Membrane Binding and Insertion of a pHLIP Peptide Studied by All-Atom Molecular Dynamics Simulations


Abstract
:
1. Introduction
2. Results and Discussion
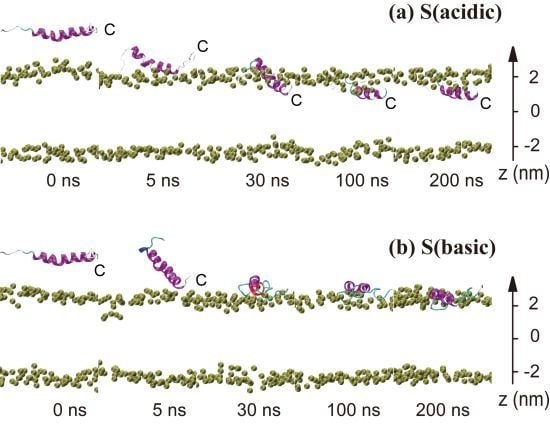
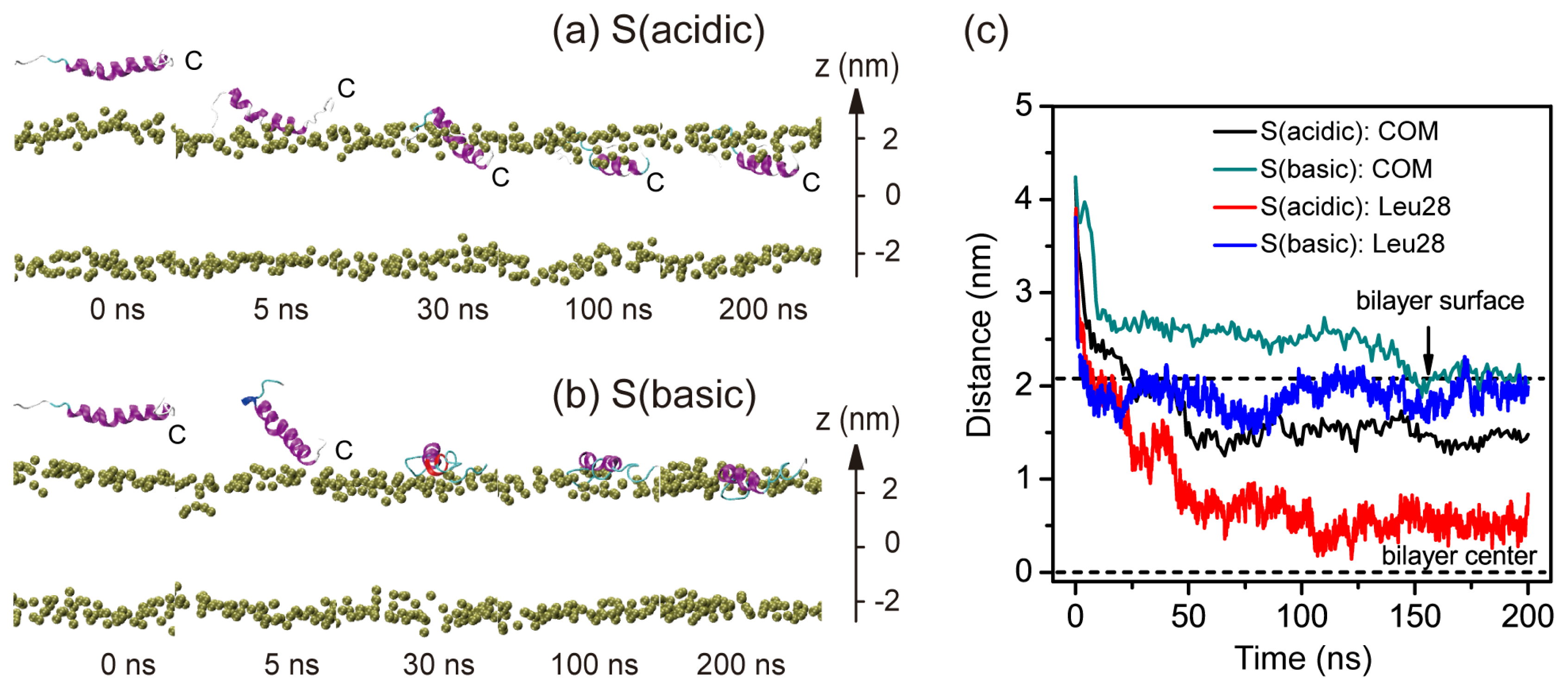
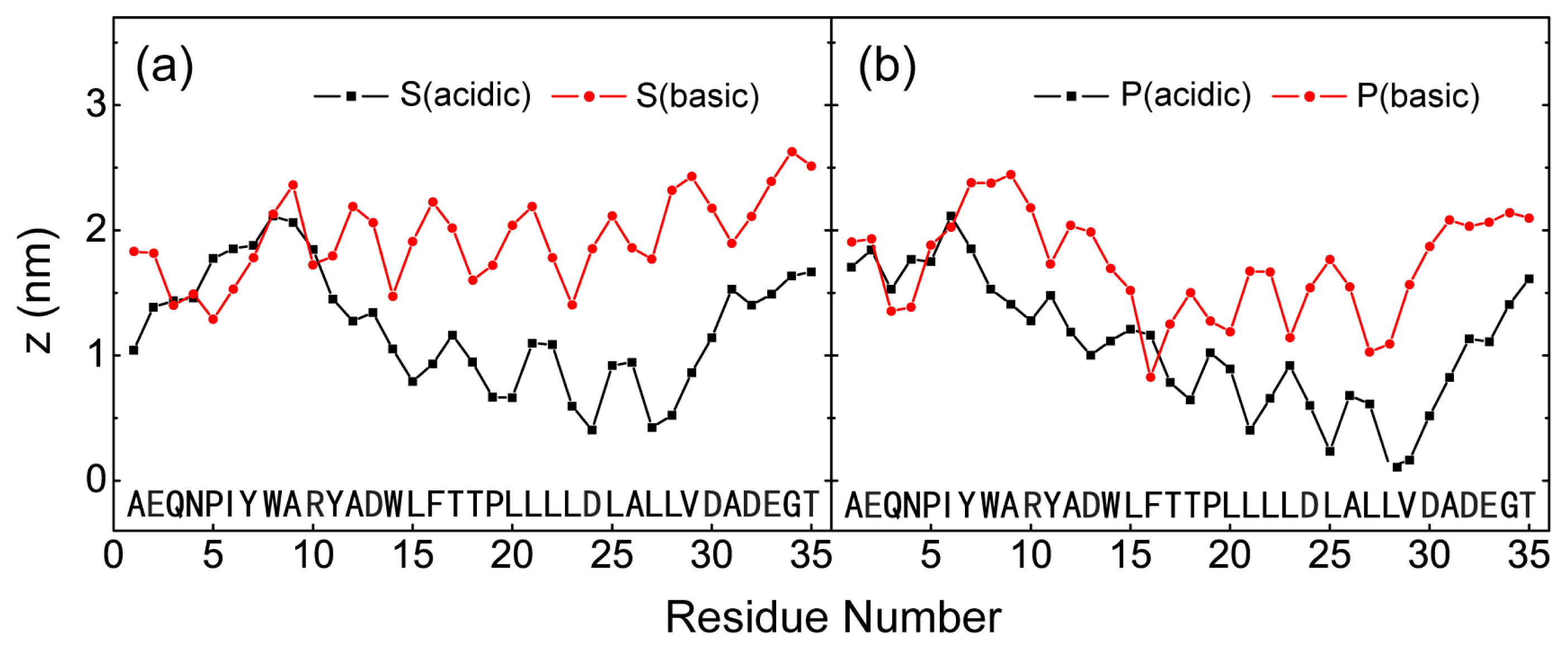
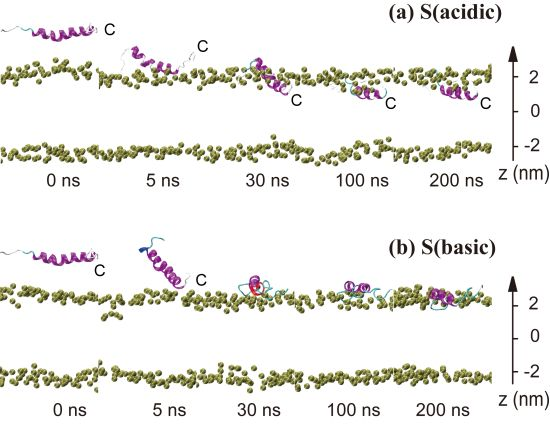
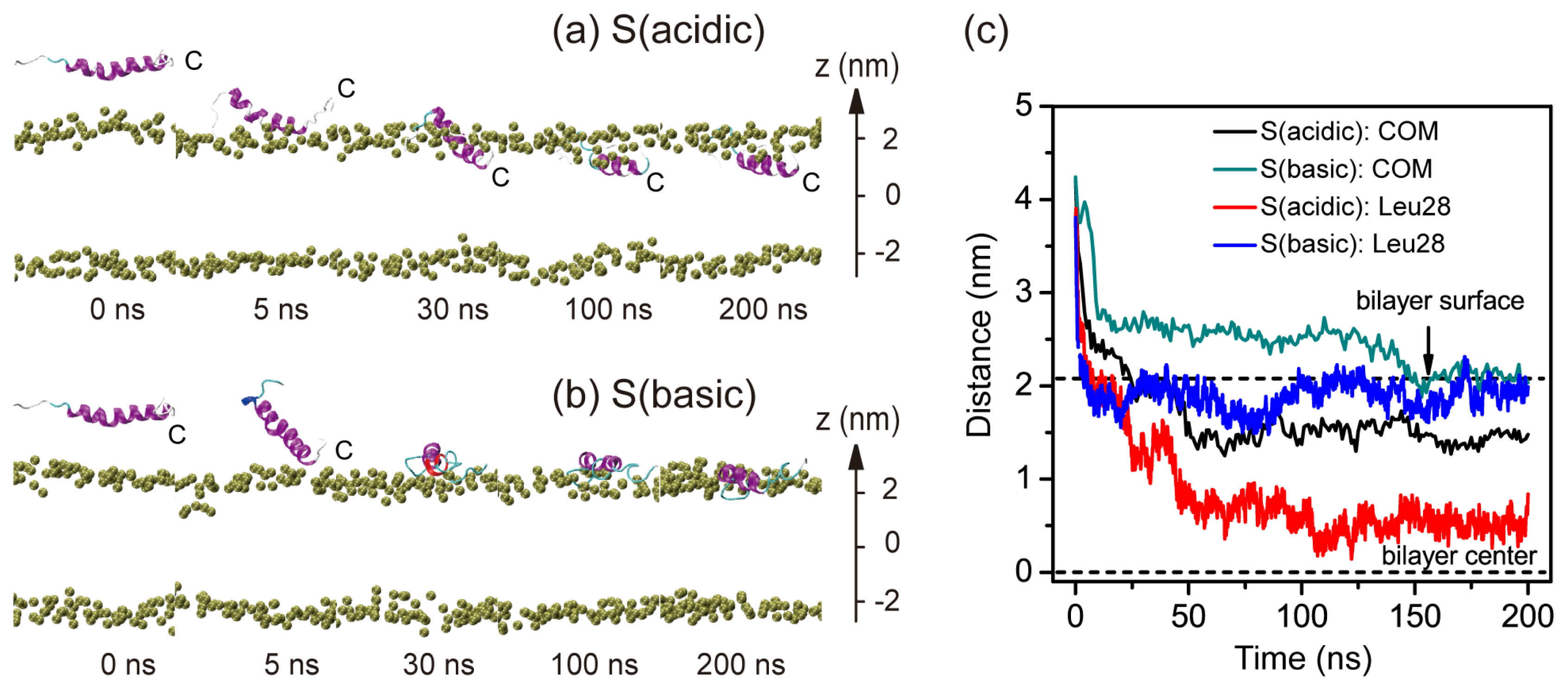
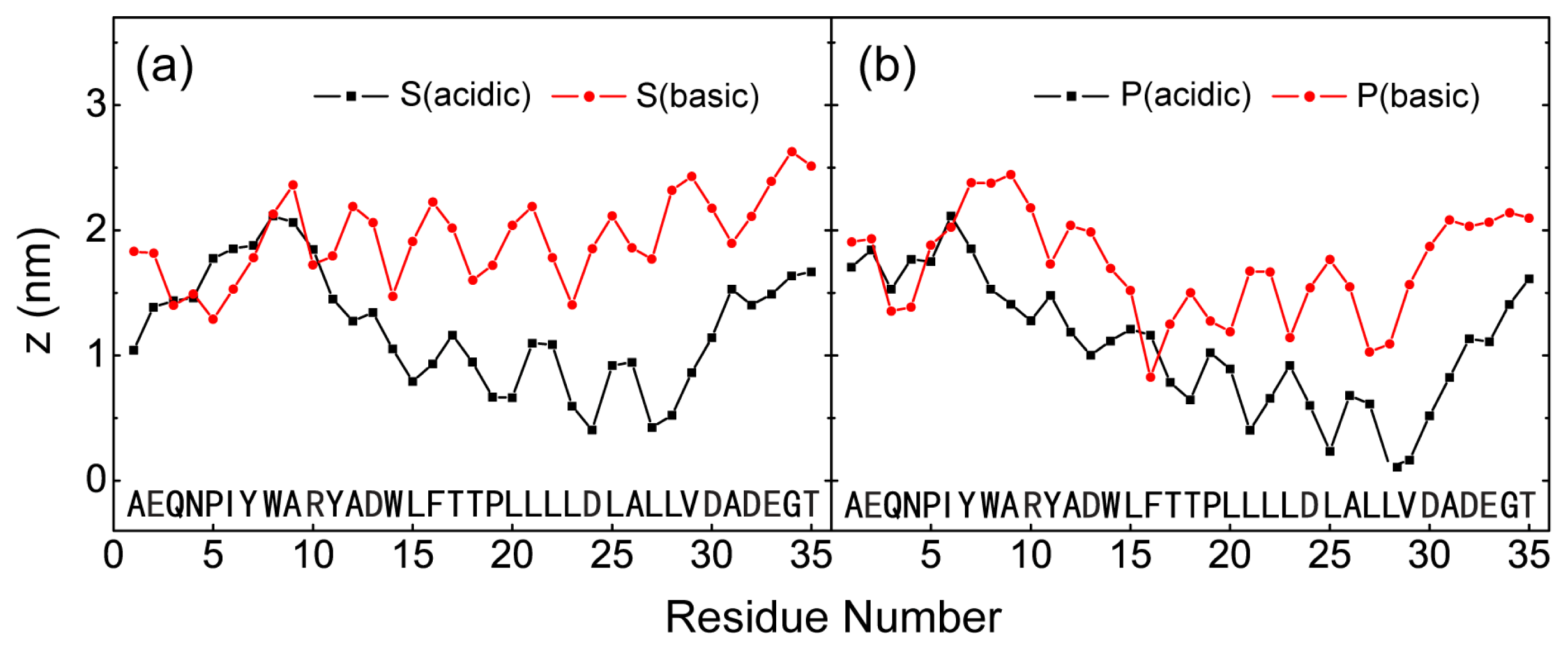
2.1. In Aqueous Solution, pHLIP Can Spontaneously Insert into POPC Bilayer at Acidic pH, while Binds to the Membrane Surface without Inserting at Basic pH
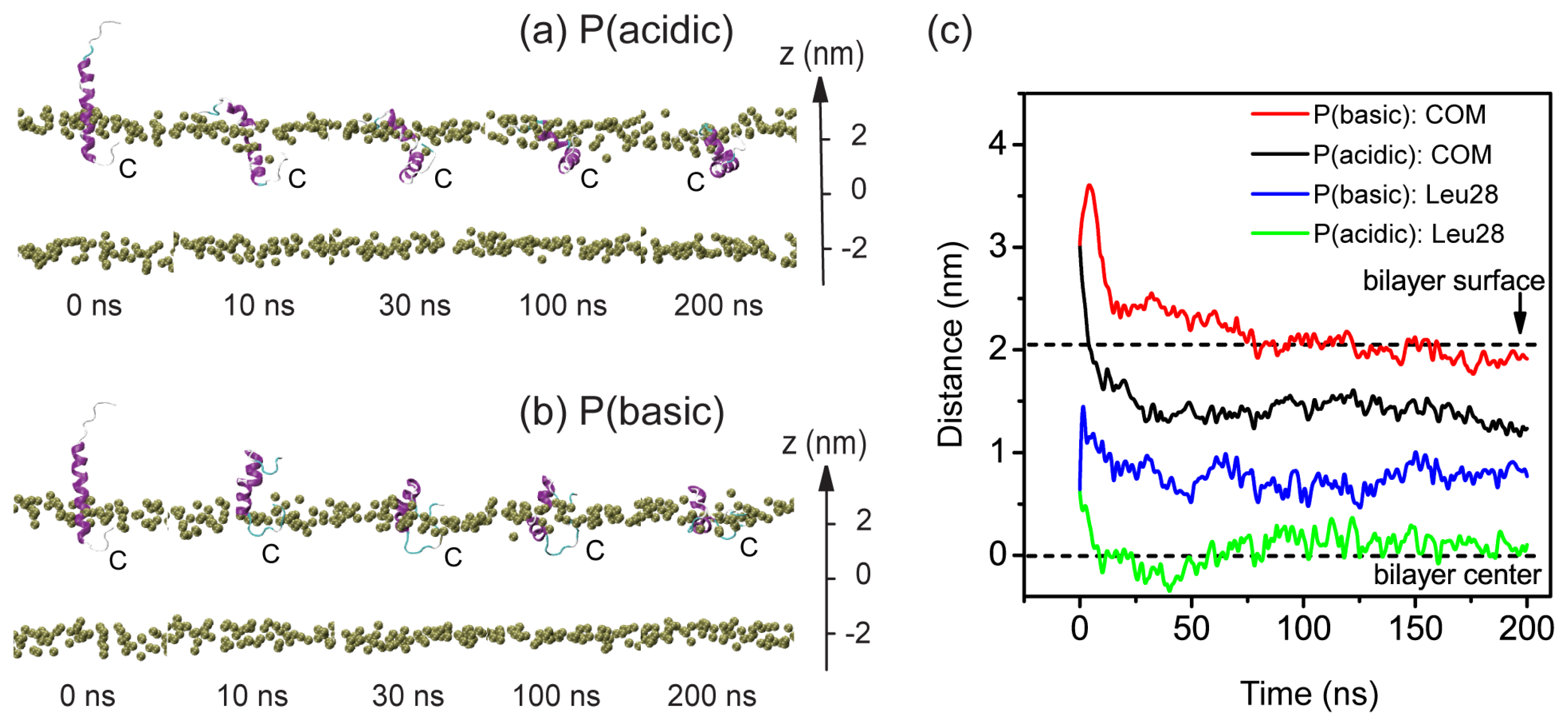
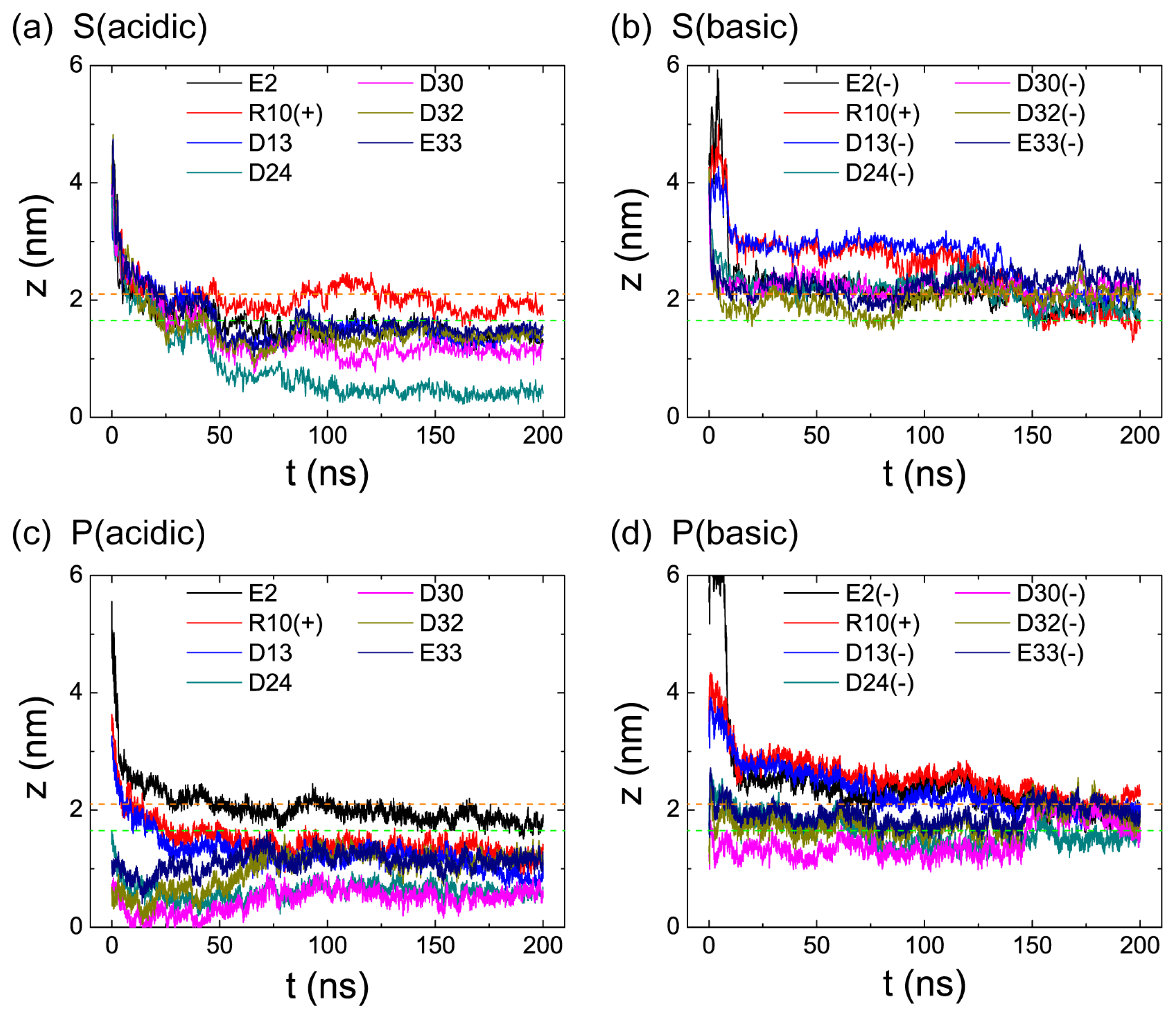
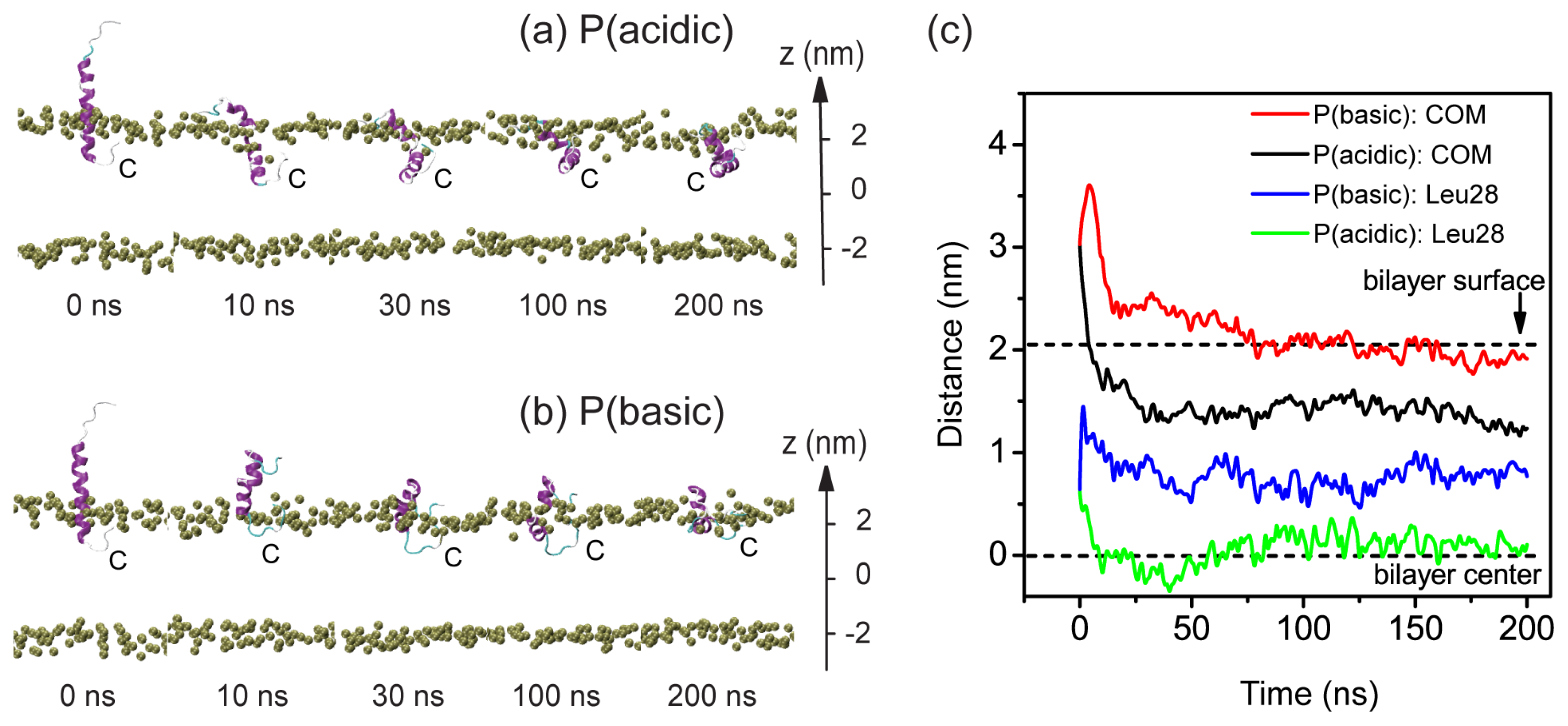
2.2. When Partially Pre-Inserted in POPC Bilayer, pHLIP Can Insert Deep into the Bilayer at Acidic pH while Moves towards the Bilayer Surface at Basic pH
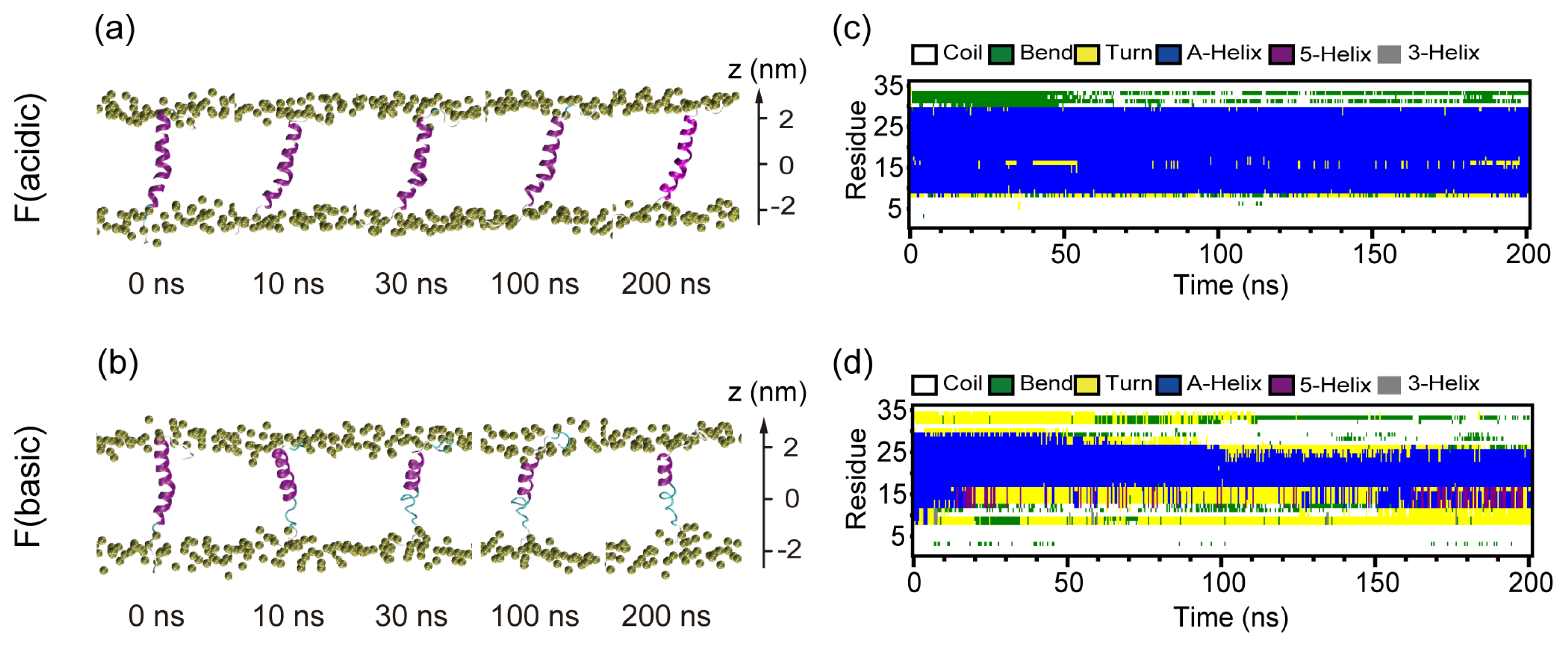
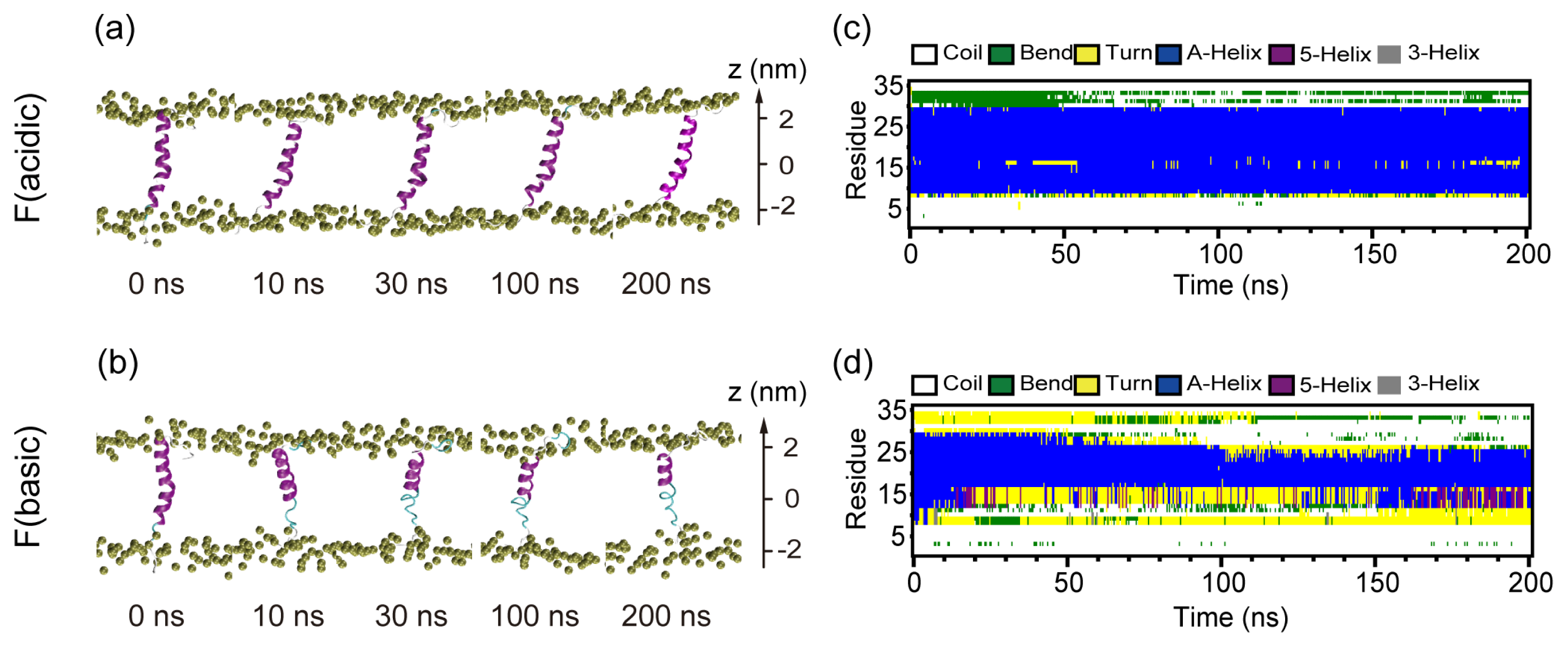
2.3. When Fully Pre-Inserted inside POPC Bilayer, pHLIP Keeps Intact α-Helical Structure at Acidic pH while the Helix Unfolds at Basic pH
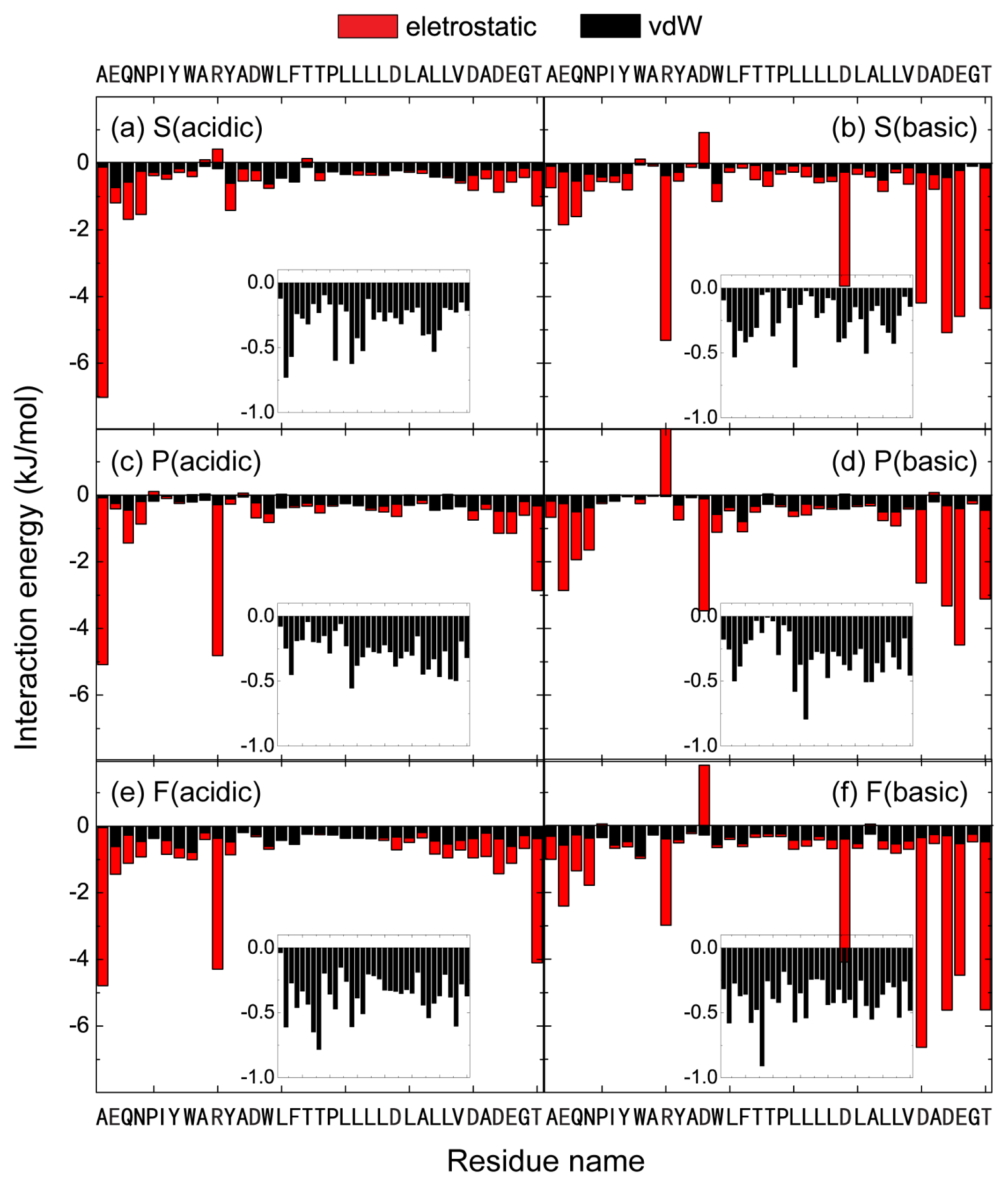
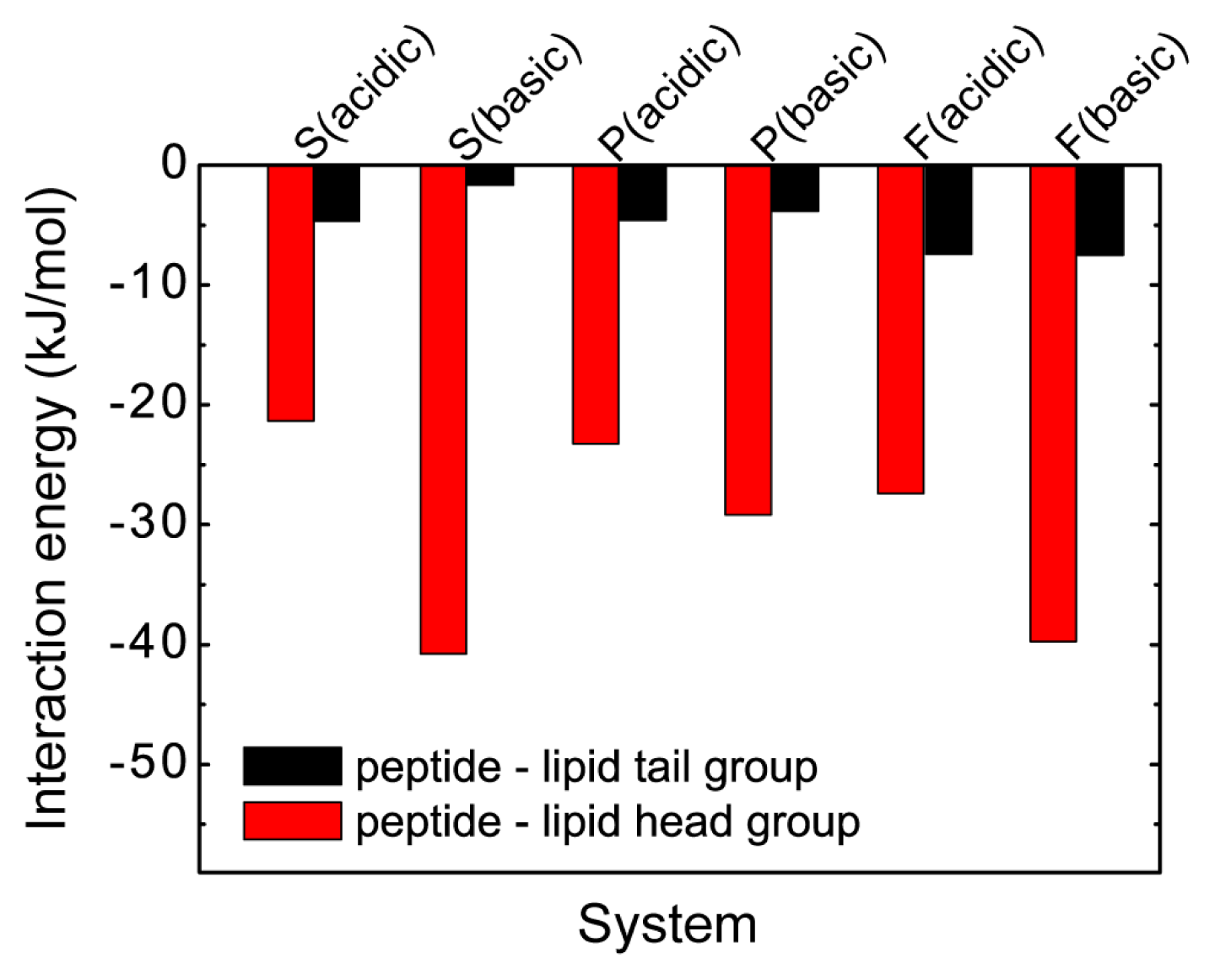
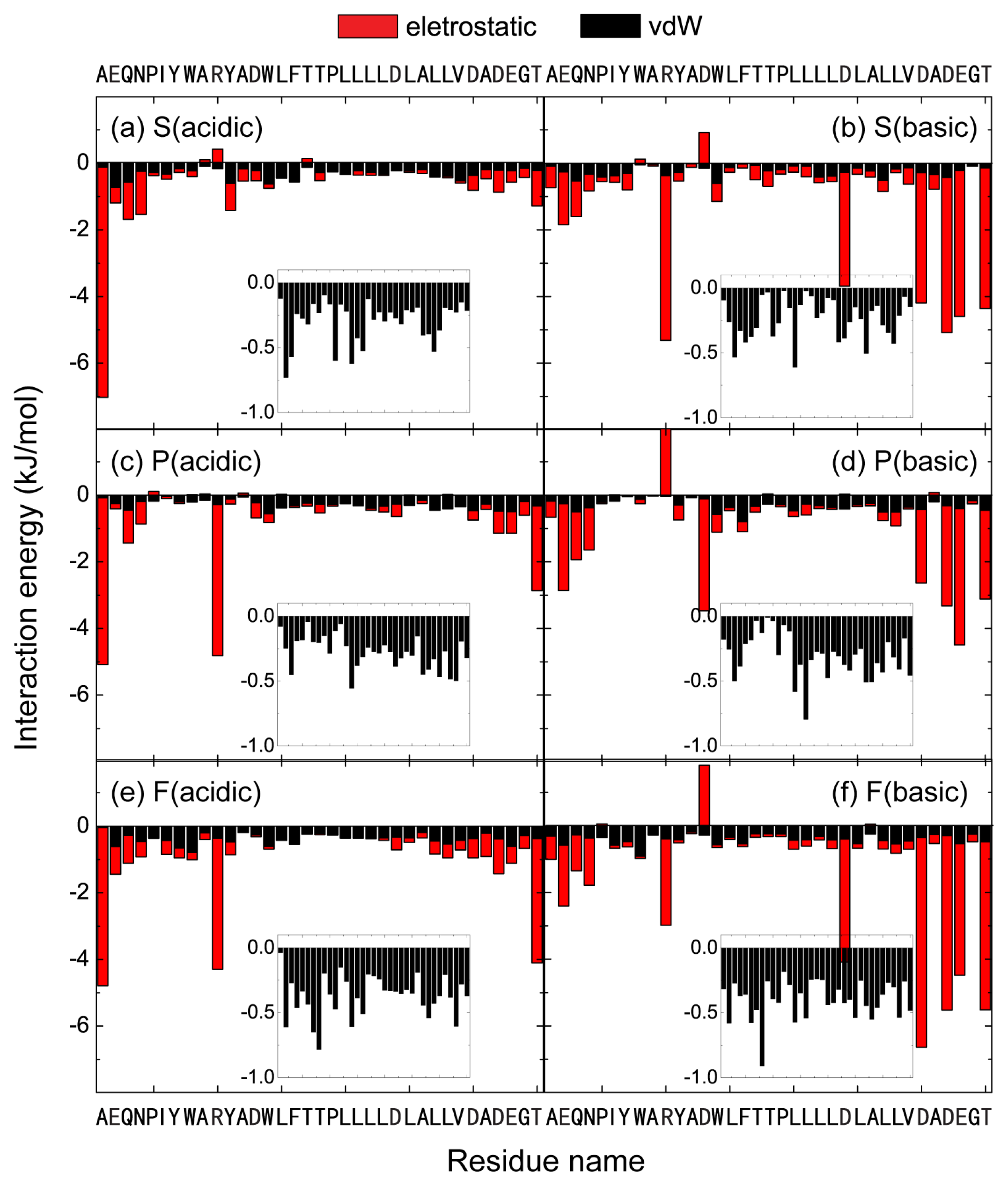
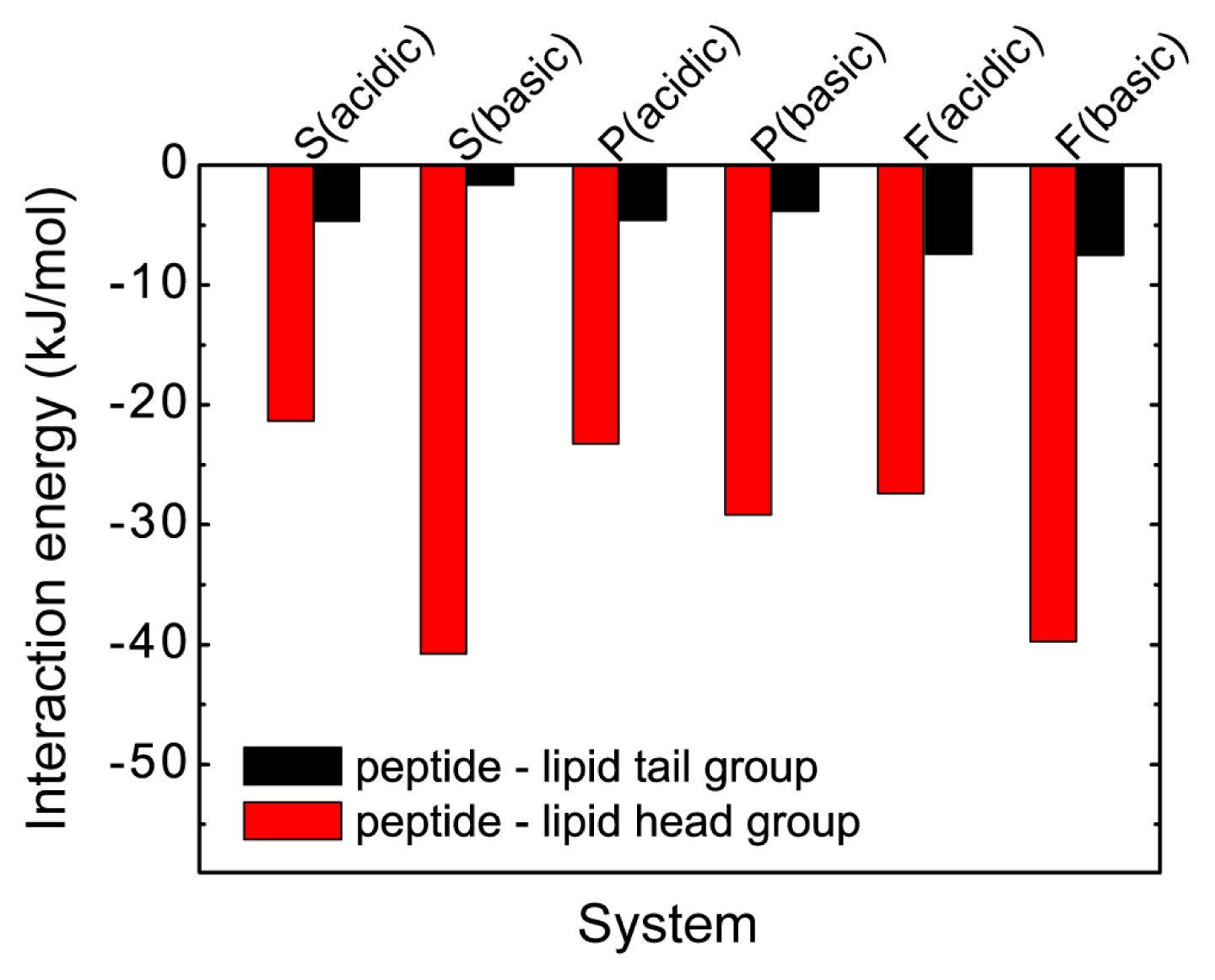
2.4. The Protonation of Transmembrane Residues Asp and the C-Terminal Residues Asp/Glu Facilitates Their Bilayer Entry Due to the Increased Hydrophobicity of pHLIP at Acidic pH, while the Strong Electrostatic Interaction between the Deprotonated Asp/Glu Residues and Lipid Headgroups Hinders the Bilayer Insertion at Basic pH
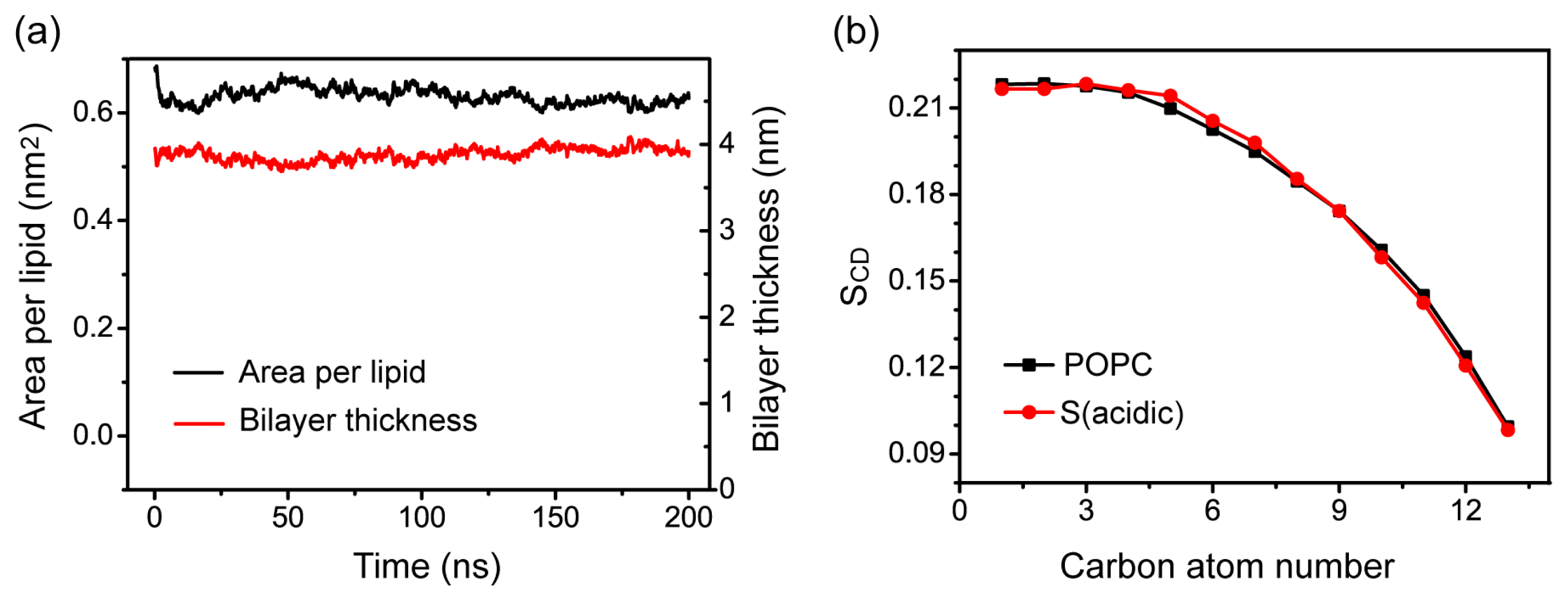
2.5. Memebrane Insertion of pHLIP Does Not Disrupt the Integrity of POPC Bilayer Structure
3. Experimental Section
3.1. pHLIP-POPC System
3.2. Molecular Dynamics Simulations
3.3. Analysis
4. Conclusions
Supplementary Information
ijms-14-14532-s001.pdfAcknowledgments
Conflict of Interest
References
- Reshetnyak, Y.K.; Andreev, O.A.; Lehnert, U.; Engelman, D.M. Translocation of molecules into cells by ph-dependent insertion of a transmembrane helix. Proc. Natl. Acad. Sci. USA 2006, 103, 6460–6465. [Google Scholar]
- An, M.; Wijesinghe, D.; Andreev, O.A.; Reshetnyak, Y.K.; Engelman, D.M. Ph-(low)-insertion-peptide (phlip) translocation of membrane impermeable phalloidin toxin inhibits cancer cell proliferation. Proc. Natl. Acad. Sci. USA 2010, 107, 20246–20250. [Google Scholar]
- Andreev, O.A.; Karabadzhak, A.G.; Weerakkody, D.; Andreev, G.O.; Engelman, D.M.; Reshetnyak, Y.K. Ph (low) insertion peptide (phlip) inserts across a lipid bilayer as a helix and exits by a different path. Proc. Natl. Acad. Sci. USA 2010, 107, 4081–4086. [Google Scholar]
- Reshetnyak, Y.K.; Andreev, O.A.; Segala, M.; Markin, V.S.; Engelman, D.M. Energetics of peptide (phlip) binding to and folding across a lipid bilayer membrane. Proc. Natl. Acad. Sci. USA 2008, 105, 15340–15345. [Google Scholar]
- Hunt, J.F.; Rath, P.; Rothschild, K.J.; Engelman, D.M. Spontaneous, ph-dependent membrane insertion of a transbilayer alpha-helix. Biochemistry 1997, 36, 15177–15192. [Google Scholar]
- Reshetnyak, Y.K.; Segala, M.; Andreev, O.A.; Engelman, D.M. A monomeric membrane peptide that lives in three worlds: In solution, attached to, and inserted across lipid bilayers. Biophys. J 2007, 93, 2363–2372. [Google Scholar]
- Andreev, O.A.; Dupuy, A.D.; Segala, M.; Sandugu, S.; Serra, D.A.; Chichester, C.O.; Engelman, D.M.; Reshetnyak, Y.K. Mechanism and uses of a membrane peptide that targets tumors and other acidic tissues in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 7893–7898. [Google Scholar]
- Musial-Siwek, M.; Karabadzhak, A.; Andreev, O.A.; Reshetnyak, Y.K.; Engelman, D.M. Tuning the insertion properties of phlip. Biochim. Biophys. Acta 2010, 1798, 1041–1046. [Google Scholar]
- Makovitzki, A.; Fink, A.; Shai, Y. Suppression of human solid tumor growth in mice by intratumor and systemic inoculation of histidine-rich and ph-dependent host defense-like lytic peptides. Cancer Res 2009, 69, 3458–3463. [Google Scholar]
- Zoonens, M.; Reshetnyak, Y.K.; Engelman, D.M. Bilayer interactions of phlip, a peptide that can deliver drugs and target tumors. Biophys. J 2008, 95, 225–235. [Google Scholar]
- Thevenin, D.; An, M.; Engelman, D.M. Phlip-mediated translocation of membrane-impermeable molecules into cells. Chem. Biol 2009, 16, 754–762. [Google Scholar]
- Vavere, A.L.; Biddlecombe, G.B.; Spees, W.M.; Garbow, J.R.; Wijesinghe, D.; Andreev, O.A.; Engelman, D.M.; Reshetnyak, Y.K.; Lewis, J.S. A novel technology for the imaging of acidic prostate tumors by positron emission tomography. Cancer Res 2009, 69, 4510–4516. [Google Scholar]
- Andreev, O.A.; Engelman, D.M.; Reshetnyak, Y.K. Targeting acidic diseased tissue: New technology based on use of the ph (low) insertion peptide (phlip). Chim. Oggi 2009, 27, 34–37. [Google Scholar]
- Tang, J.; Gai, F. Dissecting the membrane binding and insertion kinetics of a phlip peptide. Biochemistry 2008, 47, 8250–8252. [Google Scholar]
- Patzelt, H.; Simon, B.; terLaak, A.; Kessler, B.; Kuhne, R.; Schmieder, P.; Oesterhelt, D.; Oschkinat, H. The structures of the active center in dark-adapted bacteriorhodopsin by solution-state nmr spectroscopy. Proc. Natl. Acad. Sci. USA 2002, 99, 9765–9770. [Google Scholar]
- Gkeka, P.; Sarkisov, L. Interactions of phospholipid bilayers with several classes of amphiphilic alpha-helical peptides: Insights from coarse-grained molecular dynamics simulations. J. Phys. Chem. B 2010, 114, 826–839. [Google Scholar]
- Rychkova, A.; Vicatos, S.; Warshel, A. On the energetics of translocon-assisted insertion of charged transmembrane helices into membranes. Proc. Natl. Acad. Sci. USA 2010, 107, 17598–17603. [Google Scholar]
- Soliman, W.; Bhattacharjee, S.; Kaur, K. Interaction of an antimicrobial peptide with a model lipid bilayer using molecular dynamics simulation. Langmuir 2009, 25, 6591–6595. [Google Scholar]
- Dunkin, C.M.; Pokorny, A.; Almeida, P.F.; Lee, H.S. Molecular dynamics studies of transportan 10 (tp10) interacting with a popc lipid bilayer. J. Phys. Chem. B 2011, 115, 1188–1198. [Google Scholar]
- Chang, Z.; Luo, Y.; Zhang, Y.; Wei, G. Interactions of abeta25–35 beta-barrel-like oligomers with anionic lipid bilayer and resulting membrane leakage: An all-atom molecular dynamics study. J. Phys. Chem. B 2011, 115, 1165–1174. [Google Scholar]
- Yesylevskyy, S.; Marrink, S.J.; Mark, A.E. Alternative mechanisms for the interaction of the cell-penetrating peptides penetratin and the tat peptide with lipid bilayers. Biophys. J 2009, 97, 40–49. [Google Scholar]
- Fleming, E.; Maharaj, N.P.; Chen, J.L.; Nelson, R.B.; Elmore, D.E. Effect of lipid composition on buforin ii structure and membrane entry. Proteins 2008, 73, 480–491. [Google Scholar]
- Ulmschneider, M.B.; Doux, J.P.; Killian, J.A.; Smith, J.C.; Ulmschneider, J.P. Mechanism and kinetics of peptide partitioning into membranes from all-atom simulations of thermostable peptides. J. Am. Chem. Soc 2010, 132, 3452–3460. [Google Scholar]
- Elmore, D.E. Insights into buforin ii membrane translocation from molecular dynamics simulations. Peptides 2012, 38, 357–362. [Google Scholar]
- Tieleman, D.P.; Sansom, M.S.; Berendsen, H.J. Alamethicin helices in a bilayer and in solution: Molecular dynamics simulations. Biophys. J 1999, 76, 40–49. [Google Scholar]
- Leontiadou, H.; Mark, A.E.; Marrink, S.J. Antimicrobial peptides in action. J. Am. Chem. Soc 2006, 128, 12156–12161. [Google Scholar]
- Jang, H.; Ma, B.; Woolf, T.B.; Nussinov, R. Interaction of protegrin-1 with lipid bilayers: Membrane thinning effect. Biophys. J 2006, 91, 2848–2859. [Google Scholar]
- Zhang, Y.; Luo, Y.; Deng, Y.; Mu, Y.; Wei, G. Lipid interaction and membrane perturbation of human islet amyloid polypeptide monomer and dimer by molecular dynamics simulations. PLoS One 2012, 7, e38191. [Google Scholar]
- Yu, X.; Zheng, J. Cholesterol promotes the interaction of alzheimer beta-amyloid monomer with lipid bilayer. J. Mol. Biol 2012, 421, 561–571. [Google Scholar]
- Tofoleanu, F.; Buchete, N.V. Molecular interactions of alzheimer’s abeta protofilaments with lipid membranes. J. Mol. Biol 2012, 421, 572–586. [Google Scholar]
- Jia, Y.; Qian, Z.; Zhang, Y.; Wei, G. Adsorption and orientation of human islet amyloid polypeptide (hiapp) monomer at anionic lipid bilayers: Implications for membrane-mediated aggregation. Int. J. Mol. Sci 2013, 14, 6241–6258. [Google Scholar]
- Kučerka, N.; Tristram-Nagle, S.; Nagle, J. Structure of fully hydrated fluid phase lipid bilayers with monounsaturated chains. J. Membr. Biol 2006, 208, 193–202. [Google Scholar]
- Duan, M.; Fan, J.; Huo, S. Conformations of islet amyloid polypeptide monomers in a membrane environment: Implications for fibril formation. PLoS One 2012, 7, e47150. [Google Scholar]
- Hoff, B.; Strandberg, E.; Ulrich, A.S.; Tieleman, D.P.; Posten, C. 2h-nmr study and molecular dynamics simulation of the location, alignment, and mobility of pyrene in popc bilayers. Biophys. J 2005, 88, 1818–1827. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. Vmd: Visual molecular dynamics. J. Mol. Graph 1996, 14, 33–38. [Google Scholar]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. Gromacs: Fast, flexible, and free. J. Comput. Chem 2005, 26, 1701–1718. [Google Scholar]
- Piccinini, E.; Ceccarelli, M.; Affinito, F.; Brunetti, R.; Jacoboni, C. Biased molecular simulations for free-energy mapping: A comparison on the kcsa channel as a test case. J. Chem. Theory Comput 2007, 4, 173–183. [Google Scholar]
- Berendsen, H.J.C.; Postma, J.P.M.; Gunsteren, W.F.V.; Hermans, J. Interaction Models for Water in Relation to Protein Hydration; D. Reidel Pulishing Co: Dordrecht, The Netherlands, 1981; pp. 331–342. [Google Scholar]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. Lincs: A linear constraint solver for molecular simulations. J. Comput. Chem 1997, 18, 1463–1472. [Google Scholar]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the shake and rattle algorithm for rigid water models. J. Comput. Chem 1992, 13, 952–962. [Google Scholar]
- Darden, T.; York, D.; Pedersen, L. Particle mesh ewald: An n·log(n) method for ewald sums in large systems. J. Chem. Phys 1993, 98, 10089–10092. [Google Scholar]
- Kandt, C.; Ash, W.L.; Tieleman, D.P. Setting up and running molecular dynamics simulations of membrane proteins. Methods 2007, 41, 475–488. [Google Scholar]
- Patra, M.; Karttunen, M.; Hyvonen, M.T.; Falck, E.; Lindqvist, P.; Vattulainen, I. Molecular dynamics simulations of lipid bilayers: Major artifacts due to truncating electrostatic interactions. Biophys. J 2003, 84, 3636–3645. [Google Scholar]
- Vermeer, L.S.; de Groot, B.L.; Reat, V.; Milon, A.; Czaplicki, J. Acyl chain order parameter profiles in phospholipid bilayers: Computation from molecular dynamics simulations and comparison with 2h nmr experiments. Eur. Biophys. J 2007, 36, 919–931. [Google Scholar]
- Seelig, J.; Waespe-Sarcevic, N. Molecular order in cis and trans unsaturated phospholipid bilayers. Biochemistry 1978, 17, 3310–3315. [Google Scholar]
- Chau, P.L.; Hardwick, A.J. A new order parameter for tetrahedral configurations. Mol. Phys 1998, 93, 511–518. [Google Scholar]
- Petrache, H.I.; Tu, K.; Nagle, J.F. Analysis of simulated nmr order parameters for lipid bilayer structure determination. Biophys. J 1999, 76, 2479–2487. [Google Scholar]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Insertion state of pHLIP | Name of MD run | Initial state | Number of atoms | Simulation box size (nm3) | Length of MD run (ns) |
|---|---|---|---|---|---|---|
| S | Surface | S (acidic) | Figure 1a | 27,083 | 6.6 × 6.5 × 9.0 | 200 |
| Surface | S (basic) | Figure 1b | 27,066 | 6.6 × 6.5 × 9.0 | 200 | |
| P | Partial insertion | P (acidic) | Figure 2a | 24,347 | 6.5 × 6.5 × 9.0 | 200 |
| Partial insertion | P (basic) | Figure 2b | 24,345 | 6.5 × 6.5 × 9.0 | 200 | |
| F | Full insertion | F (acidic) | Figure 4a | 16,808 | 6.4 × 6.3 × 6.5 | 200 |
| Full insertion | F (basic) | Figure 4b | 16,789 | 6.4 × 6.3 × 6.5 | 200 | |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Deng, Y.; Qian, Z.; Luo, Y.; Zhang, Y.; Mu, Y.; Wei, G. Membrane Binding and Insertion of a pHLIP Peptide Studied by All-Atom Molecular Dynamics Simulations. Int. J. Mol. Sci. 2013, 14, 14532-14549. https://doi.org/10.3390/ijms140714532
Deng Y, Qian Z, Luo Y, Zhang Y, Mu Y, Wei G. Membrane Binding and Insertion of a pHLIP Peptide Studied by All-Atom Molecular Dynamics Simulations. International Journal of Molecular Sciences. 2013; 14(7):14532-14549. https://doi.org/10.3390/ijms140714532
Chicago/Turabian StyleDeng, Yonghua, Zhenyu Qian, Yin Luo, Yun Zhang, Yuguang Mu, and Guanghong Wei. 2013. "Membrane Binding and Insertion of a pHLIP Peptide Studied by All-Atom Molecular Dynamics Simulations" International Journal of Molecular Sciences 14, no. 7: 14532-14549. https://doi.org/10.3390/ijms140714532