking and scoring. II. Enrichment factors in database screening. J. Med. Chem 2004, 47, 1750–1759. [Google Scholar]
- Ma, Y.; Wang, S.Q.; Xu, W.R.; Wang, R.L.; Chou, K.C. Design novel dual agonists for treating type-2 diabetes by targeting peroxisome proliferator-activated receptors with core hopping approach. PLoS One 2012, 7, e38546. [Google Scholar]
- Zhang, L.S.; Wang, S.Q.; Xu, W.R.; Wang, R.L.; Wang, J.F. Scaffold-based pan-agonist design for the PPARalpha, PPARbeta and PPARgamma receptors. PLoS One 2012, 7, e48453. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | EPTP1B | ESHP-2 | ΔE |
|---|---|---|---|
| 13 | −46.76 | −34.41 | 12.35 |
| 20 | −42.37 | −32.98 | 9.38 |
| 6 | −41.03 | −32.30 | 8.73 |
| 15 | −40.83 | −29.03 | 11.80 |
| 14 | −40.75 | −37.02 | 3.73 |
| 7 | −40.75 | −36.33 | 4.42 |
| 16 | −40.36 | −31.62 | 8.74 |
| 19 | −40.24 | −33.79 | 6.44 |
| 5 | −39.99 | −40.23 | - |
| 17 | −39.50 | −32.89 | 6.61 |
| 2 | −38.57 | −39.03 | - |
| 11 | −38.35 | −34.17 | 4.18 |
| 8 | −37.89 | −31.22 | 6.67 |
| 12 | −35.78 | −27.98 | 7.80 |
| 10 | −36.79 | −38.20 | - |
| 3 | −36.43 | −29.90 | 6.52 |
| 9 | −35.57 | −36.96 | - |
| 4 | −33.76 | −34.78 | - |
| 1 | −33.03 | −34.02 | - |
| 18 | −32.78 | −28.22 | 4.56 |
| NSC659447 | −33.34 | −25.19 | 8.15 |
| IC50 (μM) | 13 | 15 | 20 |
|---|---|---|---|
| PTP1B | 37.4 | 57.3 | 63.9 |
| SHP-2 | 152 | 152 | >152 |
| PTP1B | Catalytic site of SHP-2 | ||||||
|---|---|---|---|---|---|---|---|
| RMSF c (nm) | DLB d (nm) | Catalytic site (Kcal/mol) | Site B (Kcal/mol) | RMSF c (nm) | Catalytic Site (kcal/mol) | Site B (kcal/mol) | |
| 13 | 0.07 | 0.70 ± 0.11 | −39.30 ± 2.80 | −34.30 ± 2.14 | 0.64 | −43.73 ± 5.18 | −8.3 ± 2.17 |
| 15 | 0.49 | 1.10 ± 0.28 | −42.00 ± 6.39 | −20.74 ± 4.51 | 0.85 | −45.97 ± 6.39 | −3.5 ± 1.76 |
| 20 | 0.33 | 0.86 ± 0.30 | 40.70 ± 7.92 | −24.67 ± 5.12 | 0.67 | −42.27 ± 4.92 | −4.9 ± 2.66 |
| PTP1B | Catalytic Site | Site B | ||||
|---|---|---|---|---|---|---|
| -Phe182- | P-loop e | Q-loop f | -Arg 24- | DF-loop g | RM-loop h | |
| 13 | −4.25 | −37.84 | −4.65 | −5.84 | −13.23 | −2.35 |
| 15 | −5.13 | −29.88 | −3.53 | −4.66 | −9.10 | −2.32 |
| 20 | −4.49 | −30.36 | −5.28 | −1.50 | −9.29 | −3.50 |
| SHP-2 | Catalytic Site | Site B | |||
|---|---|---|---|---|---|
| -His154- | P-loop i | Q-loop j | NF-loop k | RS-loop l | |
| 13 | −3.50 | −25.32 | −3.35 | −3.18 | −1.71 |
| 15 | −1.27 | −19.62 | −7.01 | −1.25 | −0.11 |
| 20 | −0.78 | −23.84 | −6.98 | −1.07 | −0.28 |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sun, S.-X.; Li, X.-B.; Liu, W.-B.; Ma, Y.; Wang, R.-L.; Cheng, X.-C.; Wang, S.-Q.; Liu, W. Design, Synthesis, Biological Activity and Molecular Dynamics Studies of Specific Protein Tyrosine Phosphatase 1B Inhibitors over SHP-2. Int. J. Mol. Sci. 2013, 14, 12661-12674. https://doi.org/10.3390/ijms140612661
Sun S-X, Li X-B, Liu W-B, Ma Y, Wang R-L, Cheng X-C, Wang S-Q, Liu W. Design, Synthesis, Biological Activity and Molecular Dynamics Studies of Specific Protein Tyrosine Phosphatase 1B Inhibitors over SHP-2. International Journal of Molecular Sciences. 2013; 14(6):12661-12674. https://doi.org/10.3390/ijms140612661
Chicago/Turabian StyleSun, Su-Xia, Xiao-Bo Li, Wen-Bo Liu, Ying Ma, Run-Ling Wang, Xian-Chao Cheng, Shu-Qing Wang, and Wei Liu. 2013. "Design, Synthesis, Biological Activity and Molecular Dynamics Studies of Specific Protein Tyrosine Phosphatase 1B Inhibitors over SHP-2" International Journal of Molecular Sciences 14, no. 6: 12661-12674. https://doi.org/10.3390/ijms140612661