Protective Effects of Hydrogen Sulfide in Hypoxic Human Umbilical Vein Endothelial Cells: A Possible Mitochondria-Dependent Pathway

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
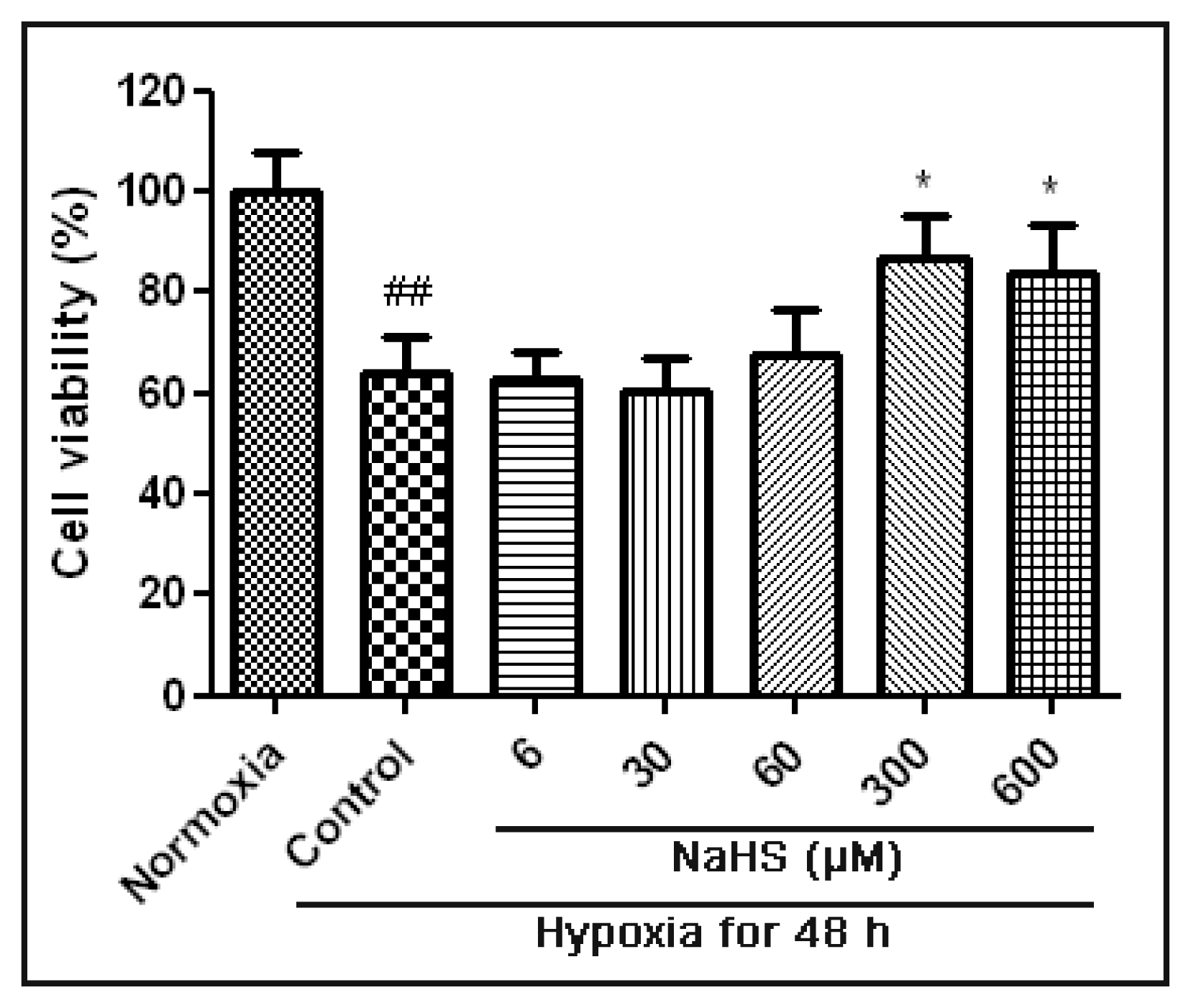
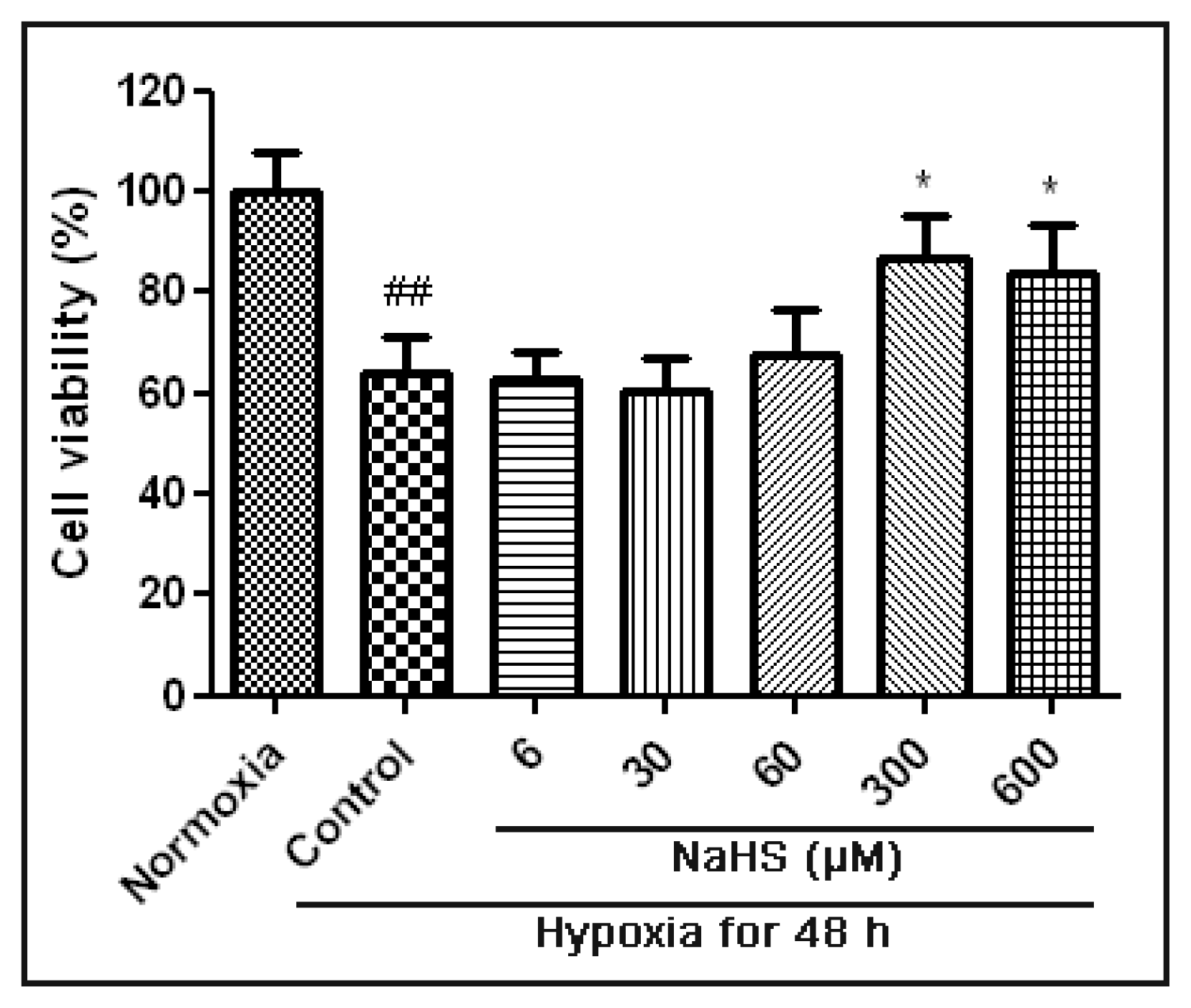
2.1. H2S Protected HUVECs under Hypoxic Conditions
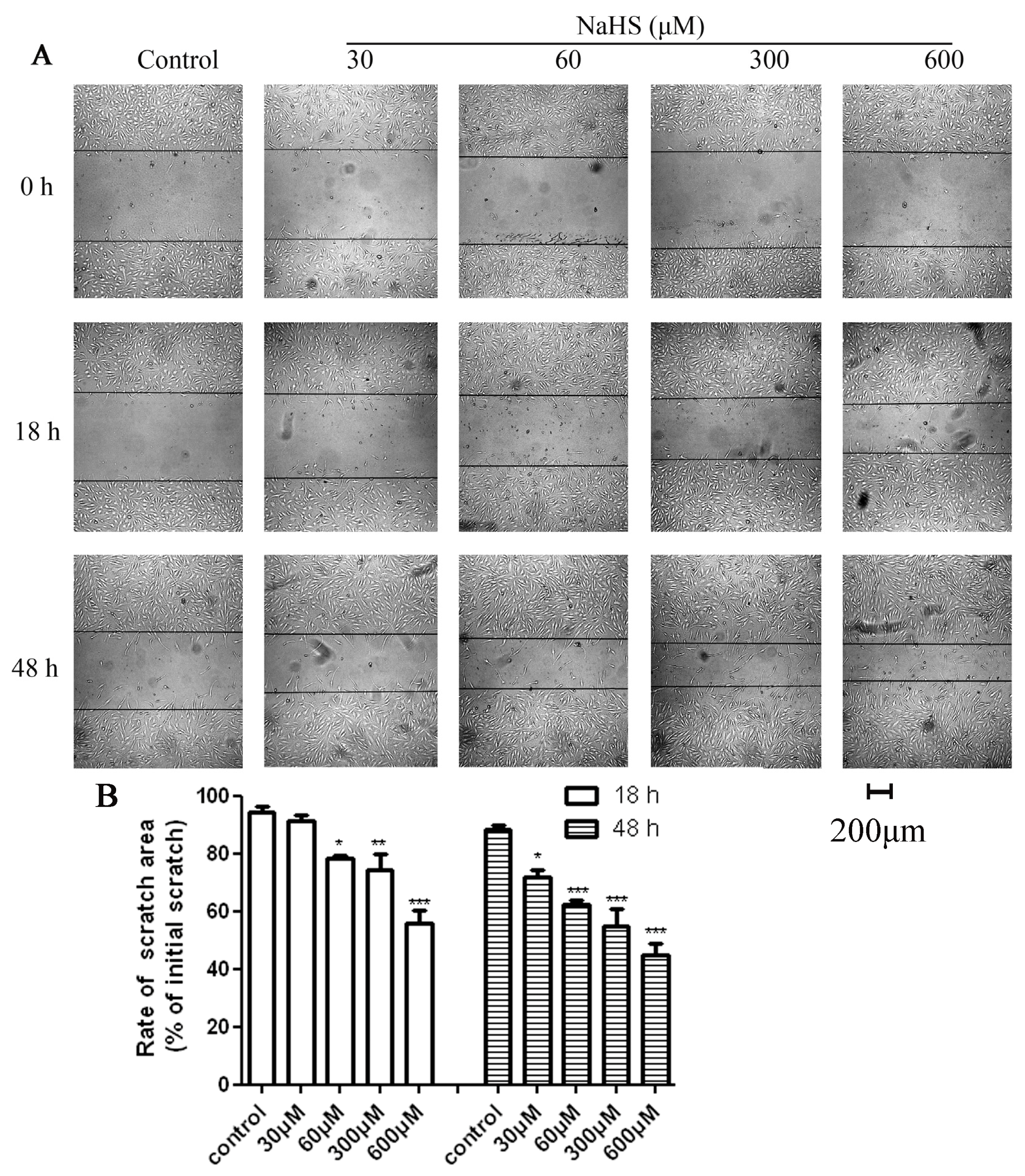
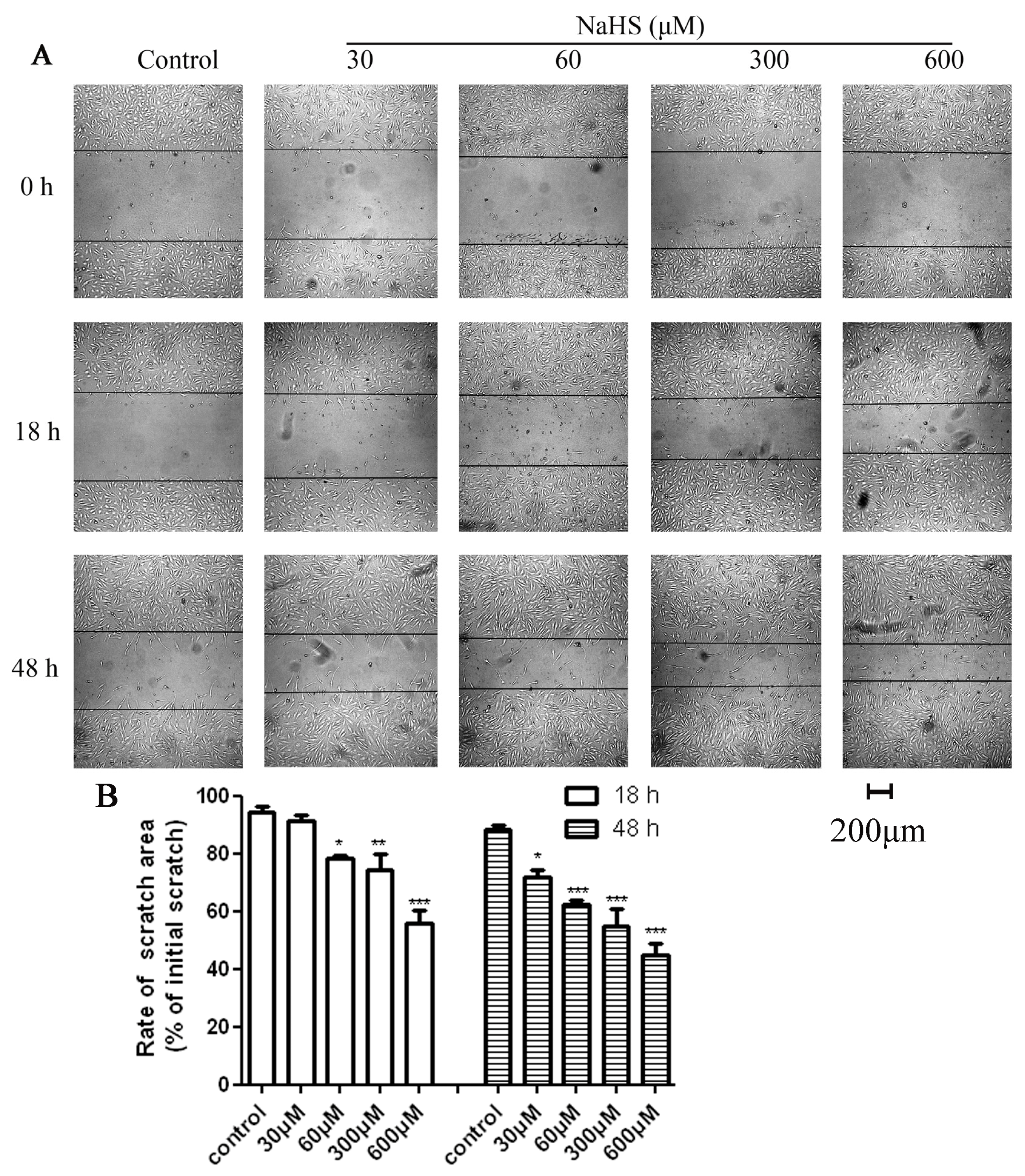
2.2. H2S Accelerated HUVECs Migration and Repaired the Scratch Damage under Hypoxic Condition
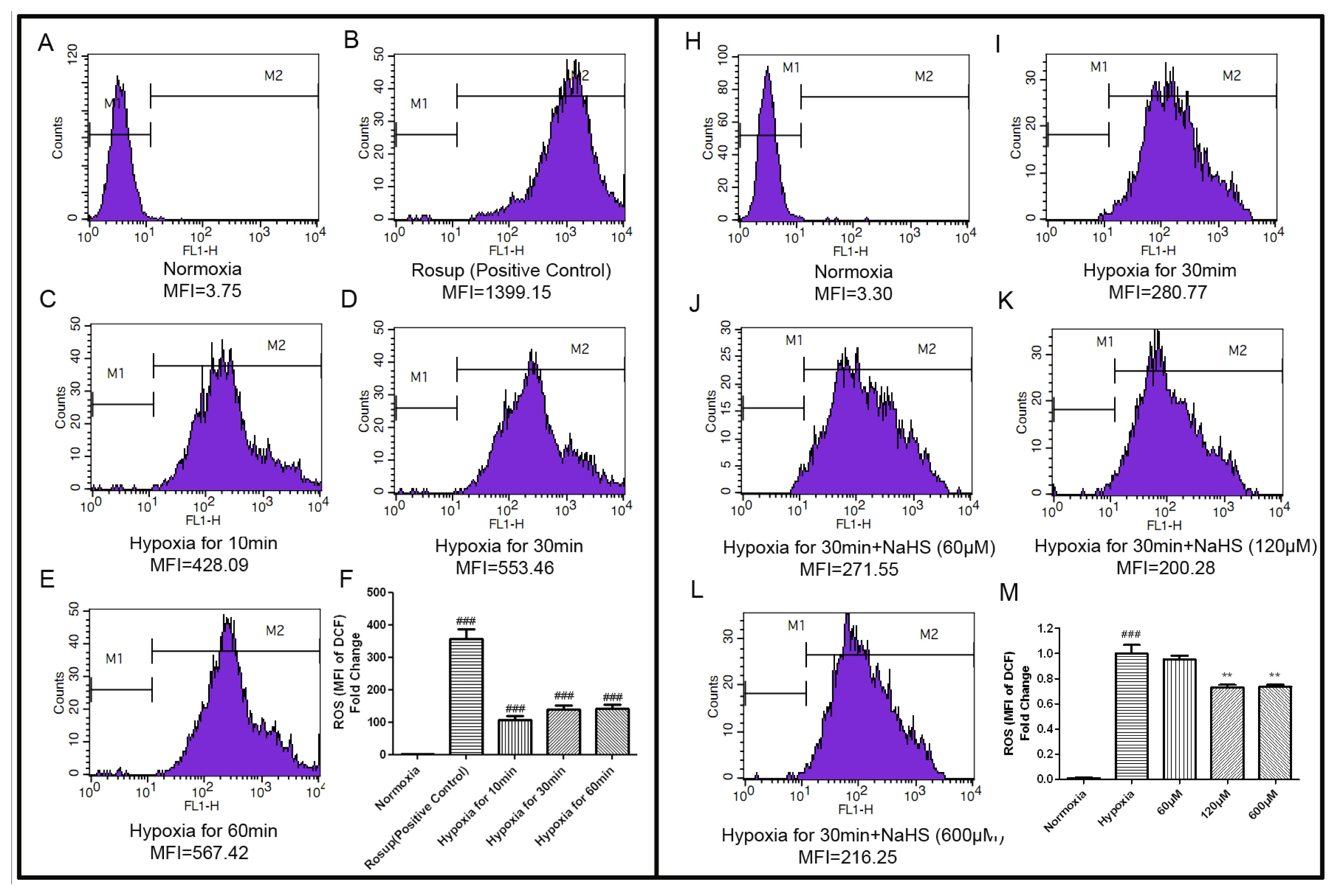
2.3. H2S Decreased the Concentration of Intracellular ROS
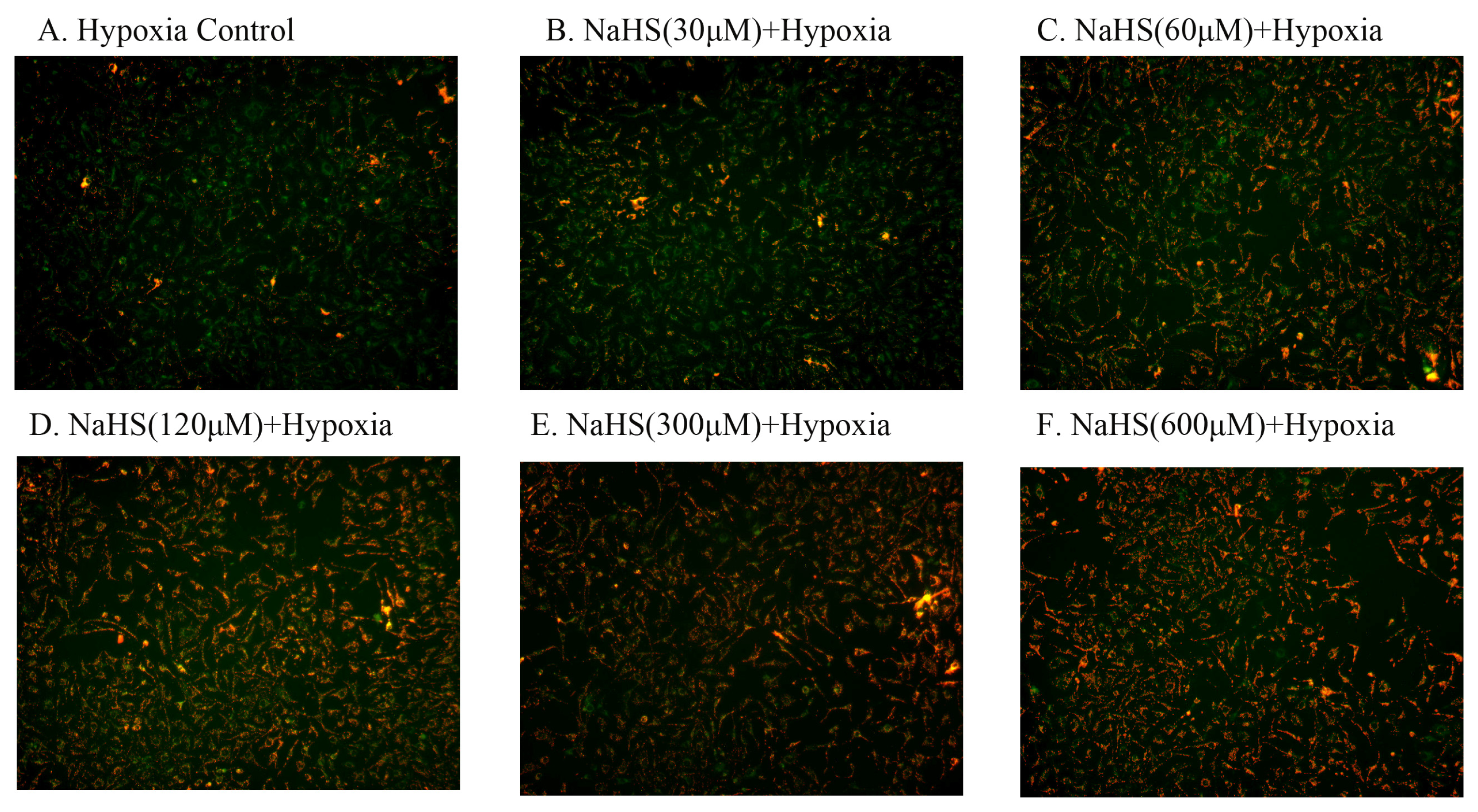
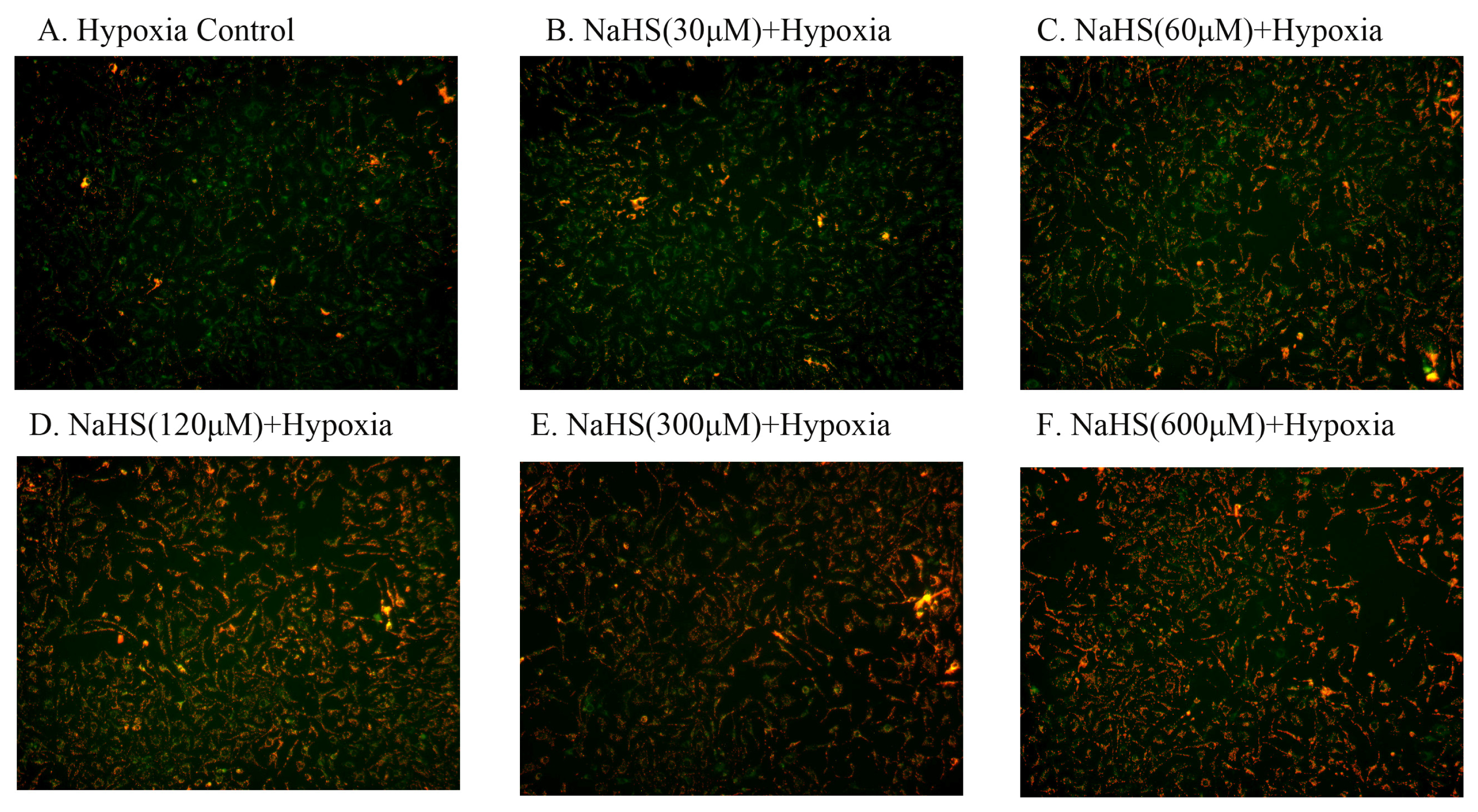
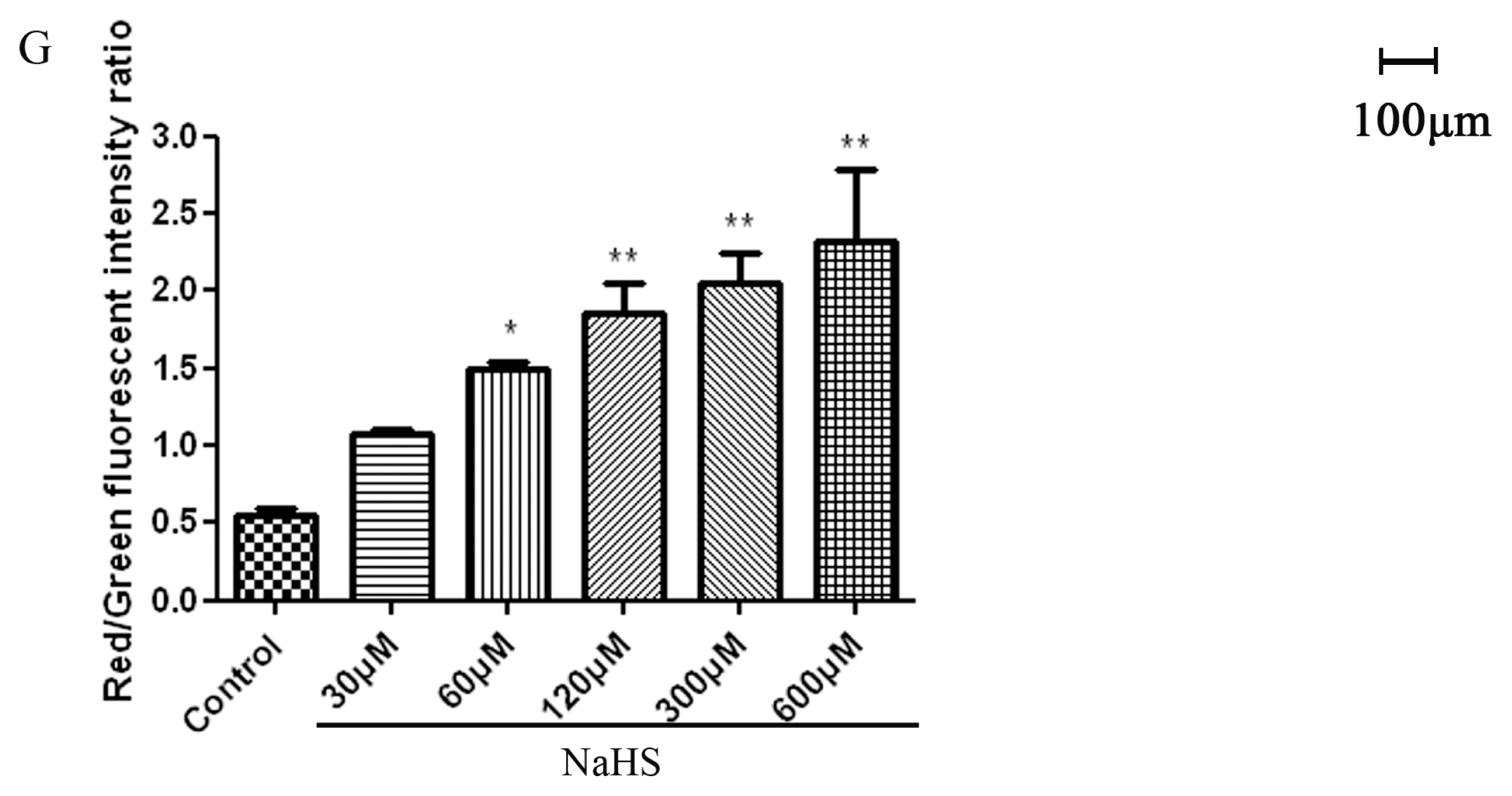
2.4. H2S Protected Mitochondrial Damage
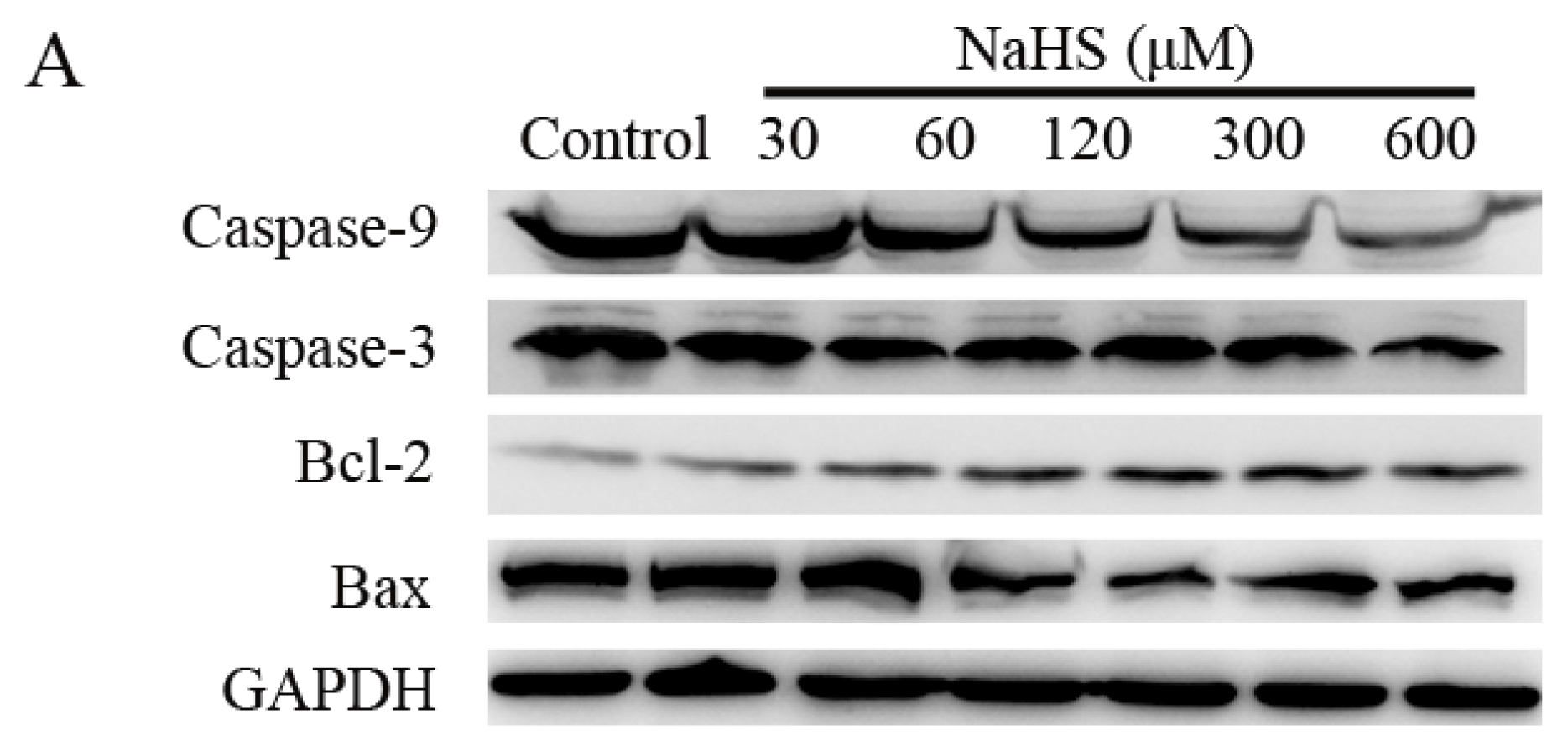
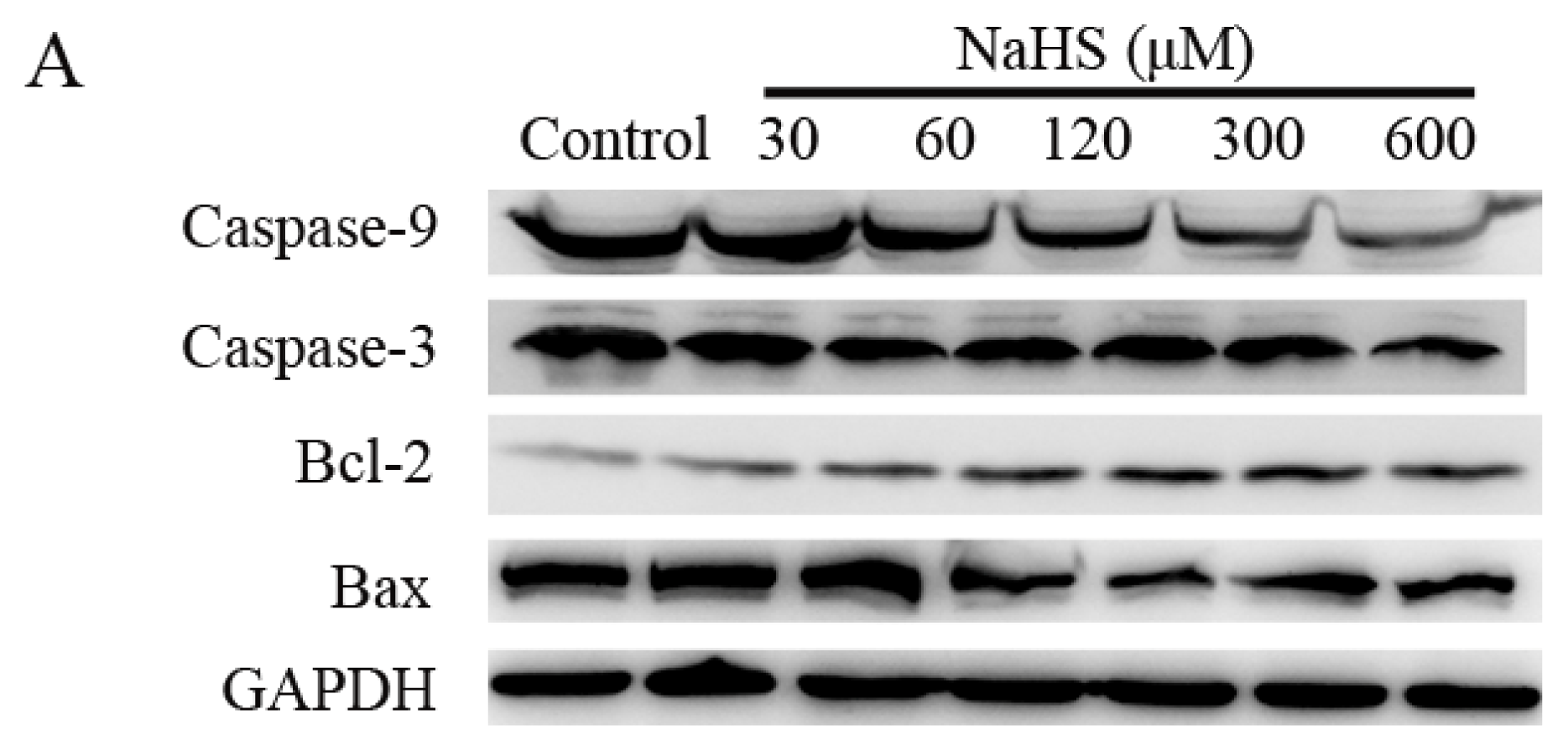
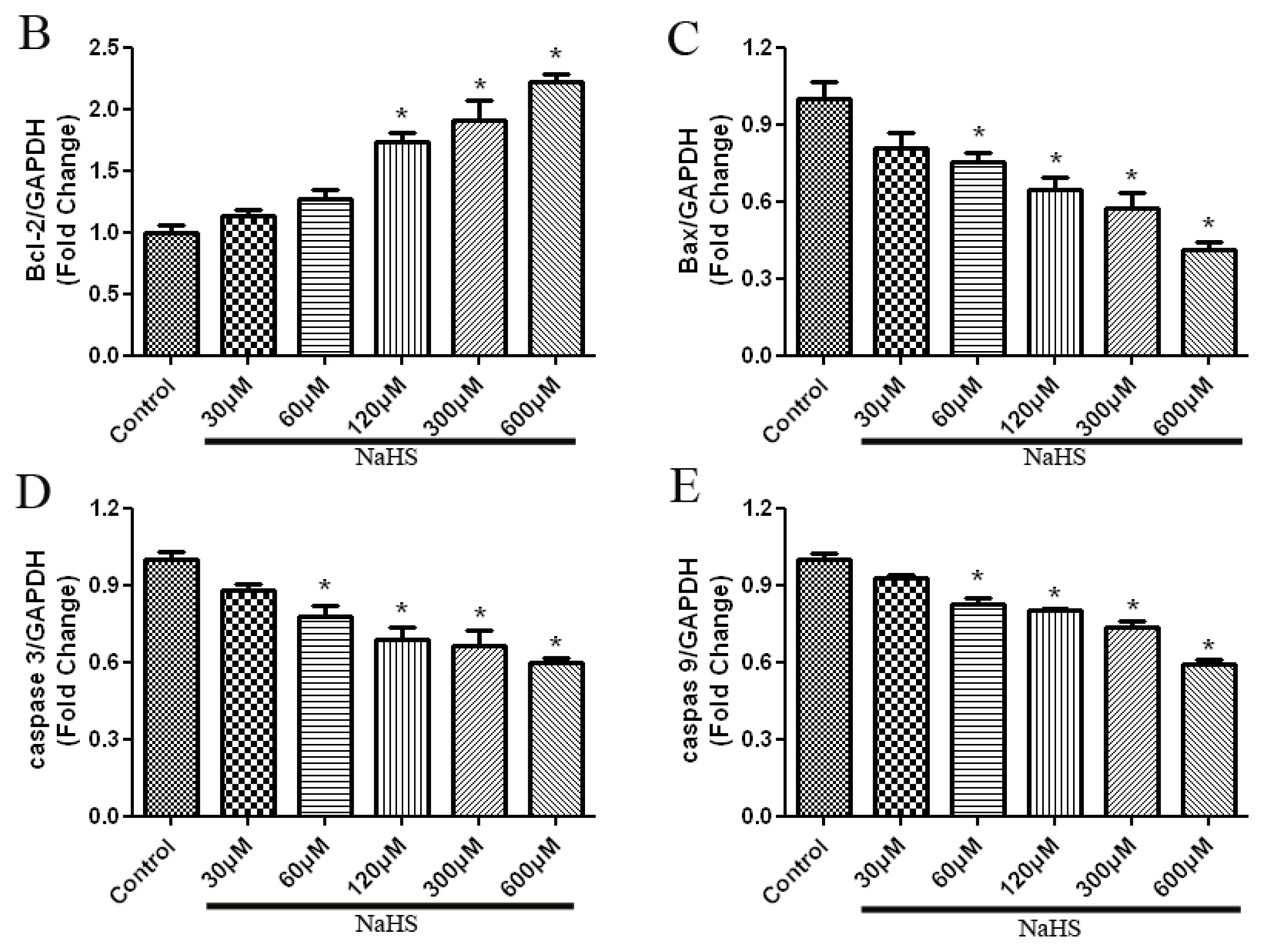
2.5. H2S Increased Anti-Apoptotic Protein Expression, and Decreased Pro-Apoptotic Protein Expression in a Dose-Dependent Way
2.6. Discussion
3. Experimental Section
3.1. Chemicals
3.2. Cell Culture
3.3. MTT Assay
3.4. Scratch Wound Healing Assay
3.5. Assay of Reactive Oxygen Species (ROS) Generation
3.6. Mitochondrial Membrane Potential Detection
3.7. Western Blot Analysis
3.8. Statistical Analysis
4. Conclusions
Acknowledgments
Abbreviations
| H2S | Hydrogen Sulfide |
| NaHS | Sodium Hydrosulfide |
| HUVEC | Human Umbilical Vein Endothelial Cell |
| ROS | Reactive Oxygen Species |
| M | mol/L |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| JC-1 | 5,5′,6,6′-Tetrachloro-1,1′,3,3′-tetraethyl-imidacarbocyanine iodide |
| LTP | Long-Term Potentiation |
| NMDA receptor | N-methyl-d-aspartate receptor |
| CRH | Corticotropin Releasing Hormone |
| ΔΨm | Mitochondrial Membrane Potential. DCFH-DA, 2′,7′-dichlorofluorescin diacetate |
| DCF | 2′,7′-dichlorofluorescein |
| MFI | Mean Fluorescent Intensity. |
Conflict of Interest
References
- Kimura, Y.; Goto, Y.-I.; Kimura, H. Hydrogen sulfide increases glutathione production and suppresses oxidative stress in mitochondria. Antioxid. Redox Signal 2010, 12, 1–13. [Google Scholar]
- Fu, M.; Zhang, W.; Wu, L.; Yang, G.; Li, H.; Wang, R. Hydrogen sulfide (H2S) metabolism in mitochondria and its regulatory role in energy production. Proc. Natl. Acad. Sci. USA 2012, 109, 2943–2948. [Google Scholar]
- Szabó, C. Hydrogen sulphide and its therapeutic potential. Nat. Rev. Drug Discov 2007, 6, 917–935. [Google Scholar]
- Kajimura, M.; Fukuda, R.; Bateman, R.M.; Yamamoto, T.; Suematsu, M. Interactions of multiple gas-transducing systems: Hallmarks and uncertainties of CO, NO, and H2S gas biology. Antioxid. Redox Signal 2010, 13, 157–192. [Google Scholar]
- Goodwin, L.R.; Francom, D.; Dieken, F.P.; Taylor, J.D.; Warenycia, M.W.; Reiffenstein, R.; Dowling, G. Determination of sulfide in brain tissue by gas dialysis/ion chromatography: Postmortem studies and two case reports. J. Anal. Toxicol 1989, 13, 105–109. [Google Scholar]
- Abe, K.; Kimura, H. The possible role of hydrogen sulfide as an endogenous neuromodulator. J. Neurosci 1996, 16, 1066–1071. [Google Scholar]
- Russo, D. Evidence that hydrogen sulphide can modulate hypothalamo-pituitary-adrenal axis function: In vitro and in vivo studies in the rat. J. Neuroendocrinol 2001, 12, 225–233. [Google Scholar]
- Zhao, W.; Zhang, J.; Lu, Y.; Wang, R. The vasorelaxant effect of H2S as a novel endogenous gaseous KATP channel opener. EMBO J 2001, 20, 6008–6016. [Google Scholar]
- Ali, M.Y.; Whiteman, M.; Low, C.M.; Moore, P.K. Hydrogen sulphide reduces insulin secretion from HIT-T15 cells by a KATP channel-dependent pathway. J. Endocrinol 2007, 195, 105–112. [Google Scholar]
- Wu, L.; Yang, W.; Jia, X.; Yang, G.; Duridanova, D.; Cao, K.; Wang, R. Pancreatic islet overproduction of H2S and suppressed insulin release in Zucker diabetic rats. Lab. Invest 2008, 89, 59–67. [Google Scholar]
- Yang, W.; Yang, G.; Jia, X.; Wu, L.; Wang, R. Activation of KATP channels by H2S in rat insulin-secreting cells and the underlying mechanisms. J. Phys 2005, 569, 519–531. [Google Scholar]
- Zhang, H.; Moochhala, S.M.; Bhatia, M. Endogenous hydrogen sulfide regulates inflammatory response by activating the ERK pathway in polymicrobial sepsis. J. Immunol 2008, 181, 4320–4331. [Google Scholar]
- Hu, L.F.; Wong, P.T.H.; Moore, P.K.; Bian, J.S. Hydrogen sulfide attenuates lipopolysaccharide-induced inflammation by inhibition of p38 mitogen-activated protein kinase in microglia. J. Neurochem 2007, 100, 1121–1128. [Google Scholar]
- Zanardo, R.C.O.; Brancaleone, V.; Distrutti, E.; Fiorucci, S.; Cirino, G.; Wallace, J.L. Hydrogen sulfide is an endogenous modulator of leukocyte-mediated inflammation. FASEB J 2006, 20, 2118–2120. [Google Scholar]
- Apel, K.; Hirt, H. Reactive oxygen species: Metabolism, oxidative stress, and signal transduction. Annu. Rev. Plant Biol 2004, 55, 373–399. [Google Scholar]
- Simon, H.U.; Haj-Yehia, A.; Levi-Schaffer, F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 2000, 5, 415–418. [Google Scholar]
- Yang, G.; Wu, L.; Jiang, B.; Yang, W.; Qi, J.; Cao, K.; Meng, Q.; Mustafa, A.K.; Mu, W.; Zhang, S.; et al. H2S as a physiologic vasorelaxant: Hypertension in mice with deletion of cystathionine-Lyase. Science 2008, 322, 587–590. [Google Scholar]
- Kimura, H. Hydrogen sulfide: Its production, release and functions. Amino Acids 2011, 41, 113–121. [Google Scholar]
- Elrod, J.W.; Calvert, J.W.; Morrison, J.; Doeller, J.E.; Kraus, D.W.; Tao, L.; Jiao, X.; Scalia, R.; Kiss, L.; Szabo, C. Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc. Natl. Acad. Sci. USA 2007, 104, 15560–15565. [Google Scholar]
- Papapetropoulos, A.; Pyriochou, A.; Altaany, Z.; Yang, G.; Marazioti, A.; Zhou, Z.; Jeschke, M.G.; Branski, L.K.; Herndon, D.N.; Wang, R. Hydrogen sulfide is an endogenous stimulator of angiogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 21972–21977. [Google Scholar]
- Wang, M.J.; Cai, W.J.; Li, N.; Ding, Y.J.; Chen, Y.; Zhu, Y.C. The hydrogen sulfide donor NaHS promotes angiogenesis in a rat model of hind limb ischemia. Antioxid. Redox Signal 2010, 12, 1065–1077. [Google Scholar]
- Dong, X.B.; Yang, C.T.; Zheng, D.D.; Mo, L.Q.; Wang, X.Y.; Lan, A.P.; Hu, F.; Chen, P.X.; Feng, J.Q.; Zhang, M.F.; et al. Inhibition of ROS-activated ERK1/2 pathway contributes to the protection of H2S against chemical hypoxia-induced injury in H9c2 cells. Mol. Cell. Biochem 2012, 362, 149–157. [Google Scholar]
- Lan, A.; Liao, X.; Mo, L.; Yang, C.; Yang, Z.; Wang, X.; Hu, F.; Chen, P.; Feng, J.; Zheng, D.; et al. Hydrogen sulfide protects against chemical hypoxia-induced injury by inhibiting ROS-activated ERK1/2 and p38MAPK signaling pathways in PC12 cells. PLoS One 2011, 6, e25921. [Google Scholar]
- Tay, A.S.; Hu, L.F.; Lu, M.; Wong, P.T.; Bian, J.S. Hydrogen sulfide protects neurons against hypoxic injury via stimulation of ATP-sensitive potassium channel/protein kinase C/extracellular signal-regulated kinase/heat shock protein 90 pathway. Neuroscience 2010, 167, 277–286. [Google Scholar]
- Xiao, L.; Lan, A.; Mo, L.; Xu, W.; Jiang, N.; Hu, F.; Feng, J.; Zhang, C. Hydrogen sulfide protects PC12 cells against reactive oxygen species and extracellular signal-regulated kinase 1/2-mediated downregulation of glutamate transporter-1 expression induced by chemical hypoxia. Int. J. Mol. Med 2012, 30, 1126–1132. [Google Scholar]
- Turcotte, S.; Desrosiers, R.R.; Béliveau, R. HIF-1α mRNA and protein upregulation involves Rho GTPase expression during hypoxia in renal cell carcinoma. J. Cell Sci 2003, 116, 2247–2260. [Google Scholar]
- Kiefmann, R.; Rifkind, J.M.; Nagababu, E.; Bhattacharya, J. Red blood cells induce hypoxic lung inflammation. Blood 2008, 111, 5205–5214. [Google Scholar]
- Rubanyi, G.M. The role of endothelium in cardiovascular homeostasis and diseases. J. Cardiovasc. Pharmacol 1993, 22, S1–S14. [Google Scholar]
- Mudau, M.; Genis, A.; Lochner, A.; Strijdom, H. Endothelial dysfunction: The early predictor of atherosclerosis. Cardiovasc. J. Afr 2012, 23, 222–231. [Google Scholar]
- Hadi, H.A.; Carr, C.S.; Al Suwaidi, J. Endothelial dysfunction: cardiovascular risk factors, therapy, and outcome. Vasc. Health Risk Manag 2005, 1, 183–198. [Google Scholar]
- Kolluru, G.K.; Bir, S.C.; Kevil, C.G. Endothelial dysfunction and diabetes: Effects on angiogenesis, vascular remodeling, and wound healing. Int. J. Vasc. Med 2012, 2012, 918267. [Google Scholar]
- Al-Magableh, M.R.; Hart, J.L. Mechanism of vasorelaxation and role of endogenous hydrogen sulfide production in mouse aorta. Naunyn-Schmied. Arch. Pharmacol 2011, 383, 403–413. [Google Scholar]
- Schleifenbaum, J.; Köhn, C.; Voblova, N.; Dubrovska, G.; Zavarirskaya, O.; Gloe, T.; Crean, C.S.; Luft, F.C.; Huang, Y.; Schubert, R. Systemic peripheral artery relaxation by KCNQ channel openers and hydrogen sulfide. J. Hypertens 2010, 28, 1875–1882. [Google Scholar]
- Vacek, T.P.; Gillespie, W.; Tyagi, N.; Vacek, J.C.; Tyagi, S.C. Hydrogen sulfide protects against vascular remodeling from endothelial damage. Amino Acids 2010, 39, 1161–1169. [Google Scholar]
- Davidson, S.M. Endothelial mitochondria and heart disease. Cardiovasc. Res 2010, 88, 58–66. [Google Scholar]
- Lemasters, J.J.; Qian, T.; He, L.; Kim, J.-S.; Elmore, S.P.; Cascio, W.E.; Brenner, D.A. Role of mitochondrial inner membrane permeabilization in necrotic cell death, apoptosis, and autophagy. Antioxid. Redox Signal 2002, 4, 769–781. [Google Scholar]
- Petronilli, V.; Penzo, D.; Scorrano, L.; Bernardi, P.; Di Lisa, F. The mitochondrial permeability transition, release of cytochrome c and cell death correlation with the duration of pore openings in situ. J. Biol. Chem 2001, 276, 12030–12034. [Google Scholar]
- Essick, E.E.; Sam, F. Oxidative stress and autophagy in cardiac disease, neurological disorders, aging and cancer. Oxid. Med. Cell. Longev 2010, 3, 168–177. [Google Scholar]
- Fiskum, G.; Starkov, A.; Polster, B.M.; Chinopoulos, C. Mitochondrial mechanisms of neural cell death and neuroprotective interventions in Parkinson’s disease. Ann. N. Y. Acad. Sci 2003, 991, 111–119. [Google Scholar]
- Knott, A.B.; Perkins, G.; Schwarzenbacher, R.; Bossy-Wetzel, E. Mitochondrial fragmentation in neurodegeneration. Nat. Rev. Neurosci 2008, 9, 505–518. [Google Scholar]
- Wen, Y.-D.; Wang, H.; Kho, S.-H.; Rinkiko, S.; Sheng, X.; Shen, H.-M.; Zhu, Y.-Z. Hydrogen sulfide protects HUVECs against hydrogen peroxide induced mitochondrial dysfunction and oxidative stress. PloS One 2013, 8, e53147. [Google Scholar]
- Lindsay, J.; Esposti, M.D.; Gilmore, A.P. Bcl-2 proteins and mitochondria—Specificity in membrane targeting for death. Biochim. Biophys. Acta 2011, 1813, 532–539. [Google Scholar]
- Susnow, N.; Zeng, L.; Margineantu, D.; Hockenbery, D.M. Bcl-2 family proteins as regulators of oxidative stress. Semin. Cancer Biol 2009, 19, 42–49. [Google Scholar]
- Breckenridge, D.G.; Xue, D. Regulation of mitochondrial membrane permeabilization by BCL-2 family proteins and caspases. Curr. Opin. Cell Biol 2004, 16, 647–652. [Google Scholar]
- Harris, M.H.; Thompson, C.B. The role of the Bcl-2 family in the regulation of outer mitochondrial membrane permeability. Cell Death and Differ 2000, 7, 1182–1191. [Google Scholar]
- Li, Z.; Jo, J.; Jia, J.-M.; Lo, S.-C.; Whitcomb, D.J.; Jiao, S.; Cho, K.; Sheng, M. Caspase-3 activation via mitochondria is required for long-term depression and AMPA receptor internalization. Cell 2010, 141, 859–871. [Google Scholar]
- Reiffenstein, R.; Hulbert, W.C.; Roth, S.H. Toxicology of hydrogen sulfide. Annu. Rev. Pharmacol. Toxicol 1992, 32, 109–134. [Google Scholar]
- Liu, X.; Chen, P.; Pan, L.; Silva, R.D.; Zhu, Y. 4-Guanidino-n-butyl syringate (Leonurine, SCM 198) protects H9c2 rat ventricular cells from hypoxia-induced apoptosis. J. Cardiovasc. Pharmacol 2009, 54, 437–444. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Shen, Y.; Guo, W.; Wang, Z.; Zhang, Y.; Zhong, L.; Zhu, Y. Protective Effects of Hydrogen Sulfide in Hypoxic Human Umbilical Vein Endothelial Cells: A Possible Mitochondria-Dependent Pathway. Int. J. Mol. Sci. 2013, 14, 13093-13108. https://doi.org/10.3390/ijms140713093
Shen Y, Guo W, Wang Z, Zhang Y, Zhong L, Zhu Y. Protective Effects of Hydrogen Sulfide in Hypoxic Human Umbilical Vein Endothelial Cells: A Possible Mitochondria-Dependent Pathway. International Journal of Molecular Sciences. 2013; 14(7):13093-13108. https://doi.org/10.3390/ijms140713093
Chicago/Turabian StyleShen, Yaqi, Wei Guo, Zhijun Wang, Yuchen Zhang, Liangjie Zhong, and Yizhun Zhu. 2013. "Protective Effects of Hydrogen Sulfide in Hypoxic Human Umbilical Vein Endothelial Cells: A Possible Mitochondria-Dependent Pathway" International Journal of Molecular Sciences 14, no. 7: 13093-13108. https://doi.org/10.3390/ijms140713093