Dependence of Interaction Free Energy between Solutes on an External Electrostatic Field

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results/Discussion
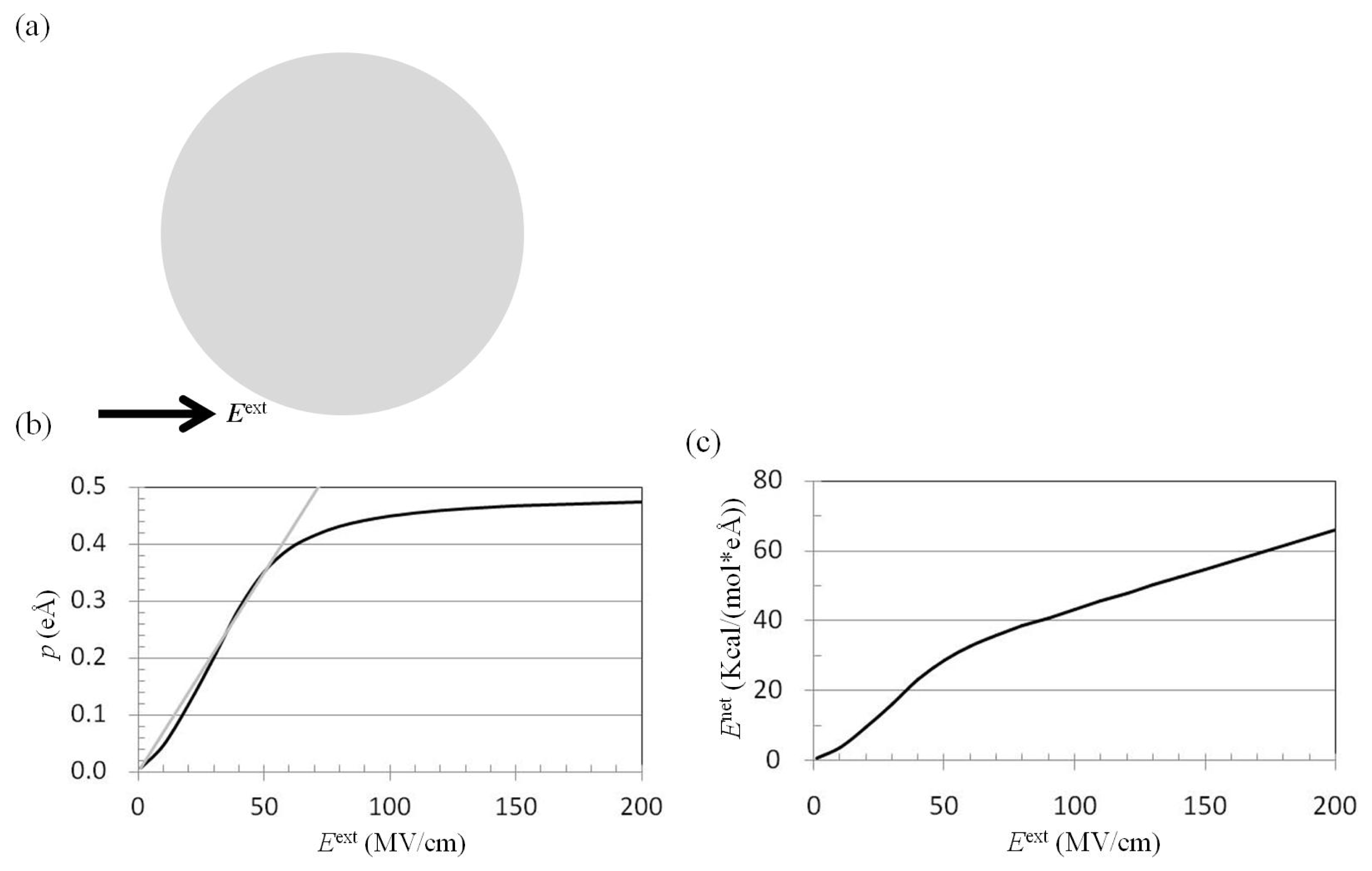
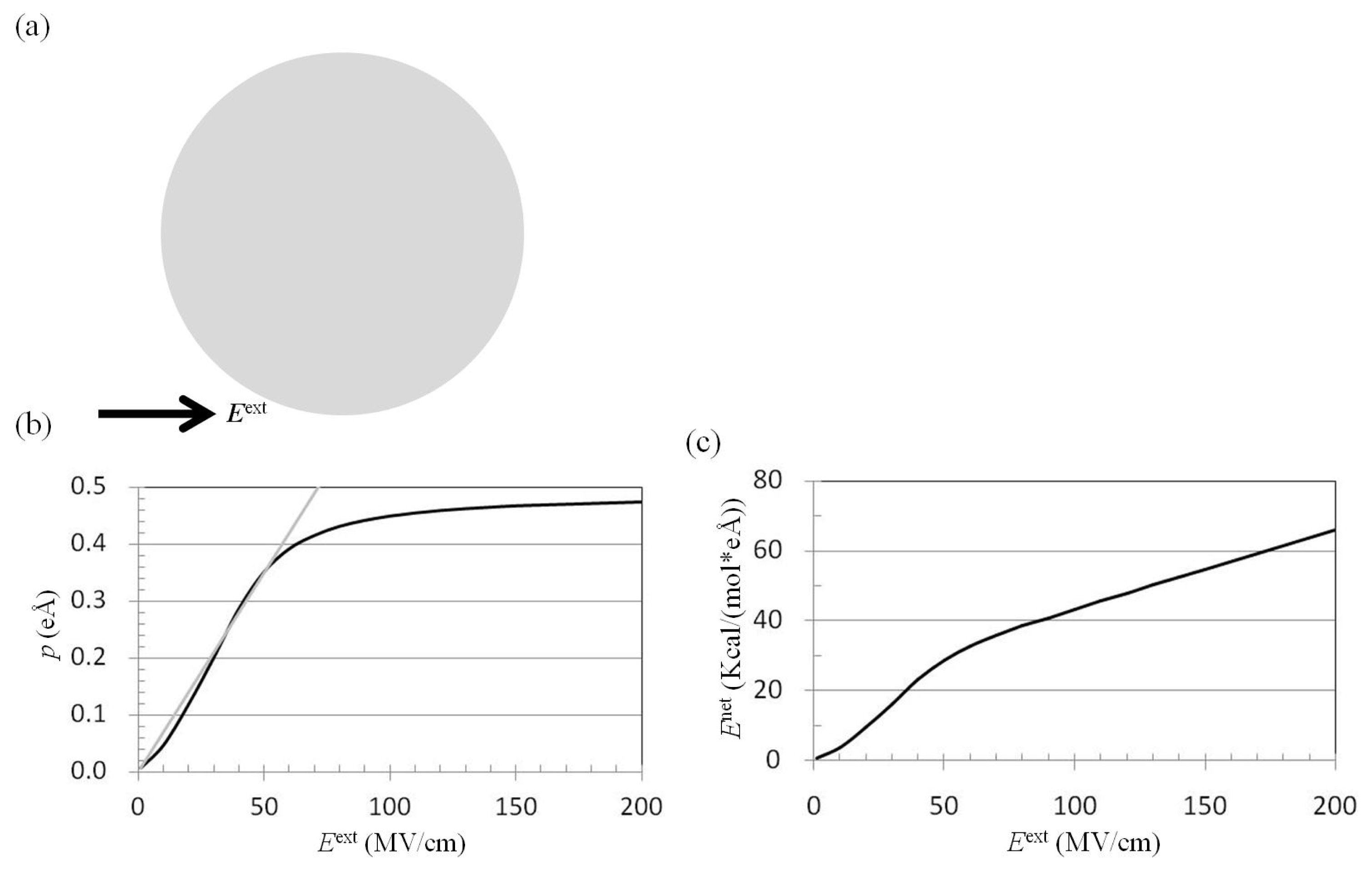
2.1. Dependence of p on Eext
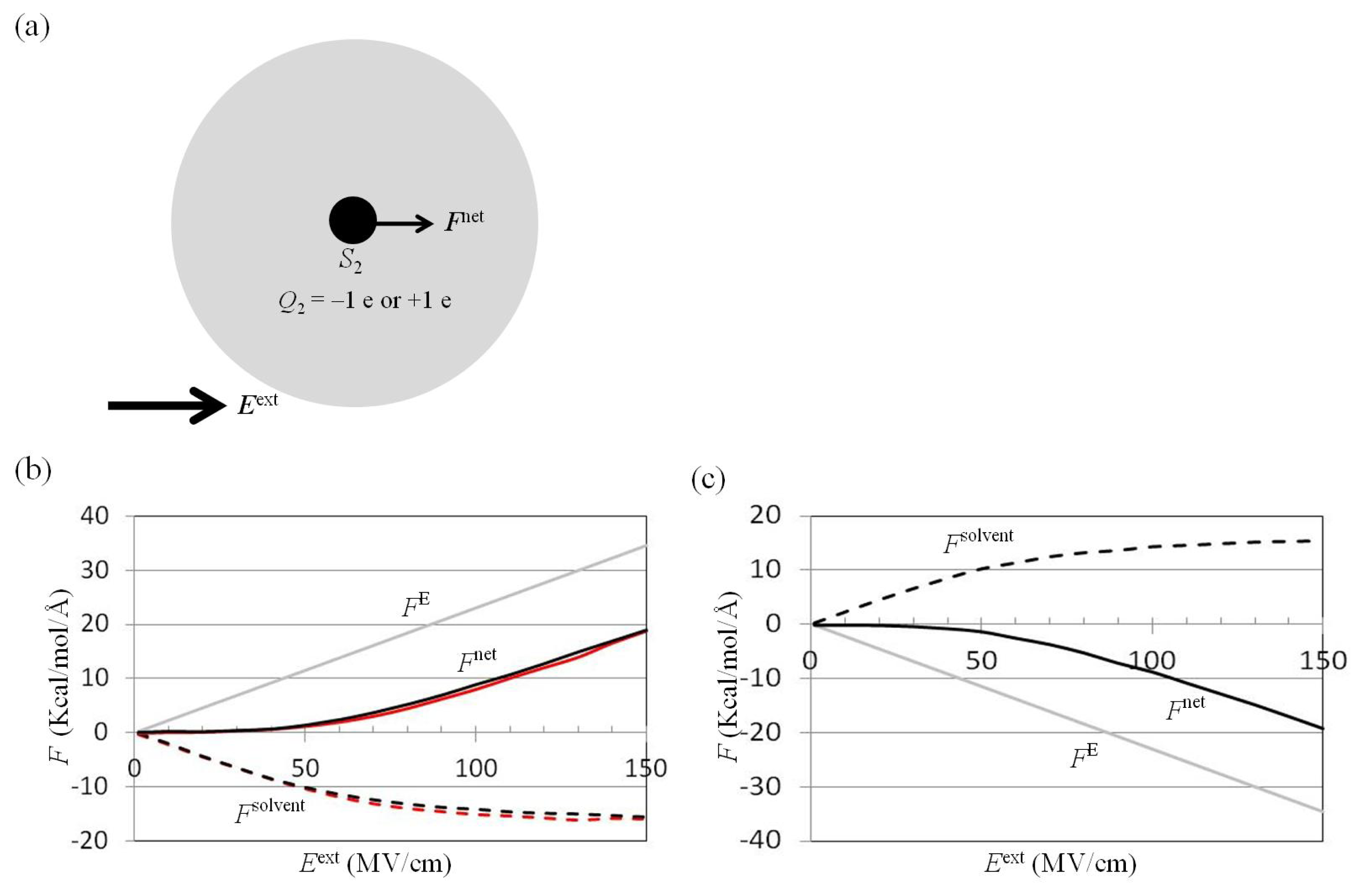
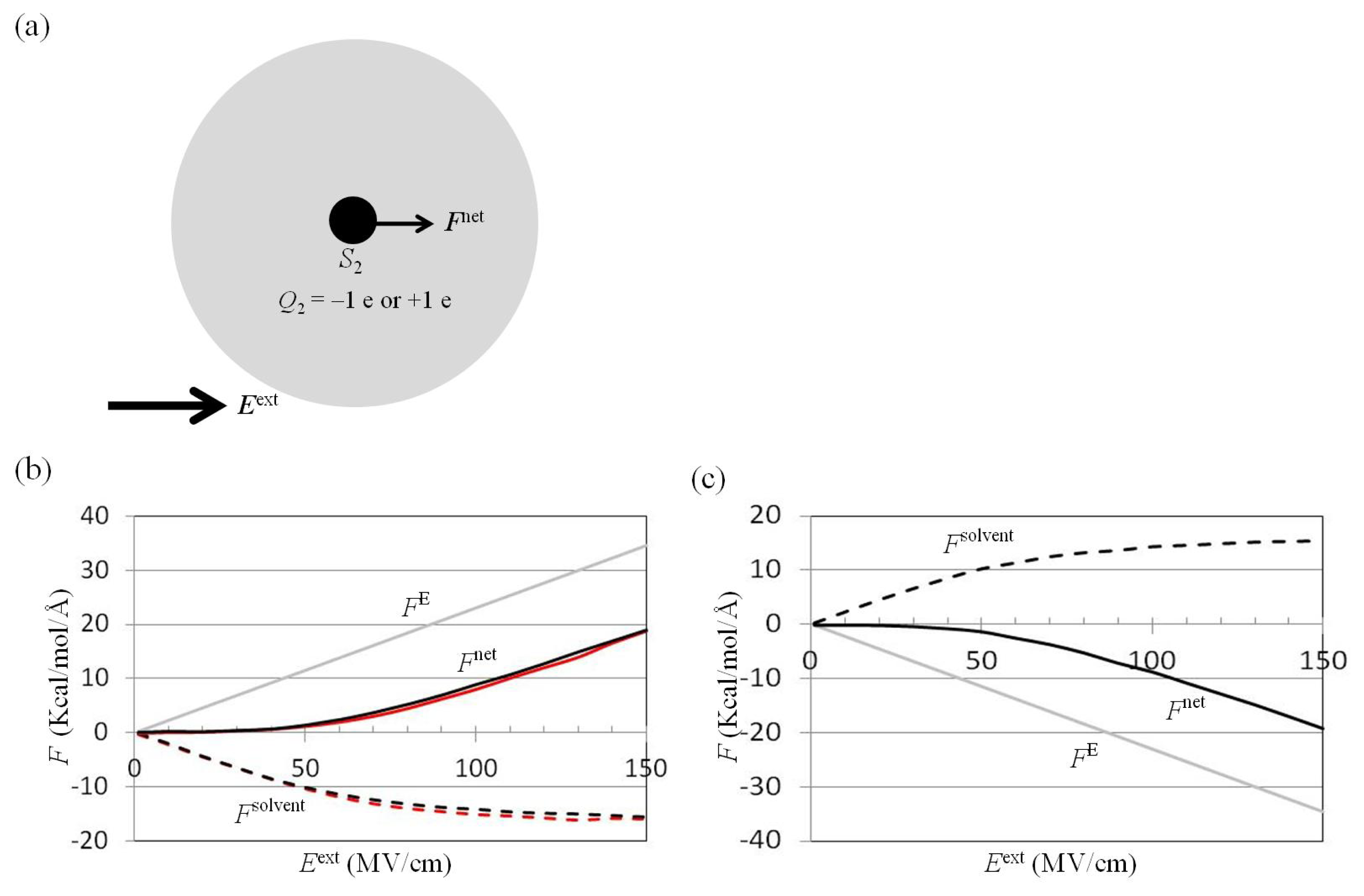
2.2. Dependence of Fnet(One_Atom) on Eext
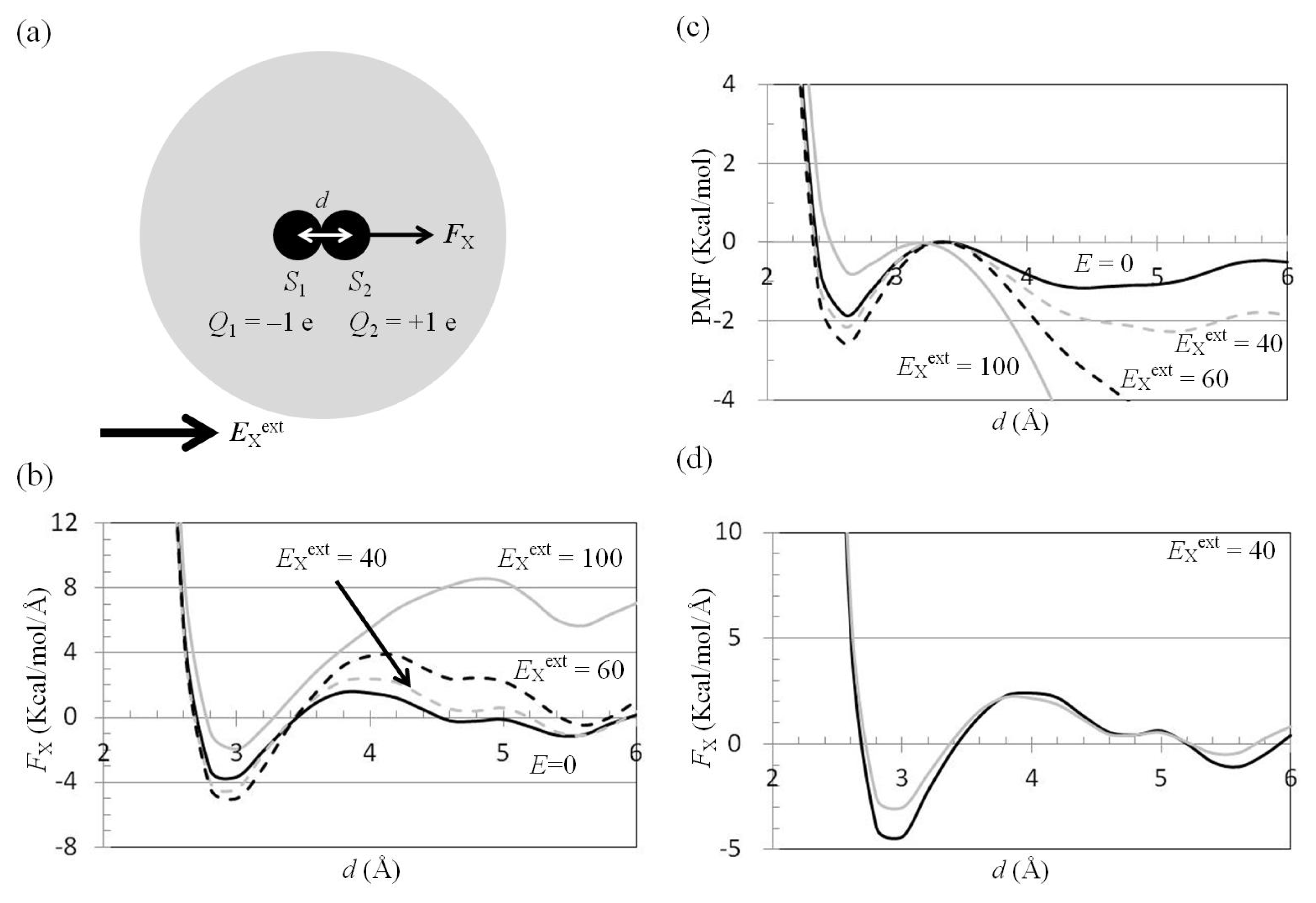
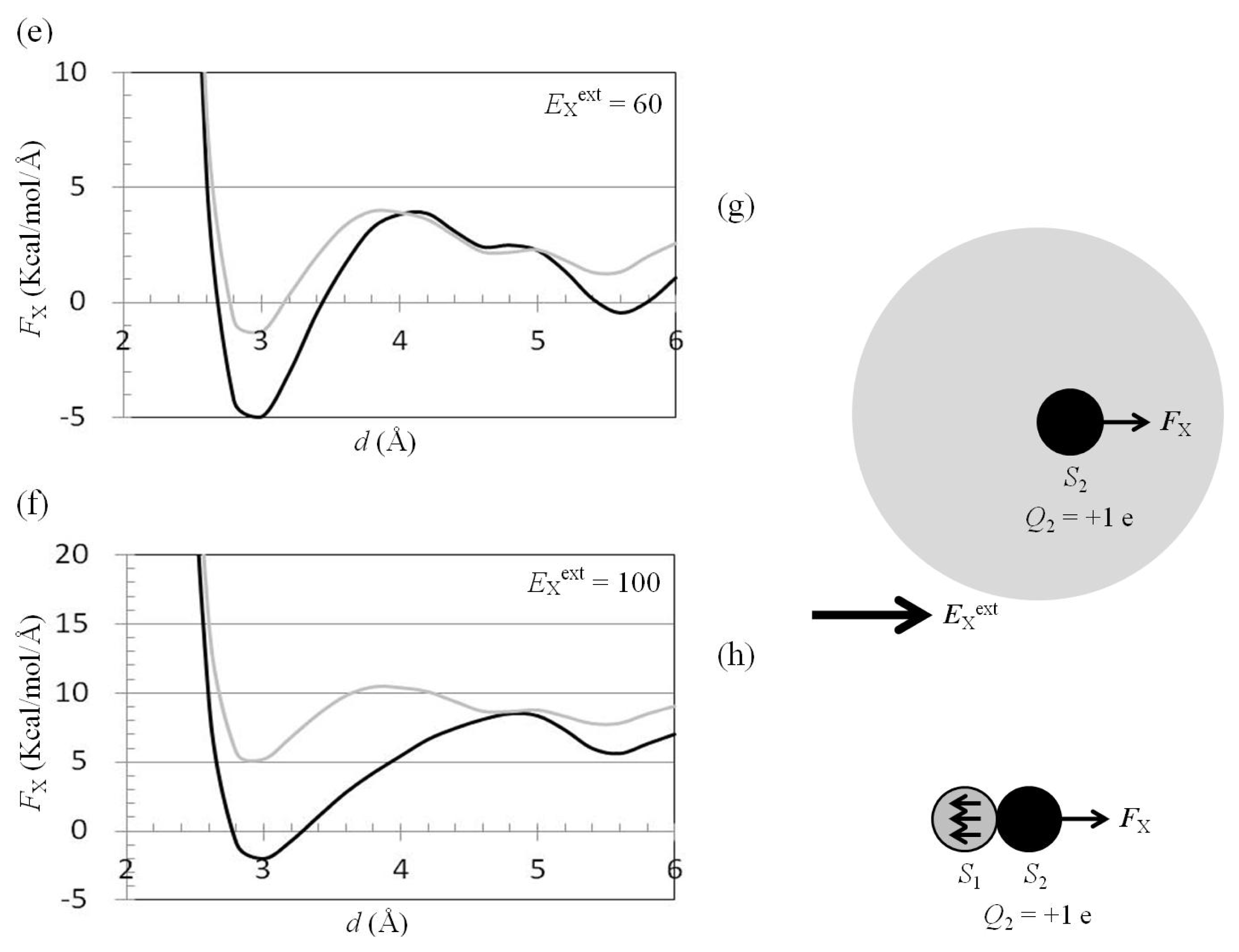
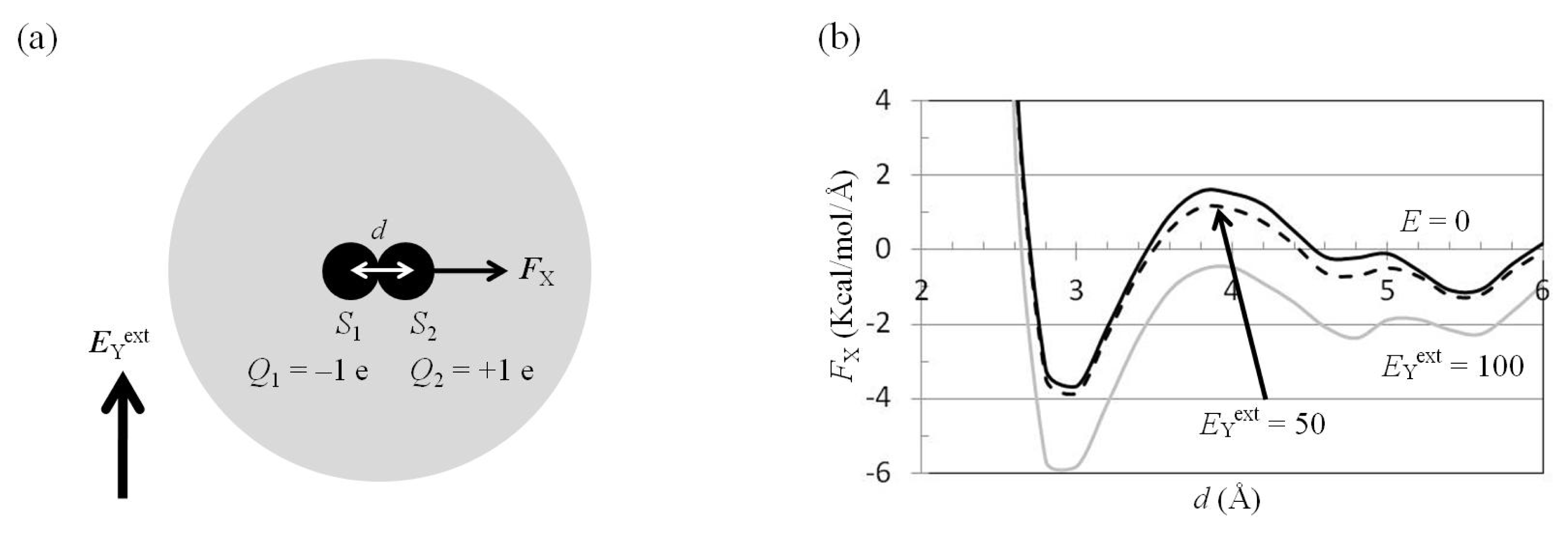
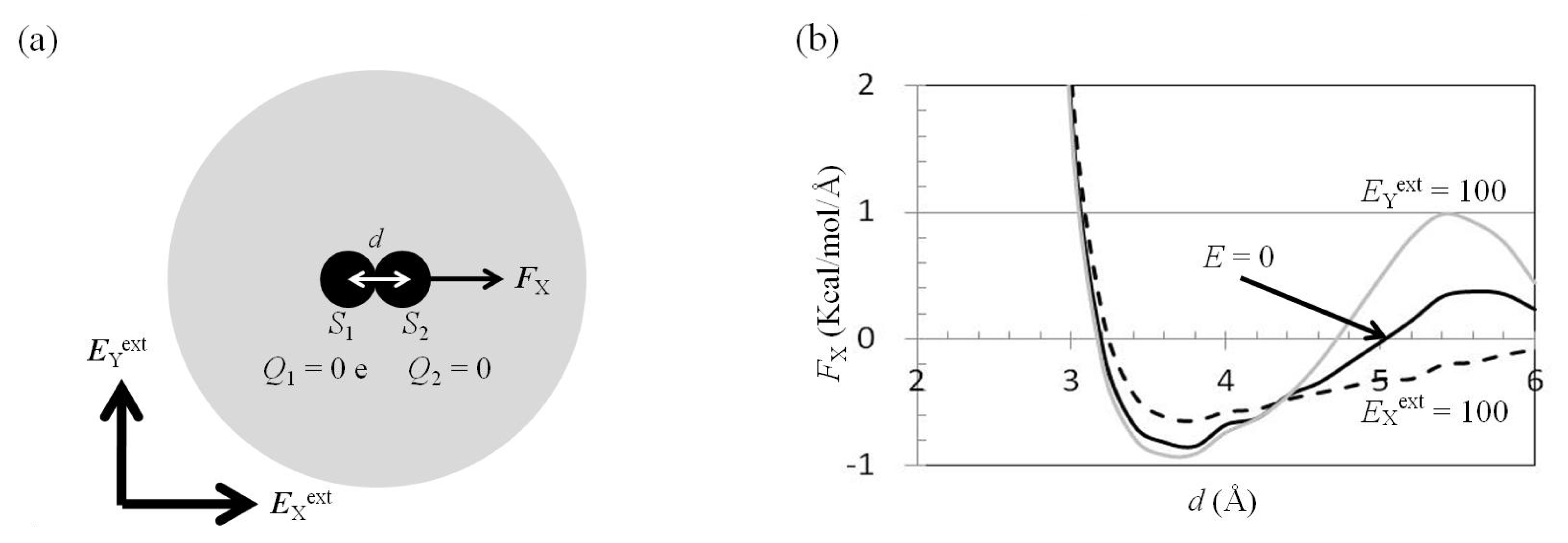
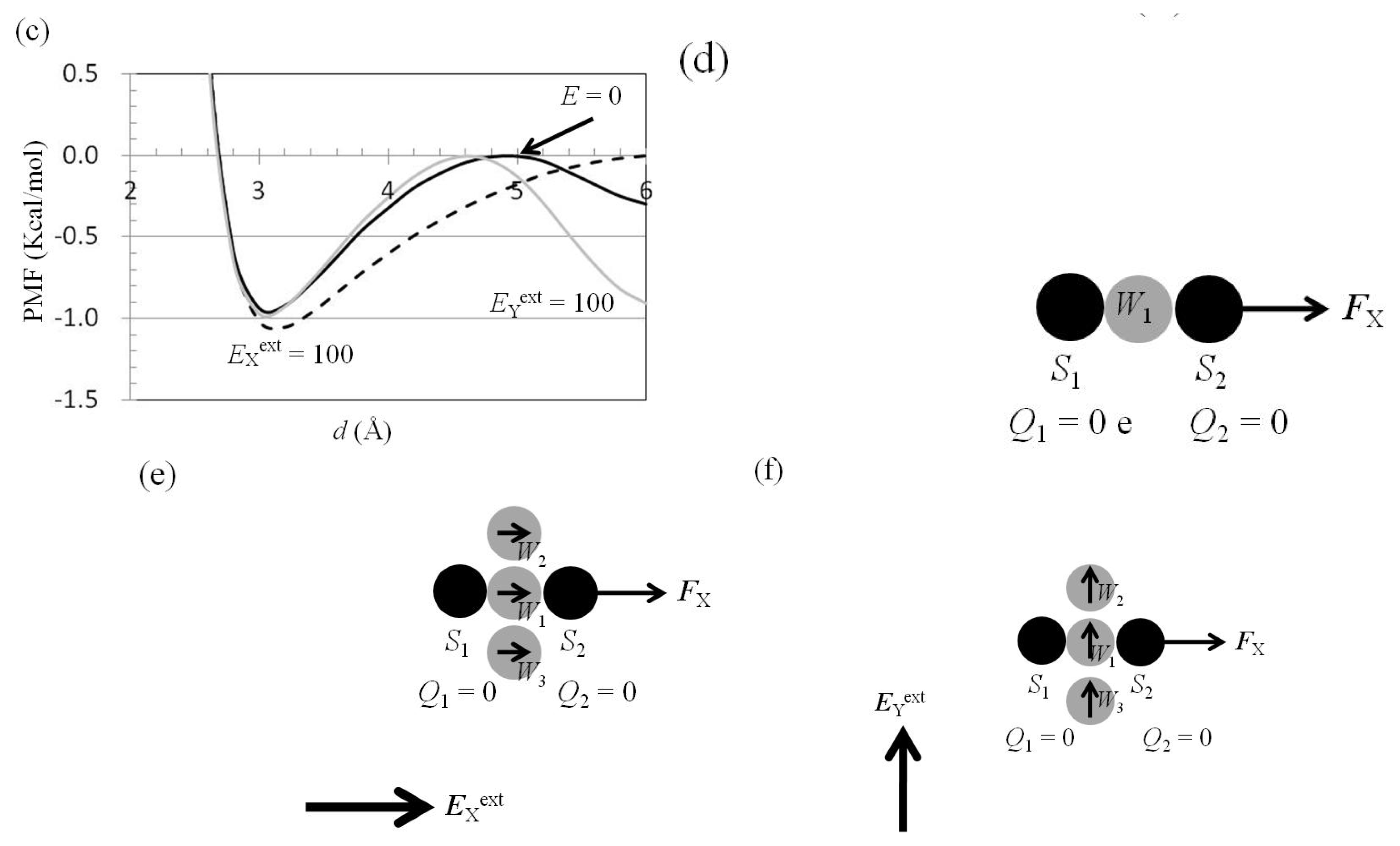
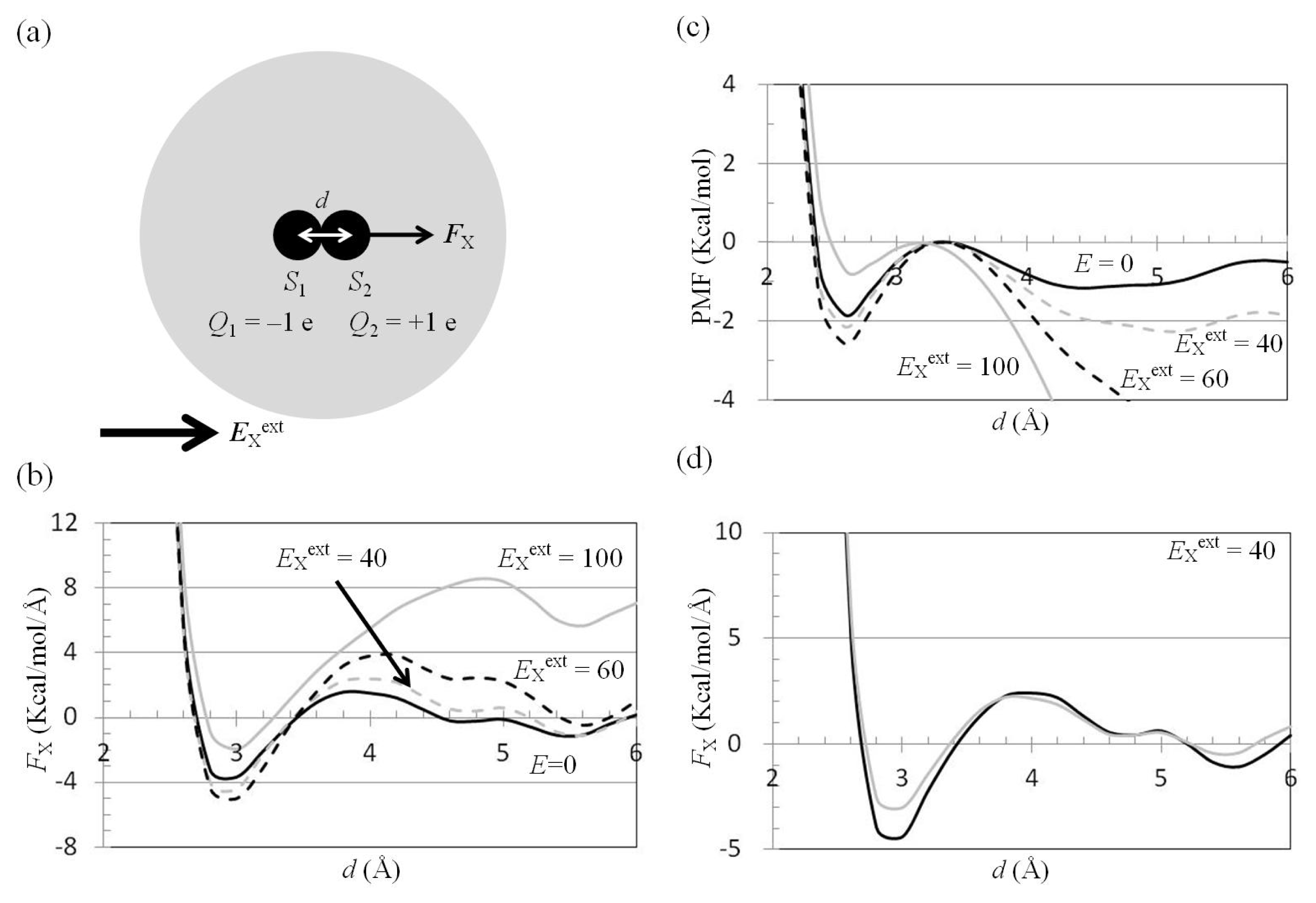
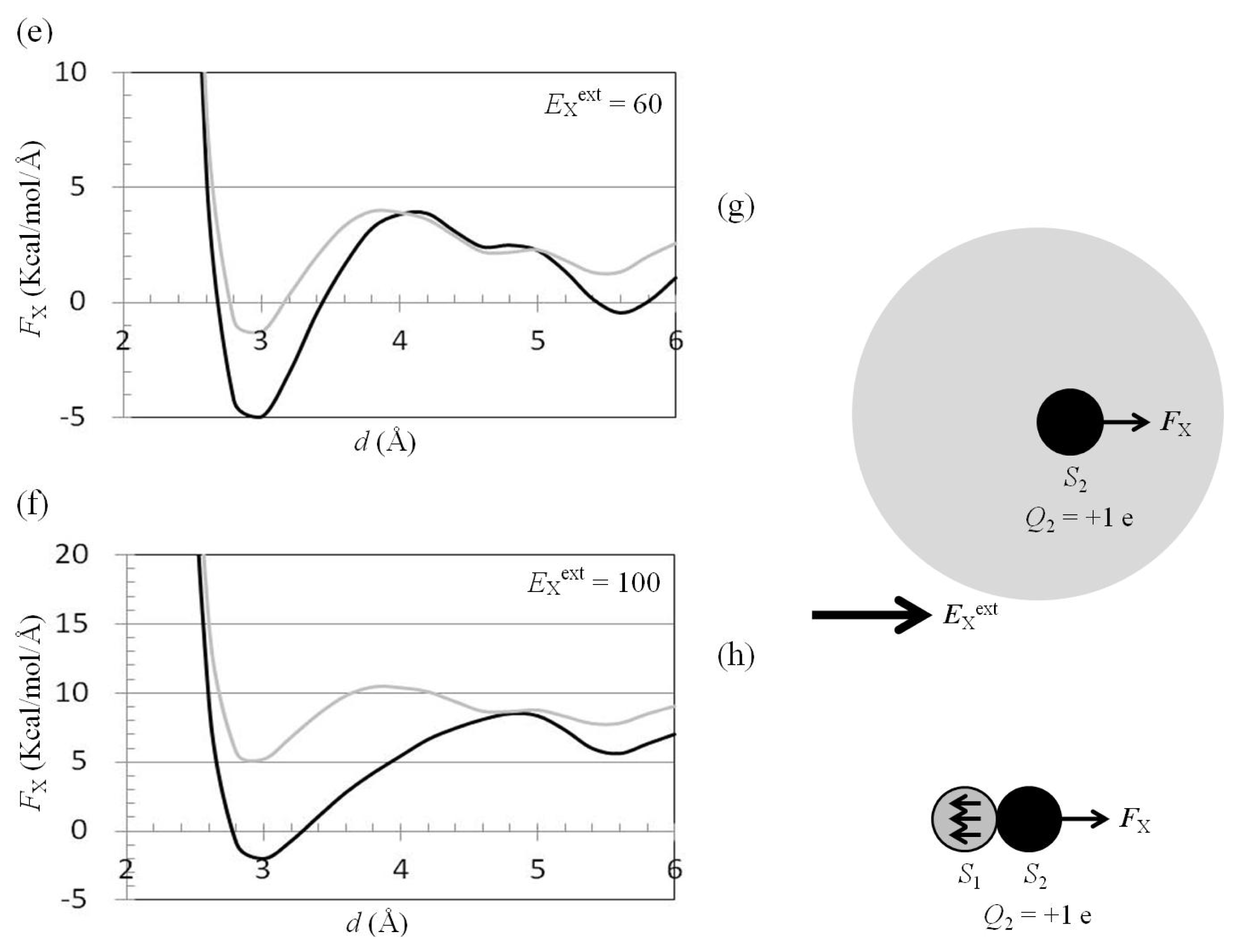
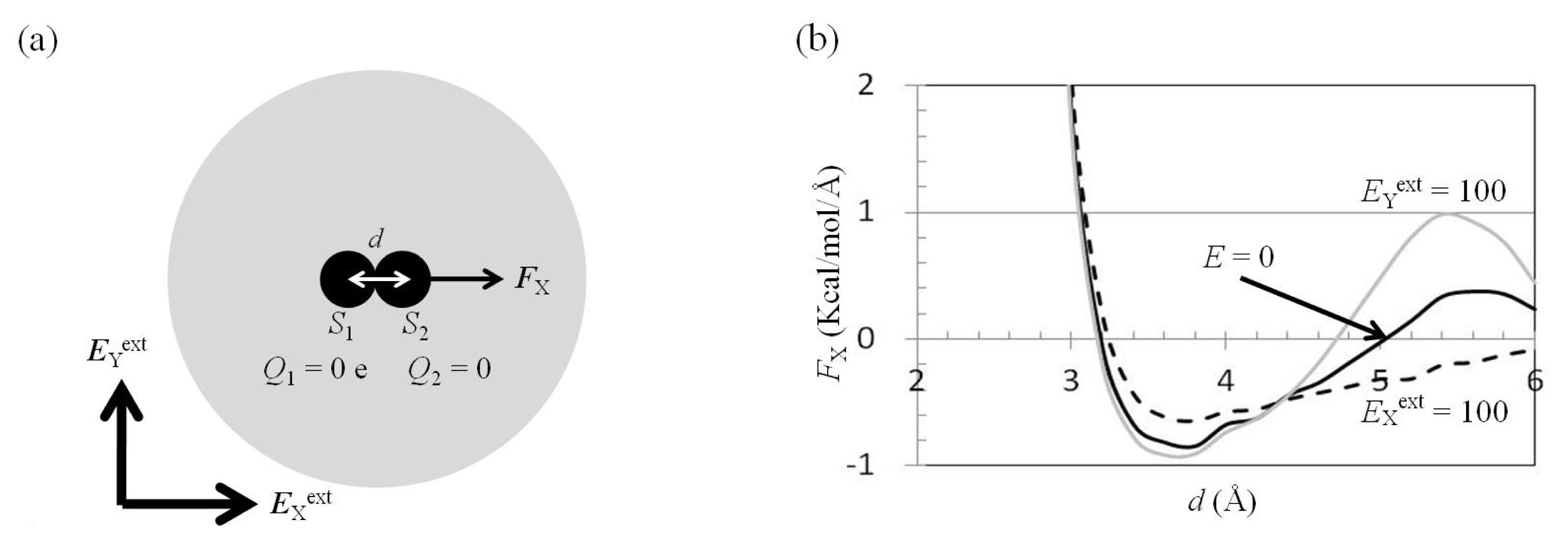
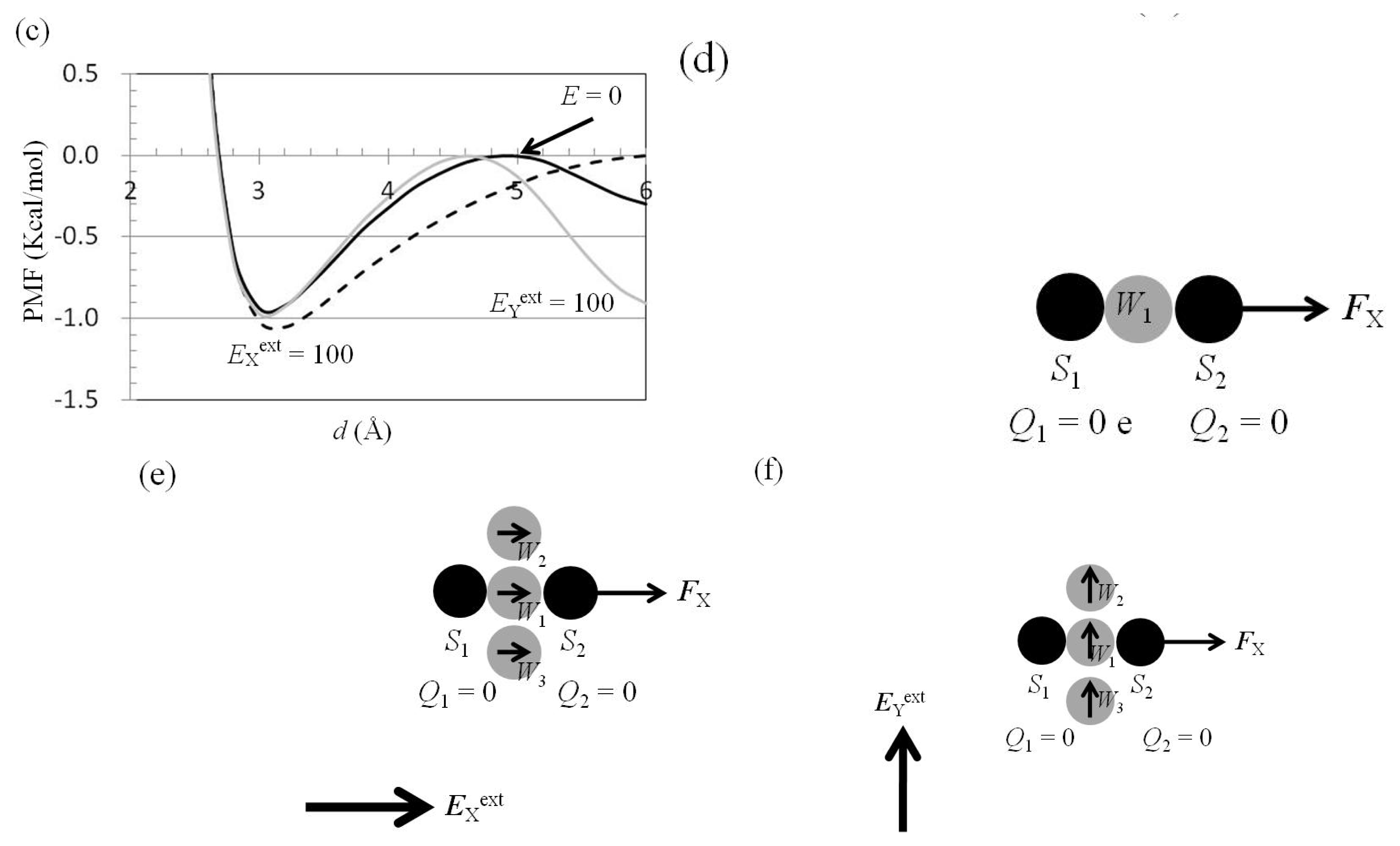
2.3. Attractive Force between S1 and S2 Could Not Be Decreased by Applying an External Electrostatic Field along the Direction of the Electric Dipole Formed by S1 and S2
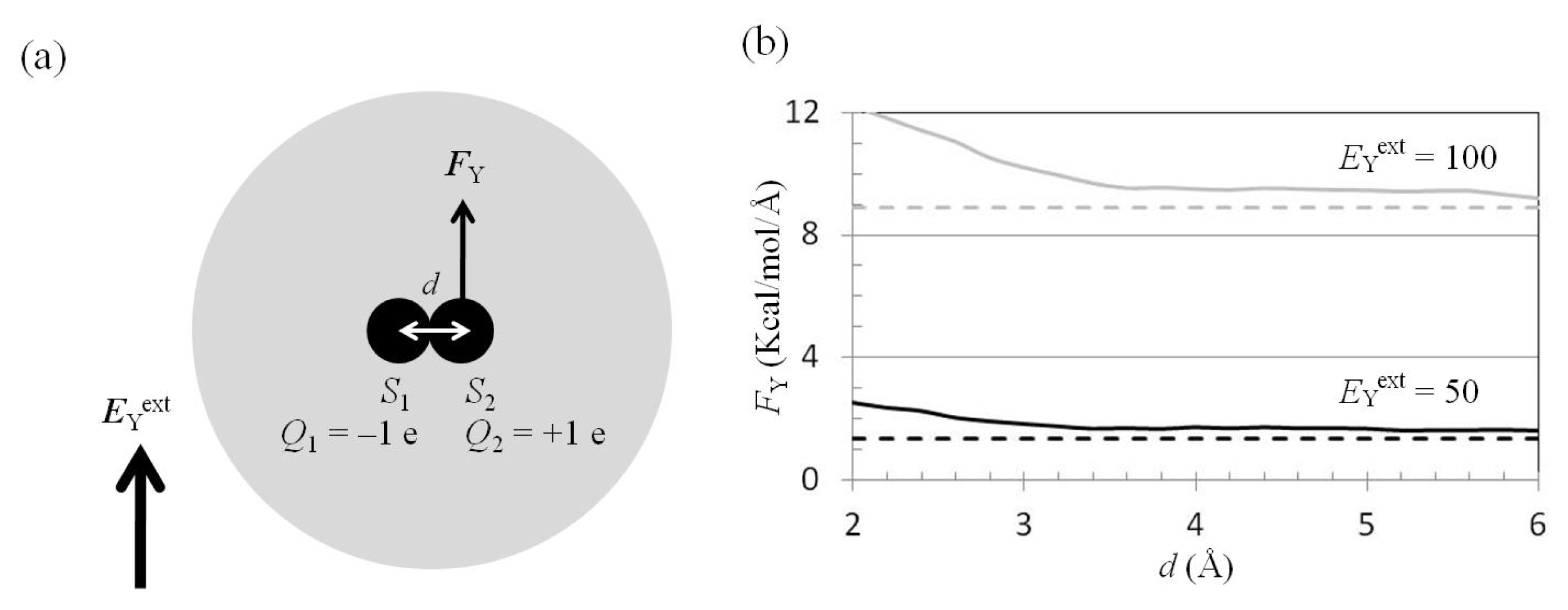
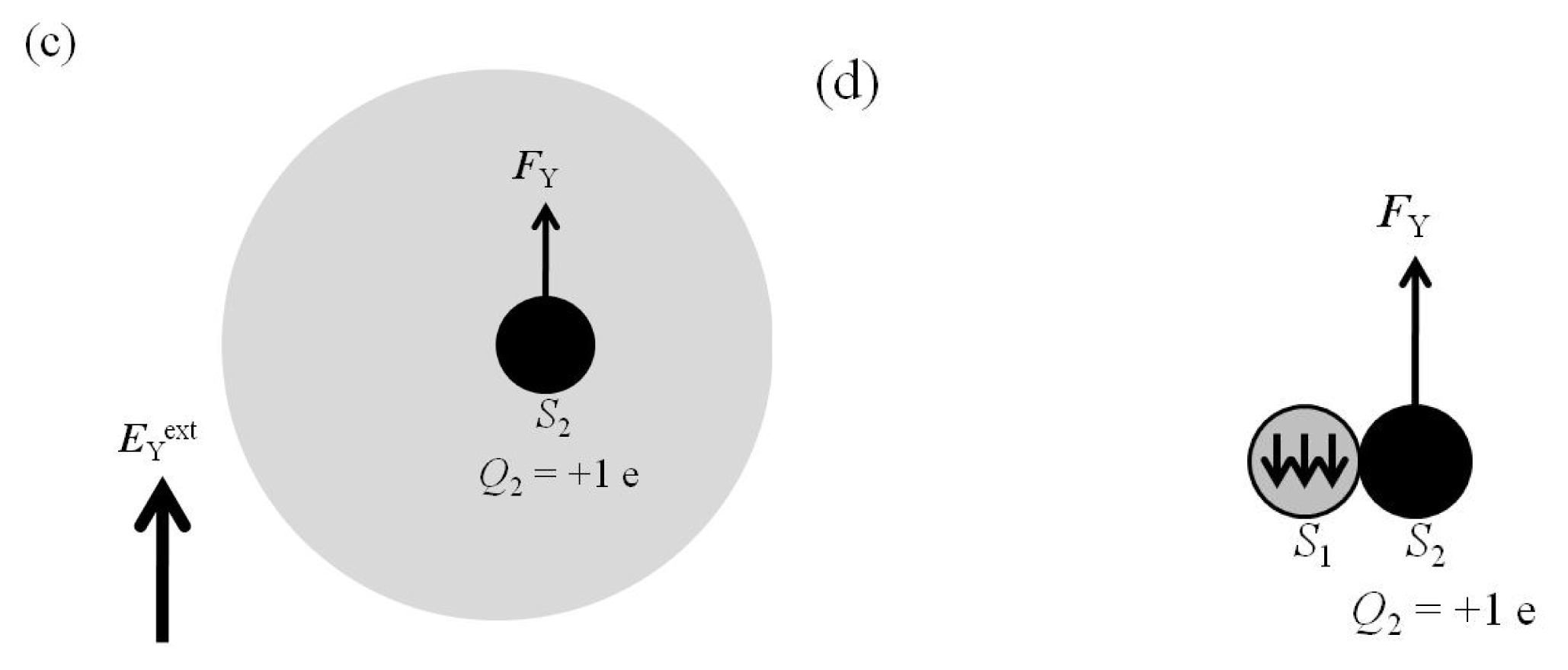
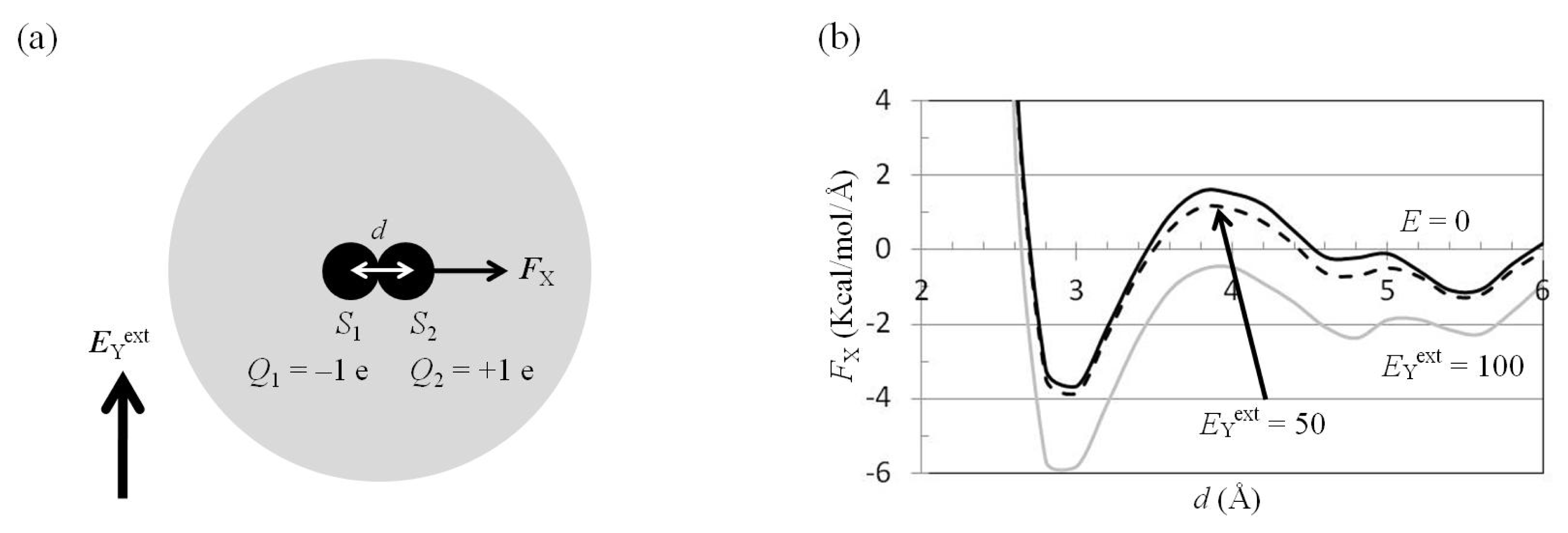
2.4. Attractive Force between S1 and S2 Was Unchanged and Increased by Applying EYext = 50 MV/cm and 100 MV/cm, Respectively
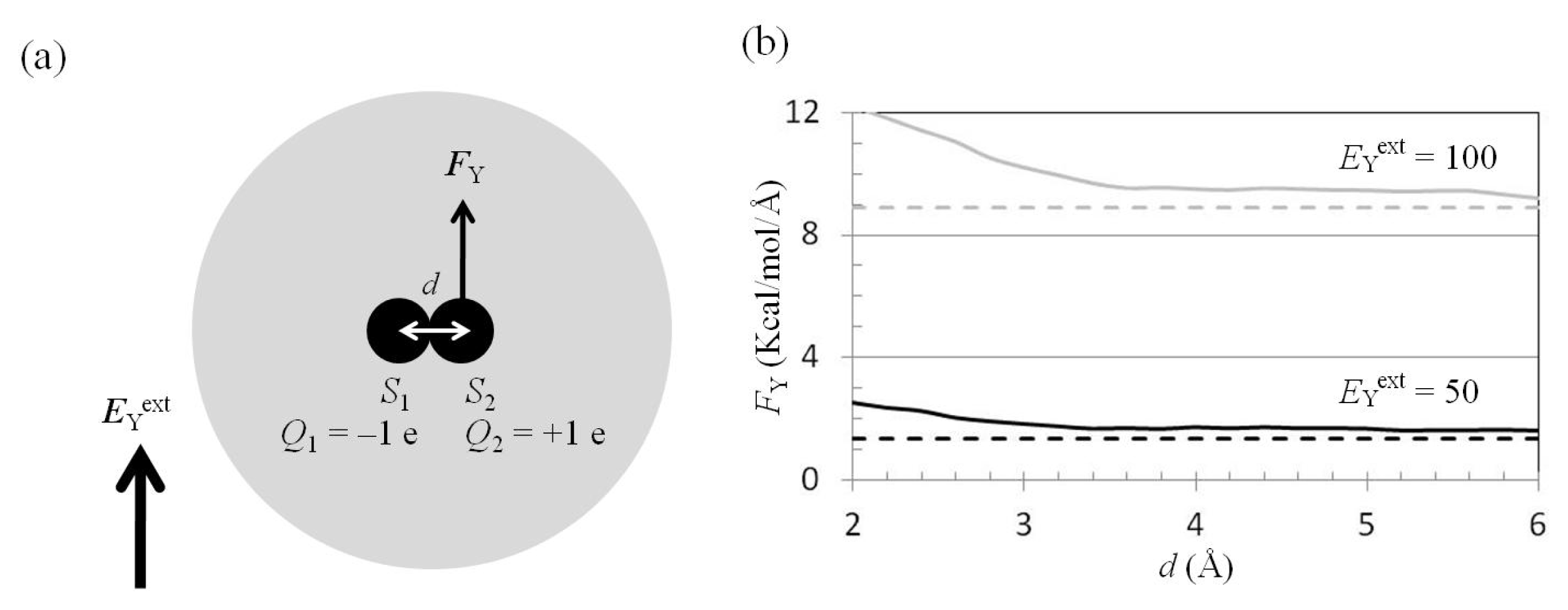
2.5. FY(Two_Atoms; EYext) Was Greater than FY(one_atom; EYext), Especially When d Was Small
2.6. Dependence of FX(Two_Neutral_Atoms) on EXext and EYext
3. Method
3.1. MD Simulations
3.2. Application of Eext to Pure Water Cluster and Calculation of p and Enet from Trajectories of MD Simulations
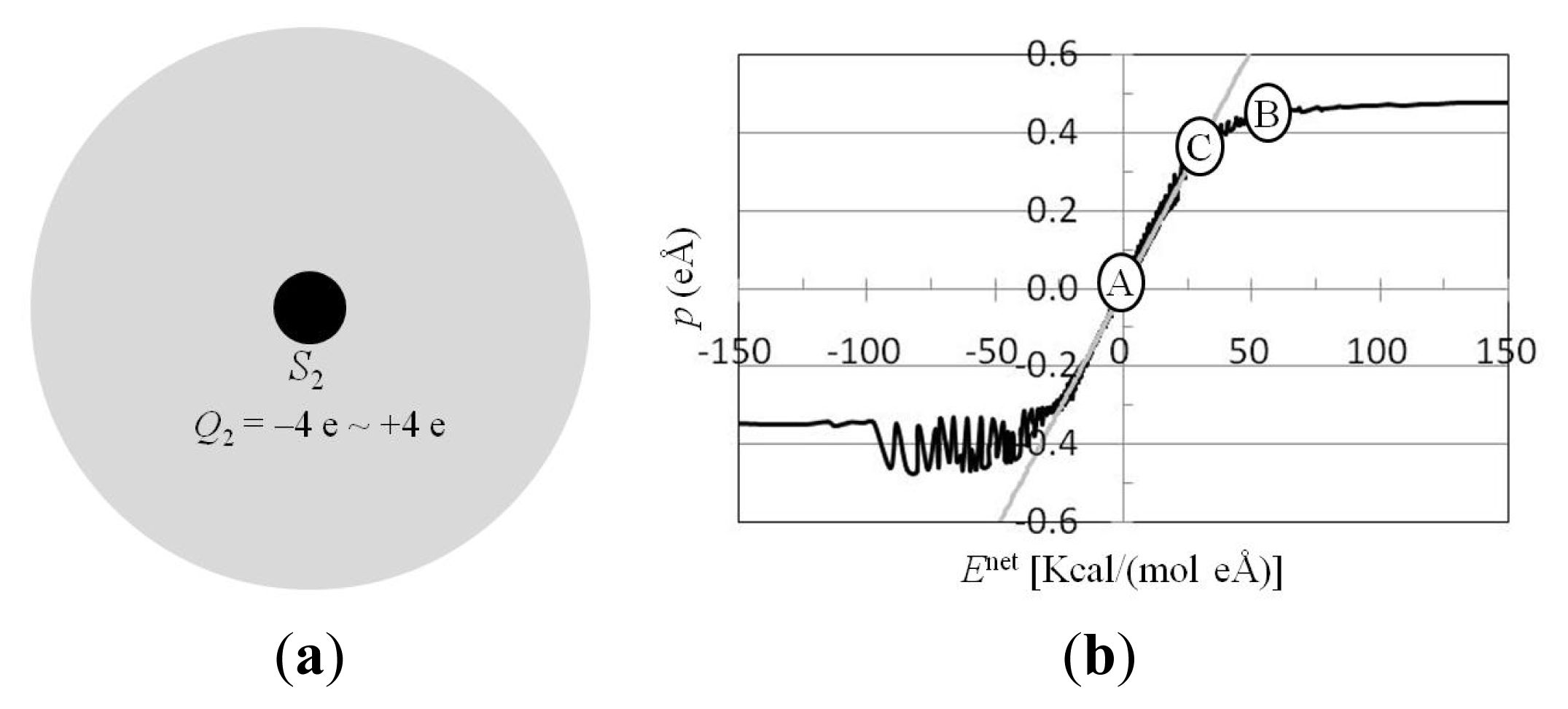
3.3. For Charged Atom in Water Cluster, Calculation of p(r) and Enet(r) from Trajectories of MD Simulations
3.4. Application of Eext to Water Cluster Containing One or Two Solutes and Calculation of Fsolv(r) and Fnet(r) from Trajectories of MD Simulations
4. Conclusions
Acknowledgements
Conflict of Interest
References
- Chou, C.K. Thirty-five years in bioelectromagnetics research. Bioelectromagnetics 2007, 28, 3–15. [Google Scholar]
- De Pomerai, D.I.; Smith, B.; Dawe, A.; North, K.; Smith, T.; Archer, D.B.; Duce, I.R.; Jones, D.; Candido, E.P.M. Microwave radiation can alter protein conformation without bulk heating. FEBS Lett 2003, 543, 93–97. [Google Scholar]
- Rahbek, U.L.; Tritsaris, K.; Dissing, S. Interactions of low frequency, pulsed electromagnetic fields with living tissue: Biochemical responses and clinical results. Oral Biosci. Med 2005, 2, 29–40. [Google Scholar]
- Tepper, O.M.; Callaghan, M.J.; Chang, E.I.; Galiano, R.D.; Bhatt, K.A.; Baharestani, S.; Gan, J.; Simon, B.; Hopper, R.A.; Levine, J.P. Electromagnetic fields increase in vitro and in vivo angiogenesis through endothelial release of FGF-2. FASEB J 2004, 18, 1231–1233. [Google Scholar]
- Mancinelli, F.; Caraglia, M.; Abbruzzese, A.; d’Ambrosio, G.; Massa, R.; Bismuto, E. Non-thermal effects of electromagnetic fields at mobile phone frequency on the refolding of an intracellular protein: Myoglobin. J. Cell. Biochem 2004, 93, 188–196. [Google Scholar]
- Shih, E.S.; Hwang, M.-J. Protein structure comparison by probability-based matching of secondary structure elements. Bioinformatics 2003, 19, 735–741. [Google Scholar]
- Copty, A.B.; Neve-Oz, Y.; Barak, I.; Golosovsky, M.; Davidov, D. Evidence for a specific microwave radiation effect on the green fluorescent protein. Biophys. J 2006, 91, 1413–1423. [Google Scholar]
- Bojjawar, T.; Jalari, M.; Aamodt, E.; Ware, M.F.; Haynie, D.T. Effect of electromagnetic nanopulses on C. elegans fertility. Bioelectromagnetics 2006, 27, 515–520. [Google Scholar]
- Cotgreave, I.A. Biological stress responses to radio frequency electromagnetic radiation: Are mobile phones really so (heat) shocking? Arch. Biochem. Biophys 2005, 435, 227–240. [Google Scholar]
- Richard, D.; Lange, S.; Viergutz, T.; Kriehuber, R.; Weiss, D.G.; Simkó, M. Influence of 50 Hz electromagnetic fields in combination with a tumour promoting phorbol ester on protein kinase C and cell cycle in human cells. Mol. Cell. Biochem 2002, 232, 133–141. [Google Scholar]
- Panagopoulos, D.J.; Chavdoula, E.D.; Nezis, I.P.; Margaritis, L.H. Cell death induced by GSM 900-MHz and DCS 1800-MHz mobile telephony radiation. Mutat. Res 2007, 626, 69–78. [Google Scholar]
- Sokolovic, D.; Djindjic, B.; Nikolic, J.; Bjelakovic, G.; Pavlovic, D.; Kocic, G.; Krstic, D.; Cvetkovic, T.; Pavlovic, V. Melatonin reduces oxidative stress induced by chronic exposure of microwave radiation from mobile phones in rat brain. J. Radiat. Res 2008, 49, 579–586. [Google Scholar]
- Kundi, M.; Mild, K.H.; Hardell, L.; Mattsson, M.-O. Mobile telephones and cancer—A review of epidemiological evidence. J. Toxicol. Environ. Health Part B 2004, 7, 351–384. [Google Scholar]
- English, N.J.; Mooney, D.A. Denaturation of hen egg white lysozyme in electromagnetic fields: A molecular dynamics study. J. Chem. Phys 2007, 126, 091105. [Google Scholar]
- Calvo, F.; Dugourd, P. Folding of gas-phase polyalanines in a static electric field: Alignment, deformations, and polarization effects. Biophys. J 2008, 95, 18. [Google Scholar]
- English, N.J.; Solomentsev, G.Y.; O’Brien, P. Nonequilibrium molecular dynamics study of electric and low-frequency microwave fields on hen egg white lysozyme. J. Chem. Phys 2009, 131, 035106. [Google Scholar]
- Zhou, R. Free energy landscape of protein folding in water: Explicit vs. implicit solvent. Proteins Struct. Funct. Bioinforma 2003, 53, 148–161. [Google Scholar]
- Sorenson, J.M.; Hura, G.; Soper, A.K.; Pertsemlidis, A.; Head-Gordon, T. Determining the role of hydration forces in protein folding. J. Phys. Chem. B 1999, 103, 5413–5426. [Google Scholar]
- Wen, E.Z.; Hsieh, M.-J.; Kollman, P.A.; Luo, R. Enhanced ab initio protein folding simulations in Poisson-Boltzmann molecular dynamics with self-guiding forces. J. Mol. Graph. Model 2004, 22, 415–424. [Google Scholar]
- Lazaridis, T.; Karplus, M. Thermodynamics of protein folding: A microscopic view. Biophys. Chem 2003, 100, 367–395. [Google Scholar]
- Kentsis, A.; Mezei, M.; Osman, R. MC-PHS: A Monte Carlo implementation of the primary hydration shell for protein folding and design. Biophys. J 2003, 84, 805. [Google Scholar]
- Noskov, S.Y.; Lim, C. Free energy decomposition of protein-protein interactions. Biophys. J 2001, 81, 737–750. [Google Scholar]
- Sheinerman, F.B.; Norel, R.; Honig, B. Electrostatic aspects of protein-protein interactions. Curr. Opin. Struct. Biol 2000, 10, 153–159. [Google Scholar]
- Jiang, L.; Gao, Y.; Mao, F.; Liu, Z.; Lai, L. Potential of mean force for protein-protein interaction studies. Proteins Struct. Funct. Bioinforma 2002, 46, 190–196. [Google Scholar]
- Jackson, R.M.; Sternberg, M. A continuum model for protein-protein interactions: Application to the docking problem. J. Mol. Biol 1995, 250, 258. [Google Scholar]
- Fennell, C.J.; Kehoe, C.W.; Dill, K.A. Modeling aqueous solvation with semi-explicit assembly. Proc. Natl. Acad. Sci 2011, 108, 3234–3239. [Google Scholar]
- Yang, P.-K.; Liaw, S.-H.; Lim, C. Representing an infinite solvent system with a rectangular finite system using image charges. J. Phys. Chem. B 2002, 106, 2973–2982. [Google Scholar]
- Du, Q.; Beglov, D.; Roux, B. Solvation free energy of polar and nonpolar molecules in water: An extended interaction site integral equation theory in three dimensions. J. Phys. Chem. B 2000, 104, 796–805. [Google Scholar]
- Thompson, J.D.; Cramer, C.J.; Truhlar, D.G. New universal solvation model and comparison of the accuracy of the SM5. 42R, SM5. 43R, C-PCM, D-PCM, and IEF-PCM continuum solvation models for aqueous and organic solvation free energies and for vapor pressures. J. Phys. Chem. A 2004, 108, 6532–6542. [Google Scholar]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev 2005, 105, 2999–3094. [Google Scholar]
- Feig, M.; Onufriev, A.; Lee, M.S.; Im, W.; Case, D.A.; Brooks, C.L. Performance comparison of generalized born and Poisson methods in the calculation of electrostatic solvation energies for protein structures. J. Comput. Chem 2004, 25, 265–284. [Google Scholar]
- Zhu, J.; Shi, Y.; Liu, H. Parametrization of a generalized Born/solvent-accessible surface area model and applications to the simulation of protein dynamics. J. Phys. Chem. B 2002, 106, 4844–4853. [Google Scholar]
- Yang, P.-K.; Lim, C. The importance of excluded solvent volume effects in computing hydration free energies. J. Phys. Chem. B 2008, 112, 14863–14868. [Google Scholar]
- Babu, C.S.; Lim, C. Solvation free energies of polar molecular solutes: Application of the two-sphere Born radius in continuum models of solvation. J. Chem. Phys 2001, 114, 889. [Google Scholar]
- Hummer, G.; Pratt, L.R.; García, A.E. Free energy of ionic hydration. J. Phys. Chem 1996, 100, 1206–1215. [Google Scholar]
- Garde, S.; Hummer, G.; Paulaitis, M.E. Free energy of hydration of a molecular ionic solute: Tetramethylammonium ion. J. Chem. Phys 1998, 108, 1552. [Google Scholar]
- Yang, P.K. Discrepancy in the near-solute electric dipole moment calculated from the electric field. J. Comput. Chem 2011, 32, 2783–2799. [Google Scholar]
- Yang, P.K.; Lim, C. Strategies to model the near-solute solvent molecular density/polarization. J. Comput. Chem 2009, 30, 700–709. [Google Scholar]
- Brooks, B.R.; Bruccoleri, R.E.; Olafson, B.D.; Swaminathan, S.; Karplus, M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem 1983, 4, 187–217. [Google Scholar]
- Gavryushov, S.; Linse, P. Polarization deficiency and excess free energy of ion hydration in electric fields. J. Phys. Chem. B 2003, 107, 7135–7142. [Google Scholar]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys 1983, 79, 926. [Google Scholar]
- Yang, P.-K.; Lim, C. Reformulation of maxwell’s equations to incorporate near-solute solvent structure. J. Phys. Chem. B 2008, 112, 10791–10794. [Google Scholar]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J. Numerical integration of the Cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys 1977, 23, 327–341. [Google Scholar]
- Chelli, R.; Barducci, A.; Bellucci, L.; Schettino, V.; Procacci, P. Behavior of polarizable models in presence of strong electric fields. I. Origin of nonlinear effects in water point-charge systems. J. Chem. Phys 2005, 123, 194109. [Google Scholar]


© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yang, P.-K. Dependence of Interaction Free Energy between Solutes on an External Electrostatic Field. Int. J. Mol. Sci. 2013, 14, 14408-14425. https://doi.org/10.3390/ijms140714408
Yang P-K. Dependence of Interaction Free Energy between Solutes on an External Electrostatic Field. International Journal of Molecular Sciences. 2013; 14(7):14408-14425. https://doi.org/10.3390/ijms140714408
Chicago/Turabian StyleYang, Pei-Kun. 2013. "Dependence of Interaction Free Energy between Solutes on an External Electrostatic Field" International Journal of Molecular Sciences 14, no. 7: 14408-14425. https://doi.org/10.3390/ijms140714408