Chemical and Colloidal Stability of Carboxylated Core-Shell Magnetite Nanoparticles Designed for Biomedical Applications

 , ,
, ,
Abstract
:
1. Introduction
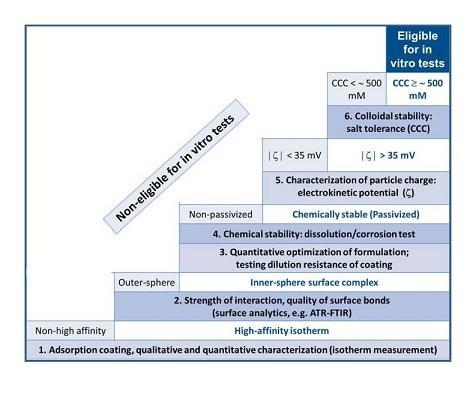
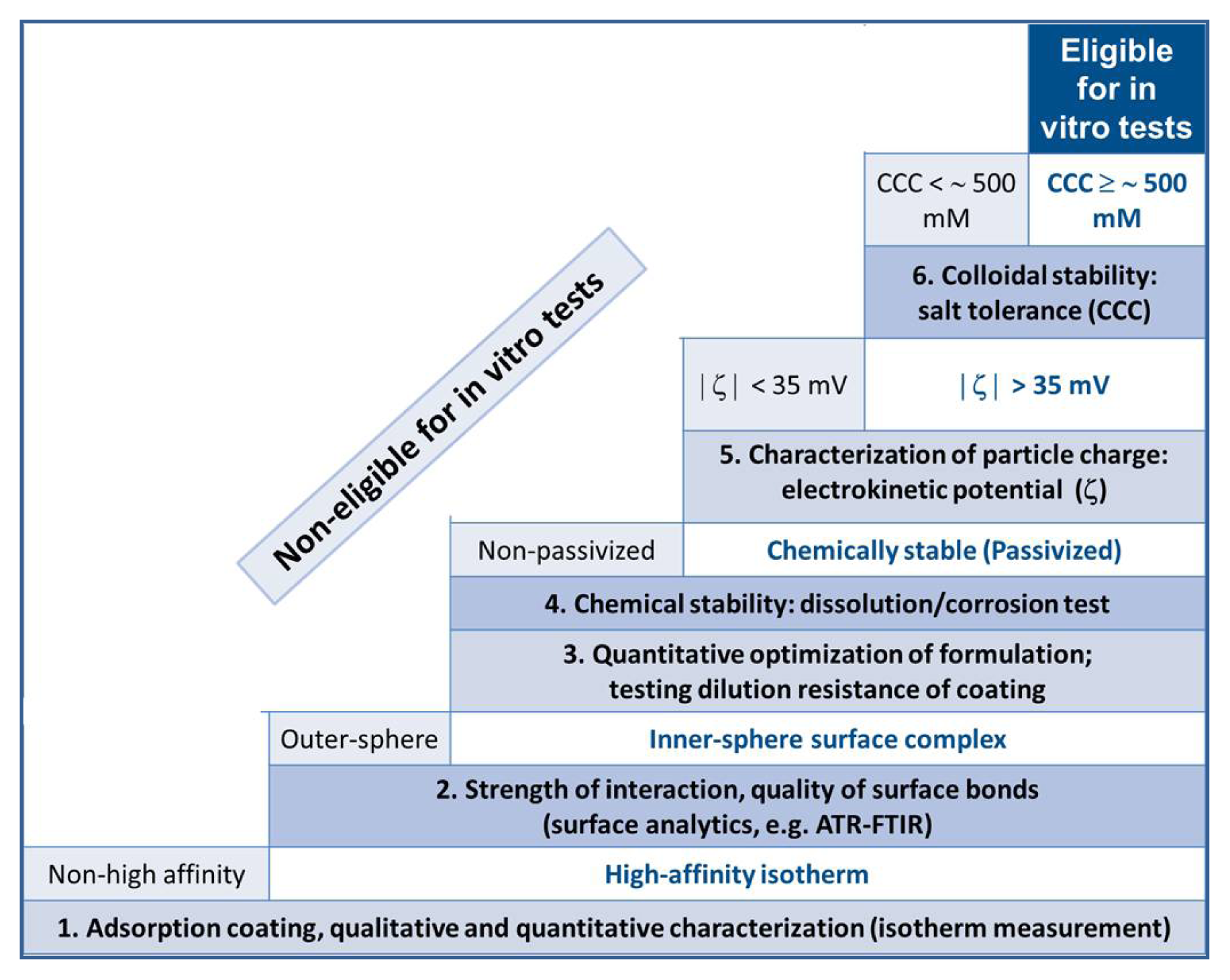
2. Results and Discussion
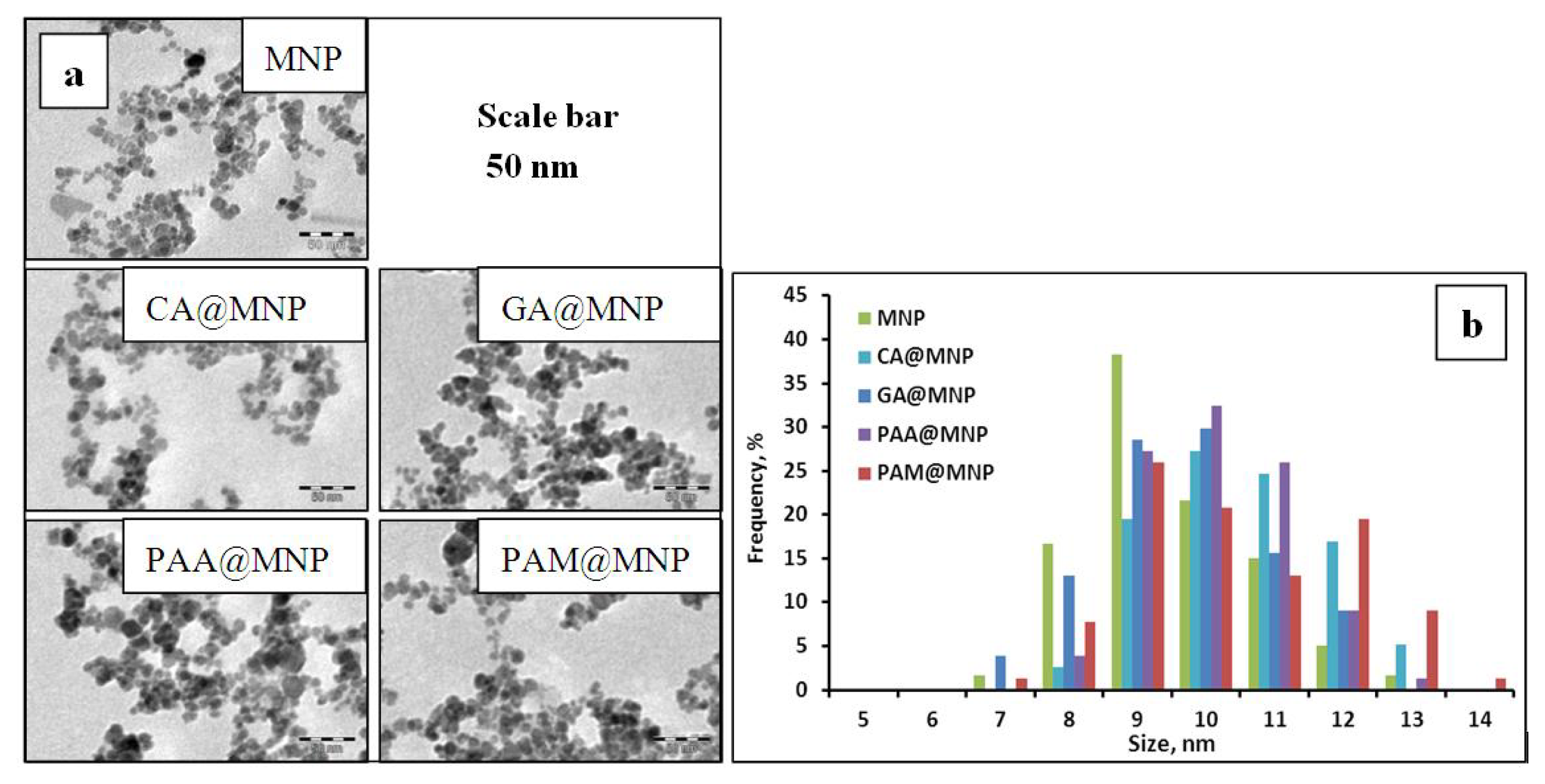
2.1. The Strength of Core-Shell Binding
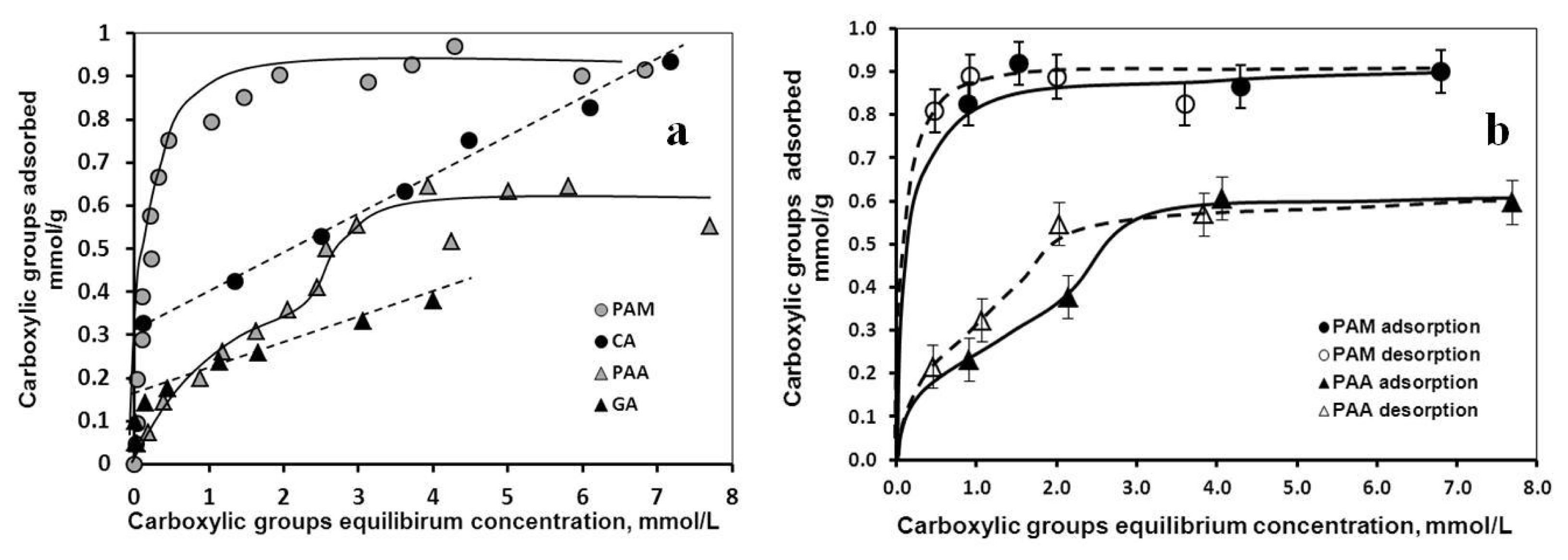
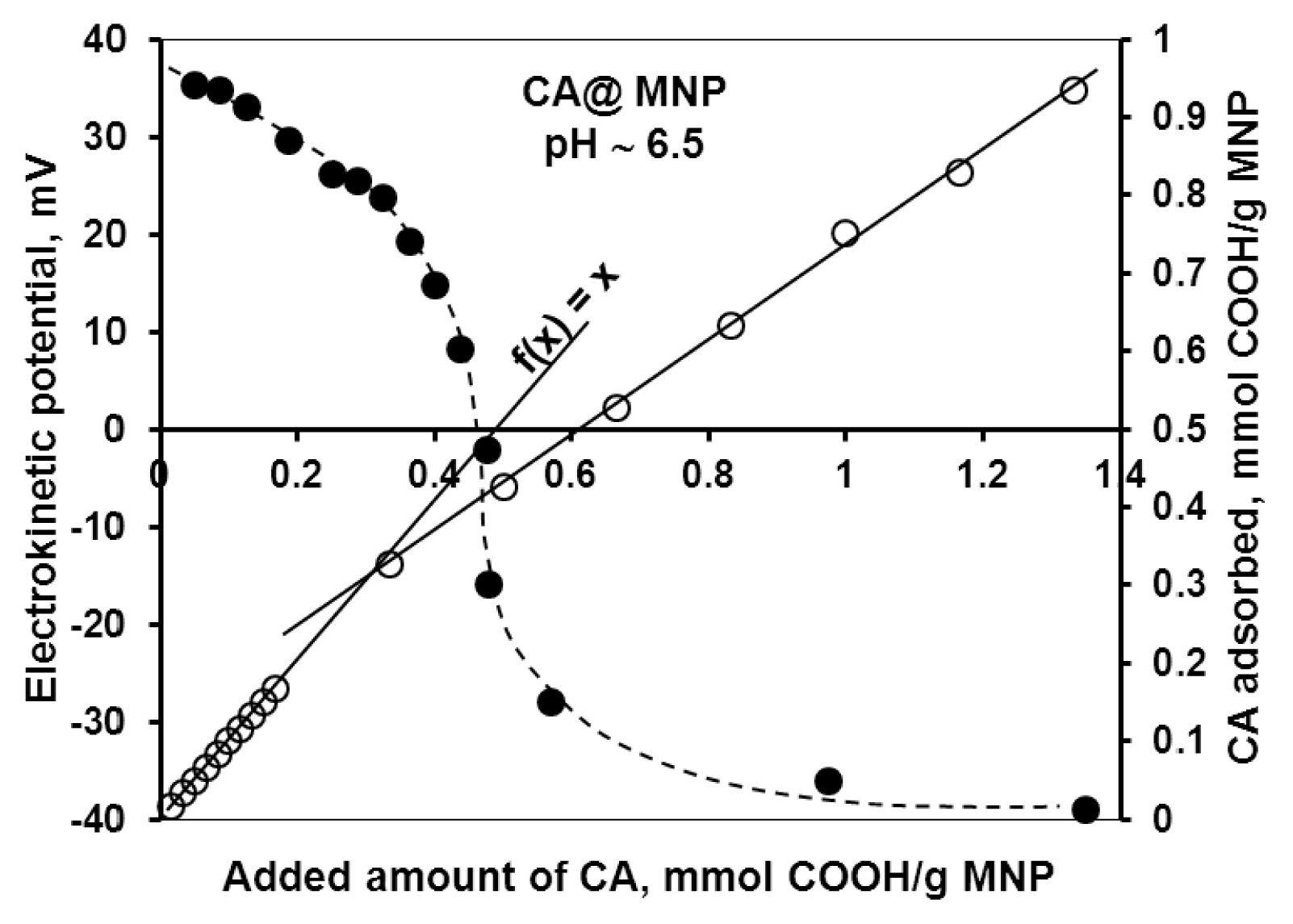
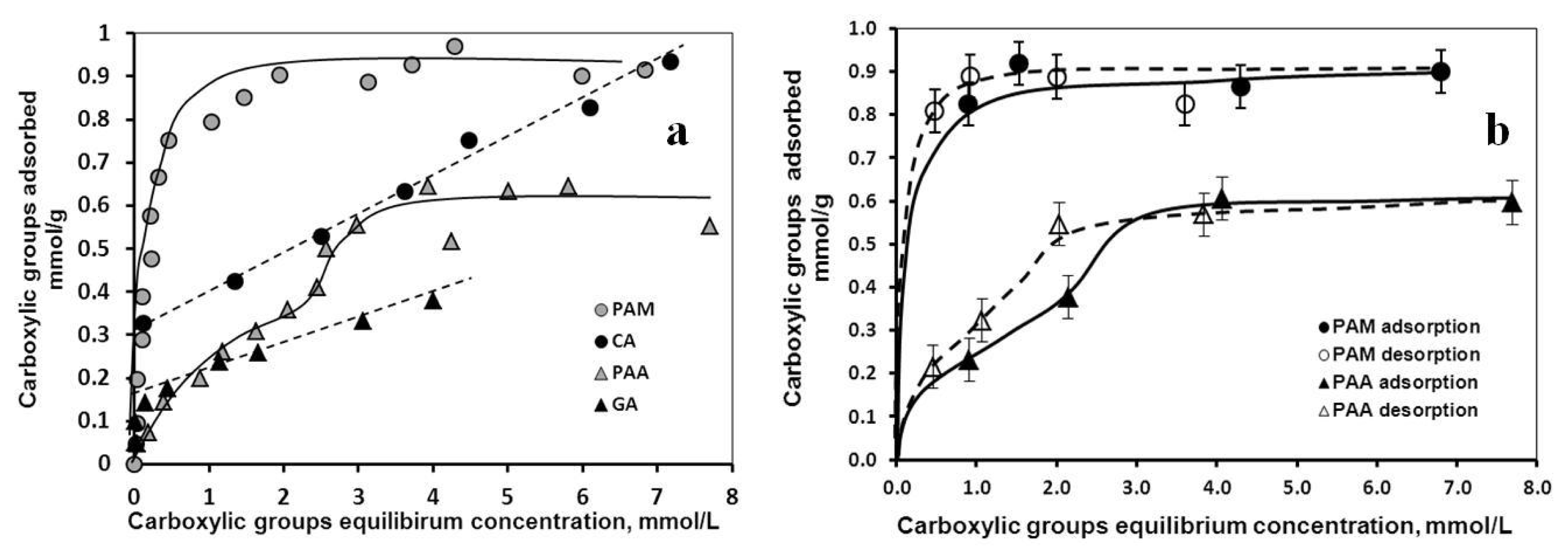
2.1.1. Adsorption and Desorption of the Carboxylates
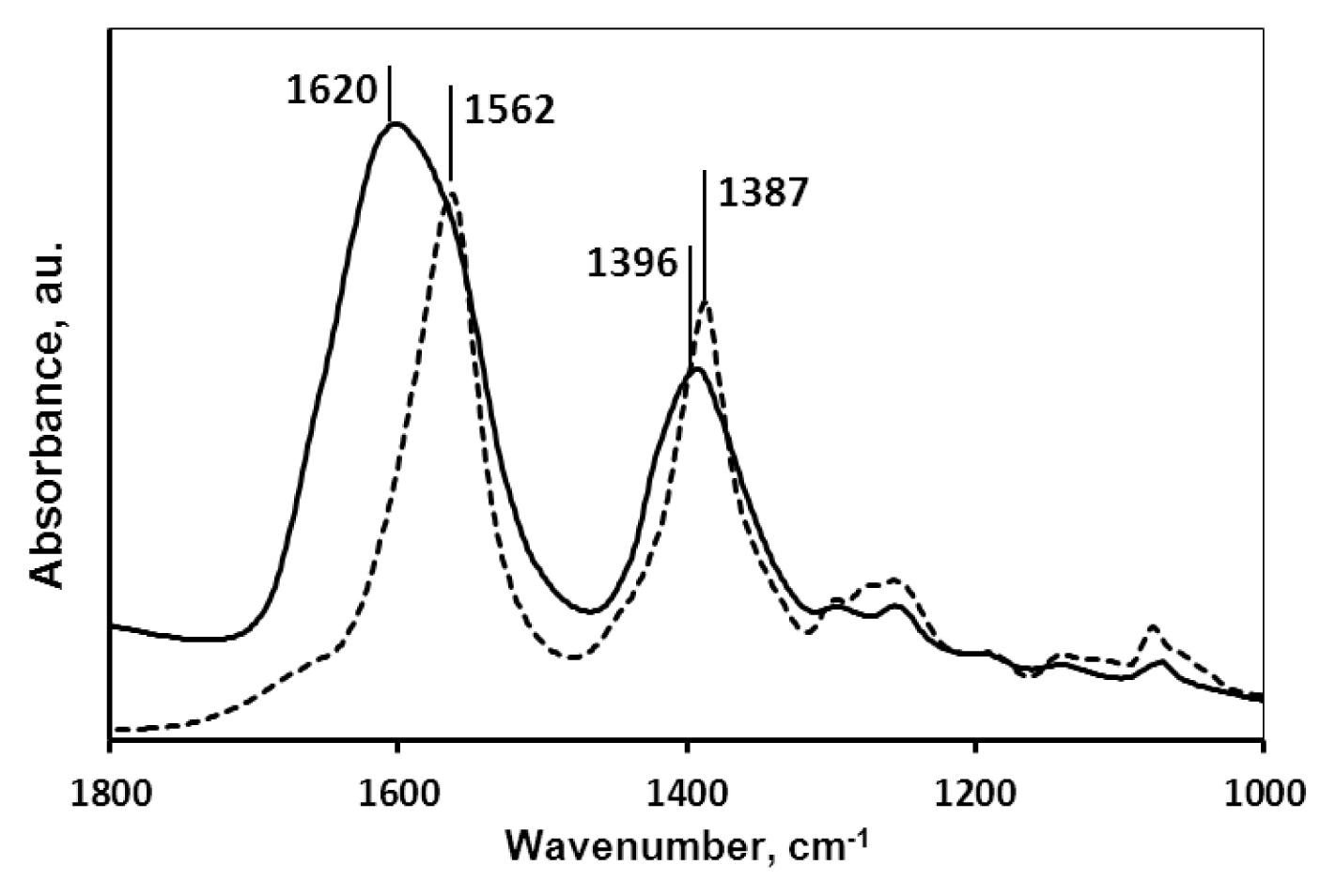
2.1.2. Molecular Mechanism of the Carboxylate-MNP Surface Interactions
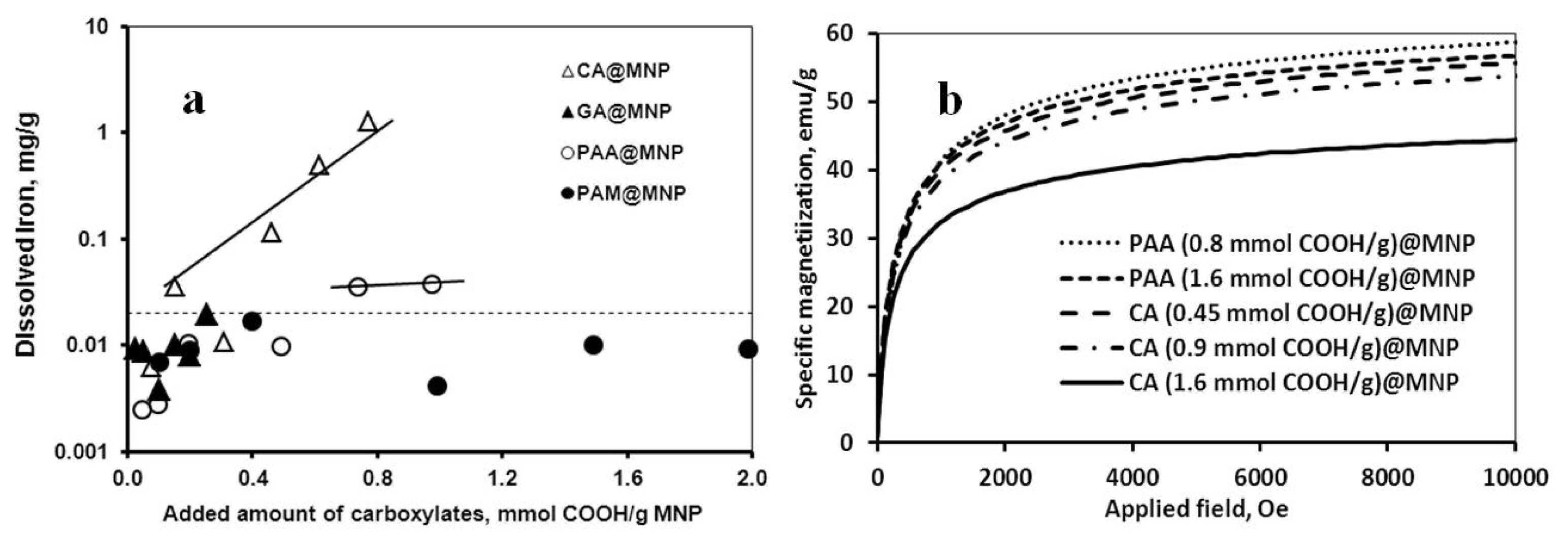
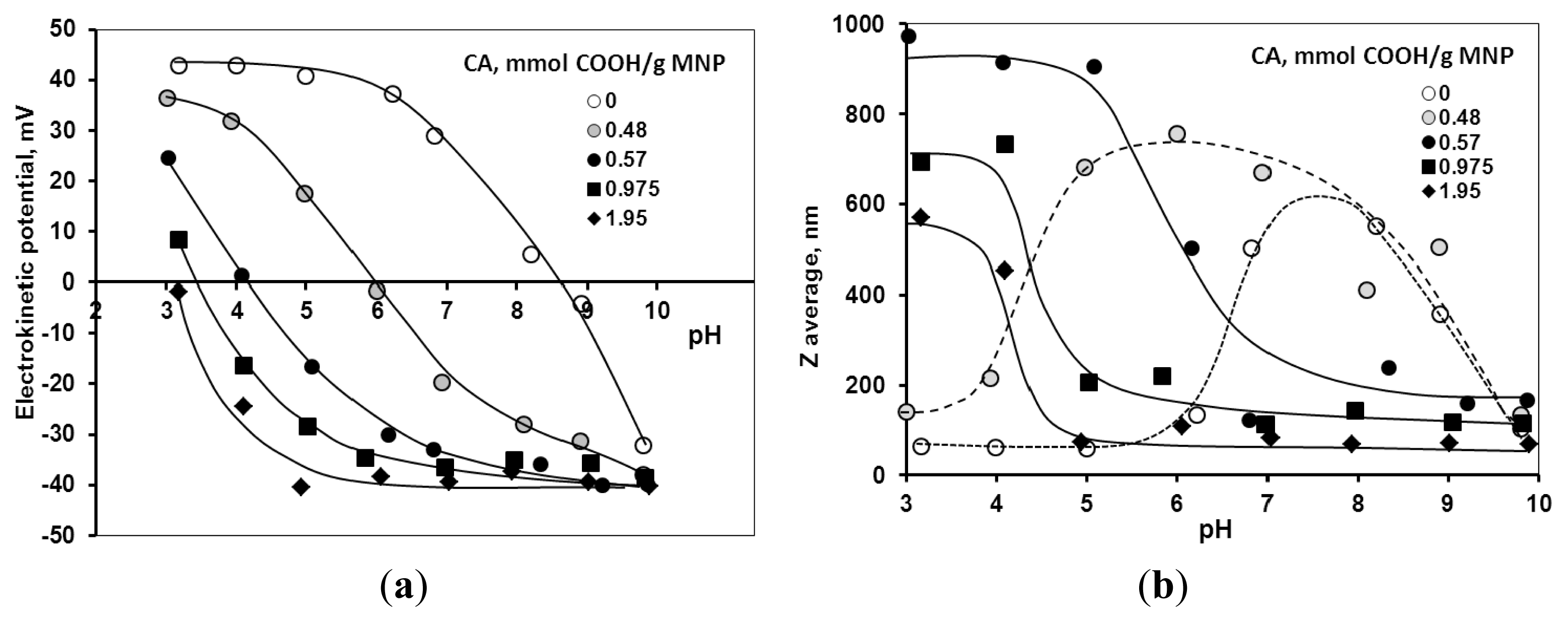
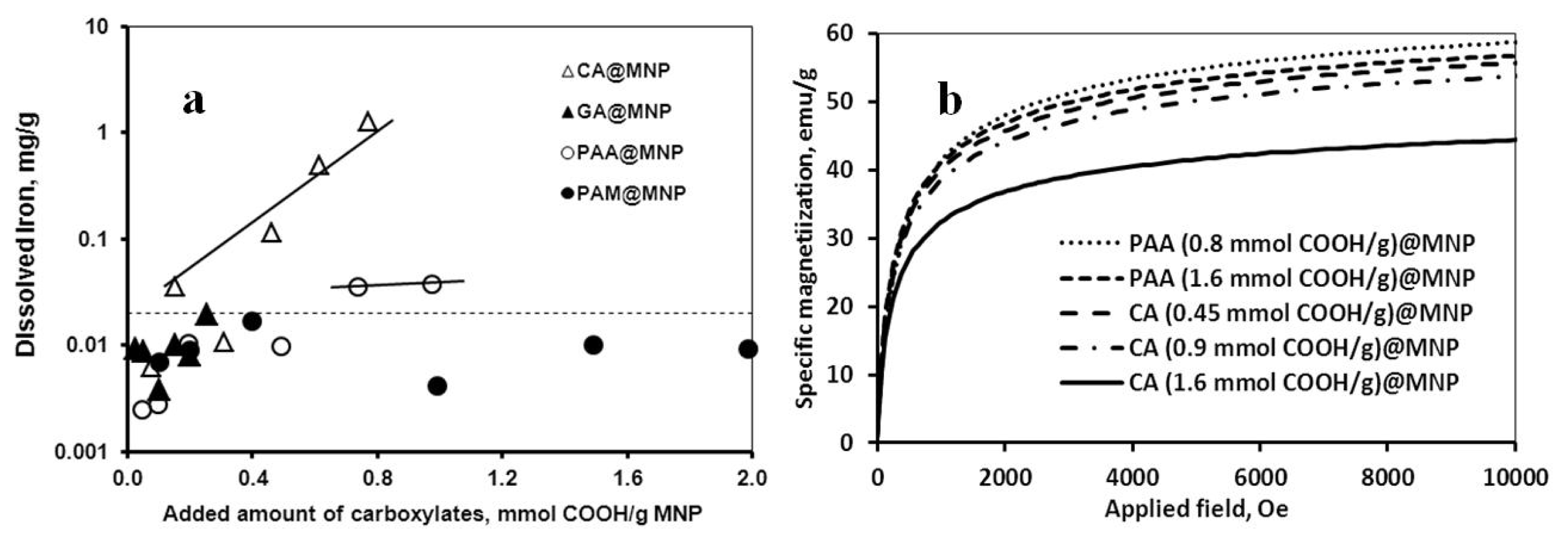
2.2. Chemical Stability of the Core-Shell MNPs
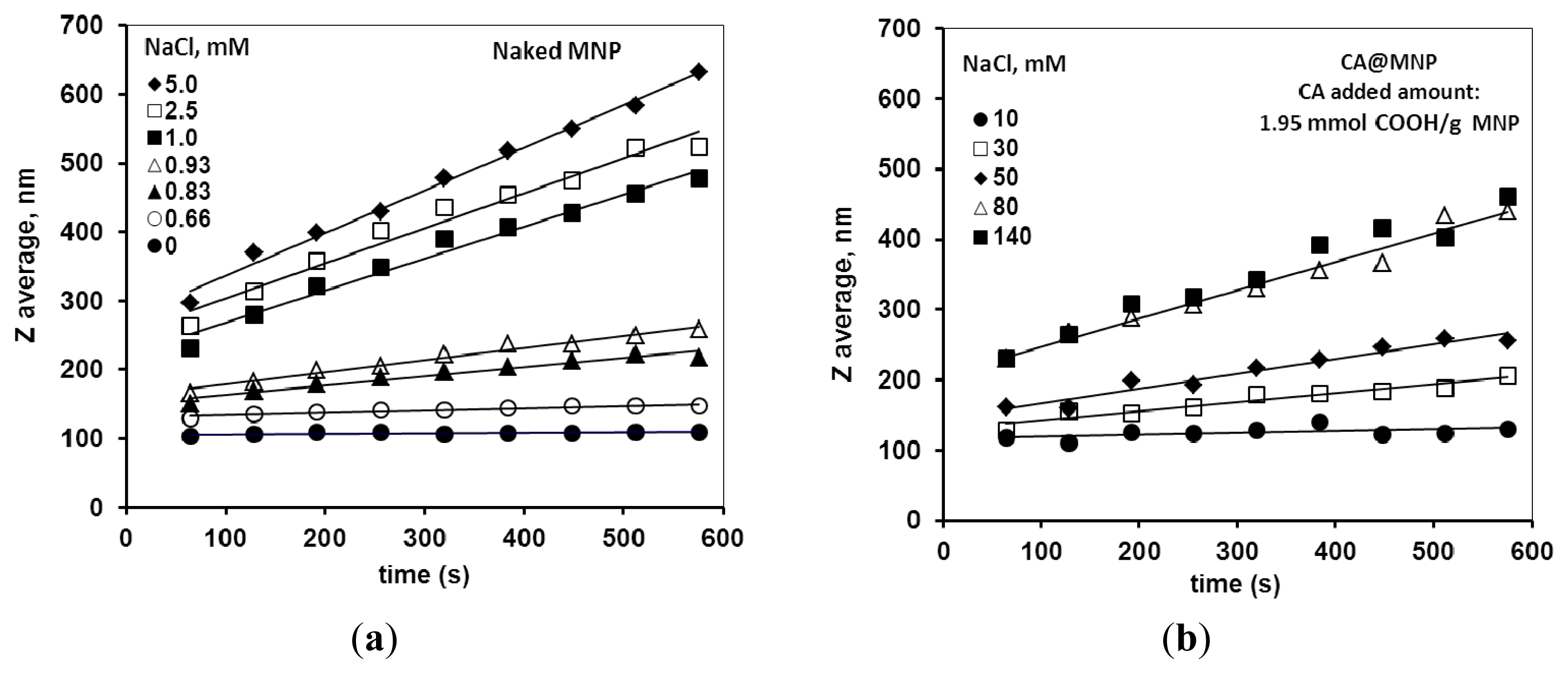
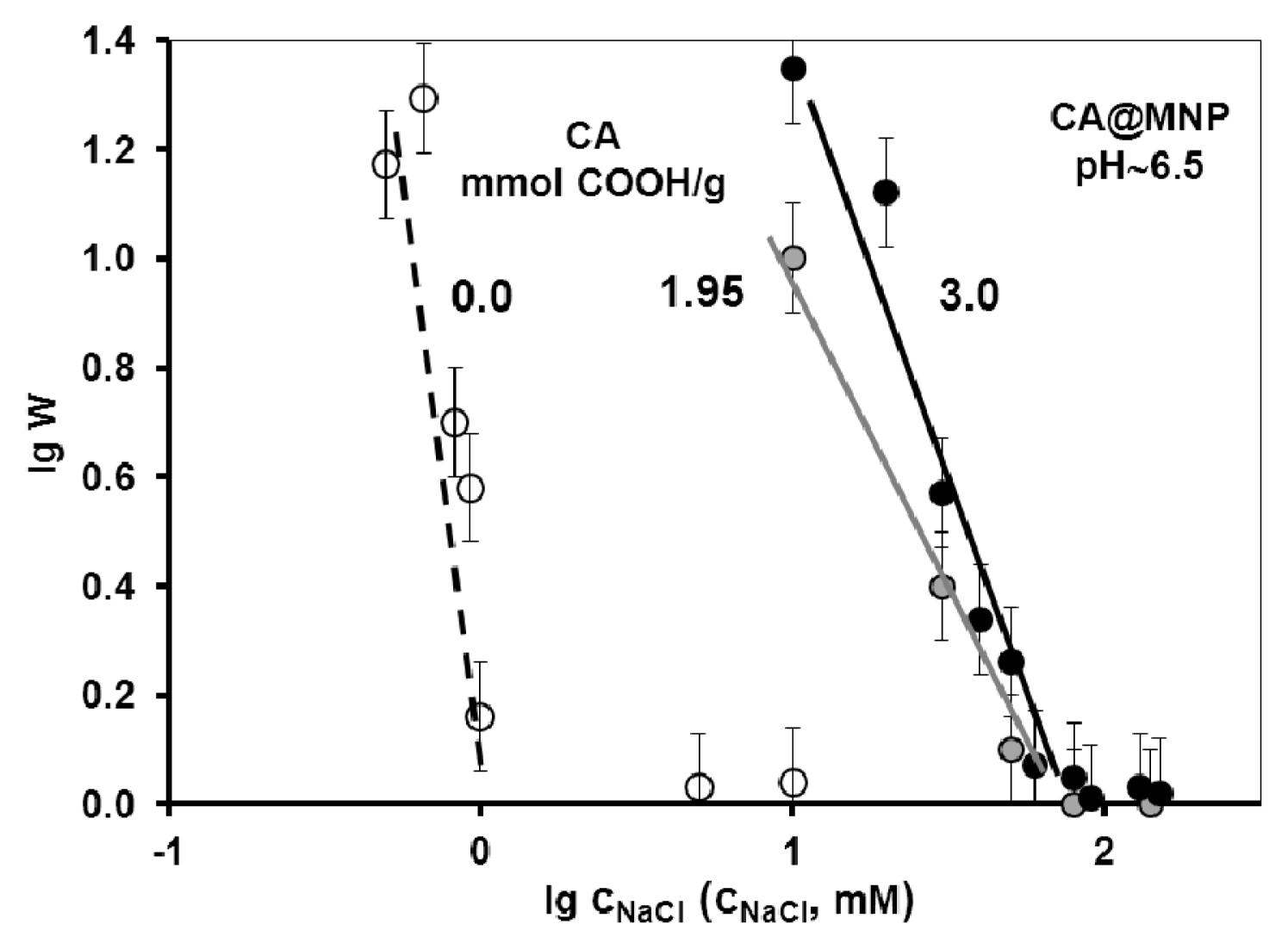
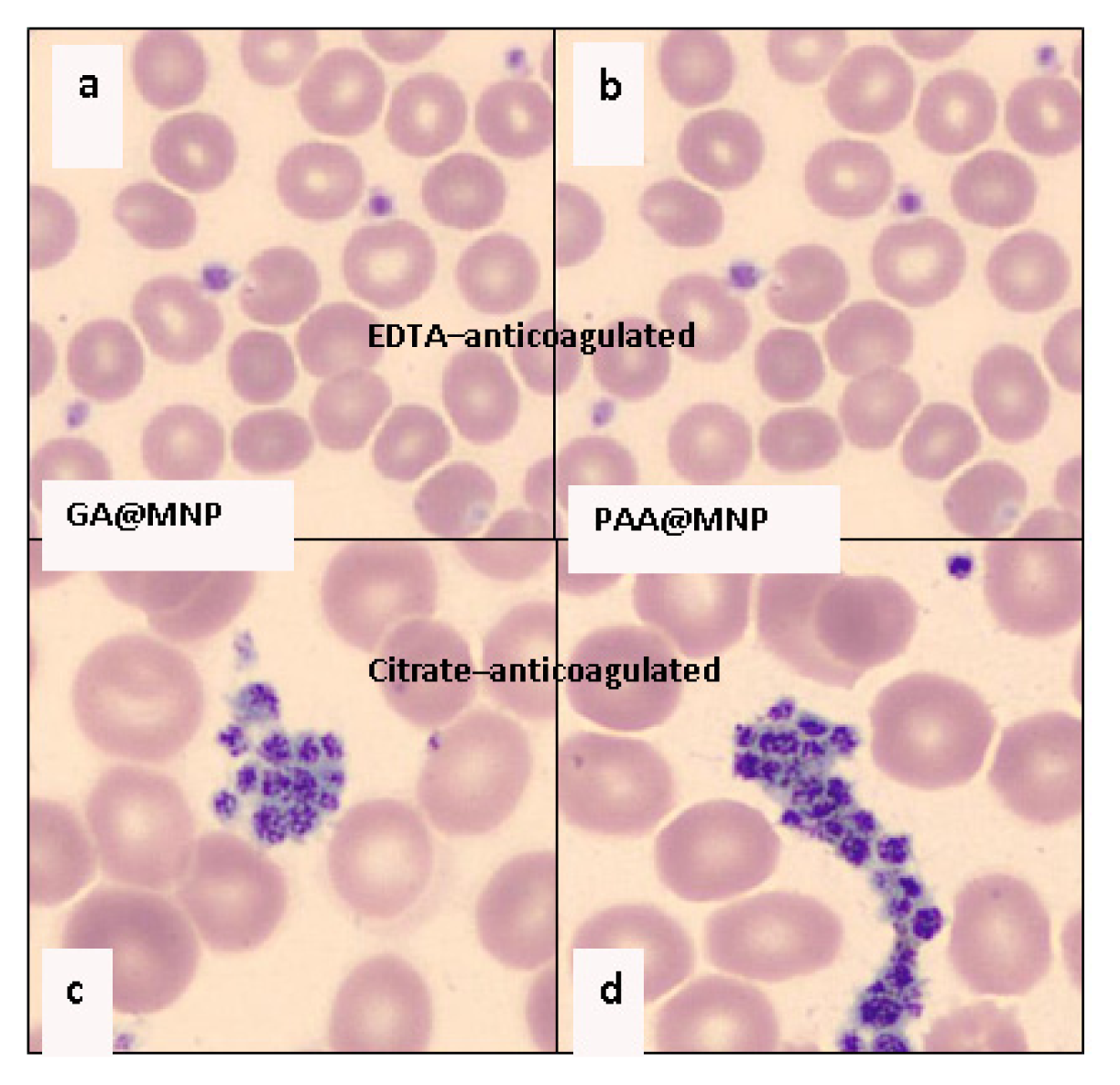
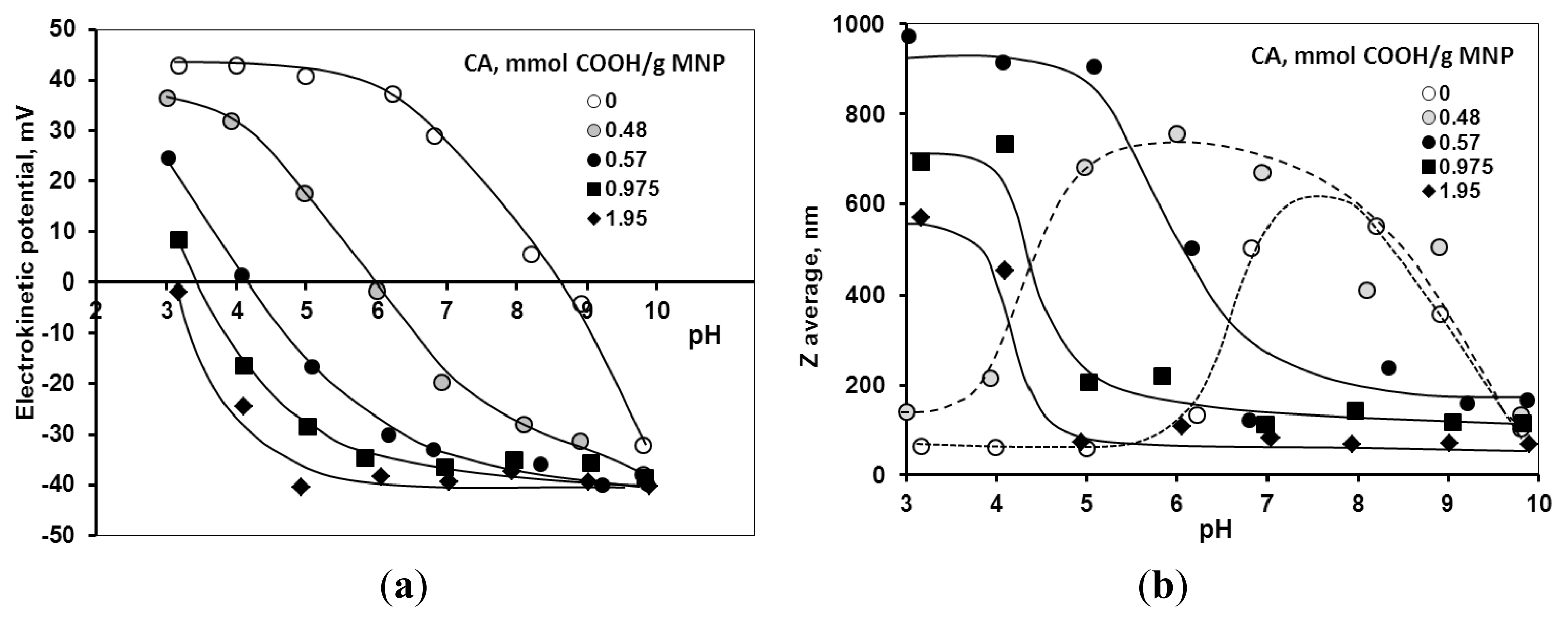
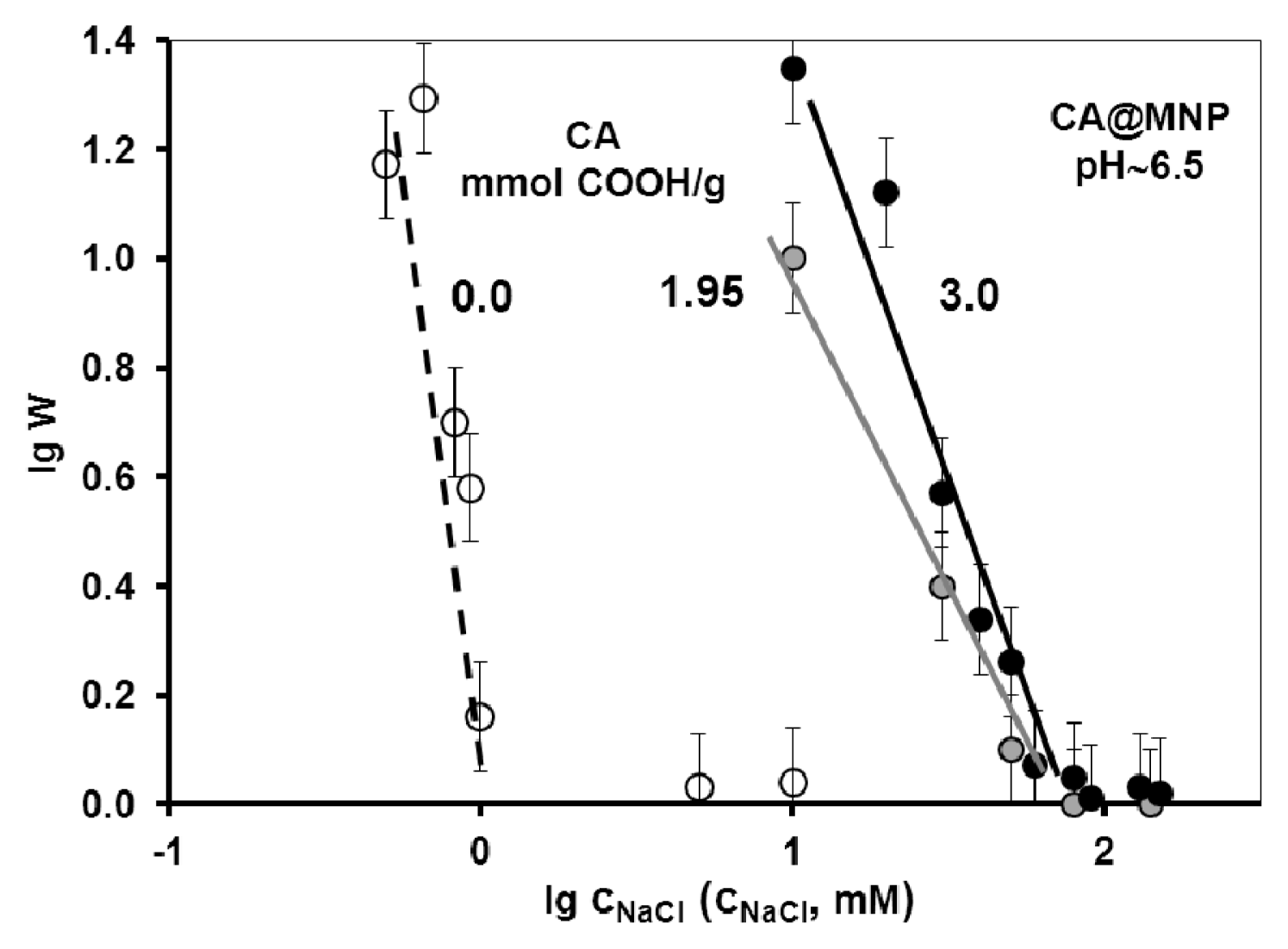
2.3. Colloidal Stability of the Core-Shell MNPs
2.4. In Vitro and in Vivo Performance of Carboxylate@MNPs
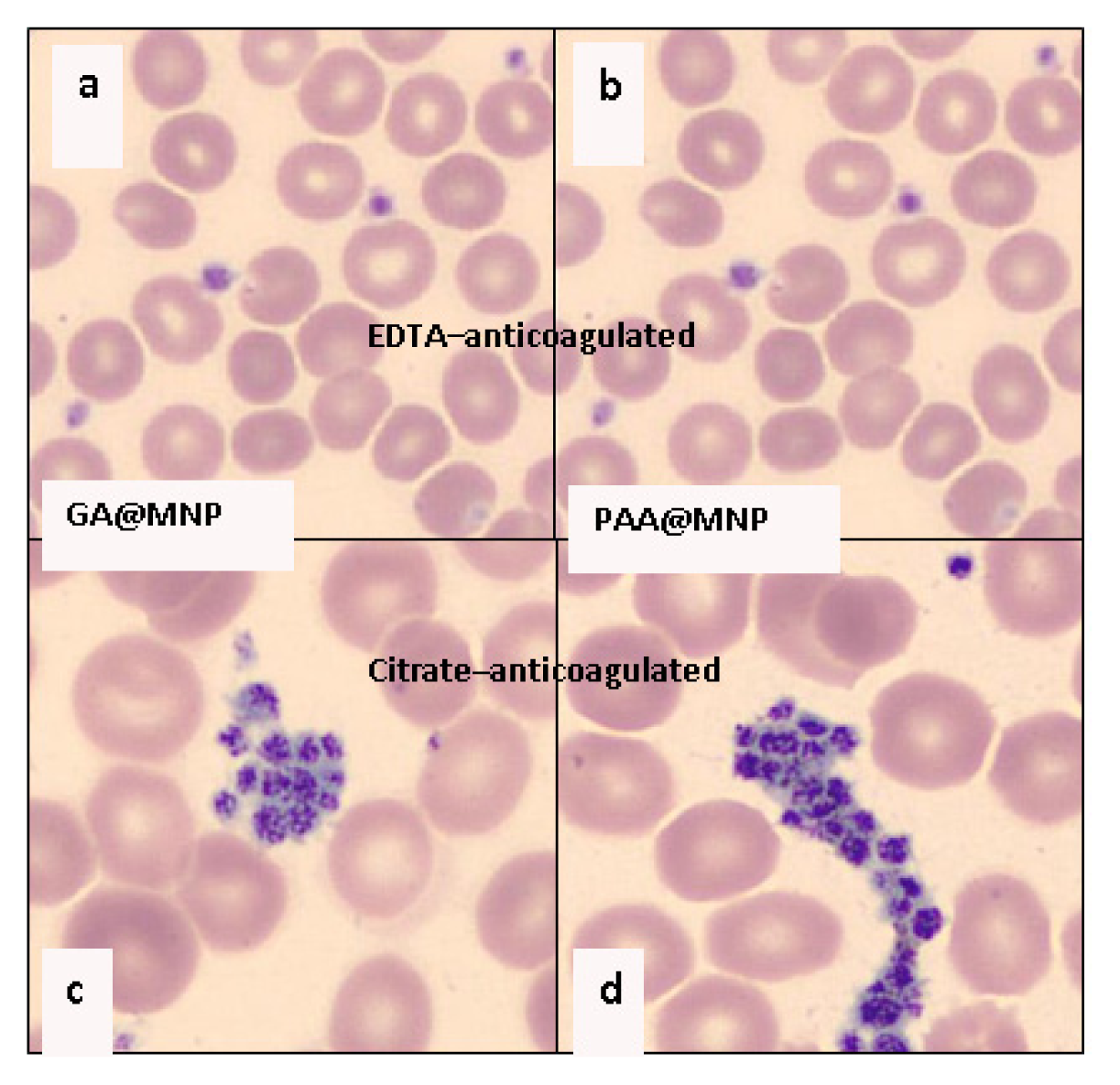
2.4.1. Biocompatibility Tests
2.4.2. Chemical Stability of Carboxylate@MNPs in Biological Media
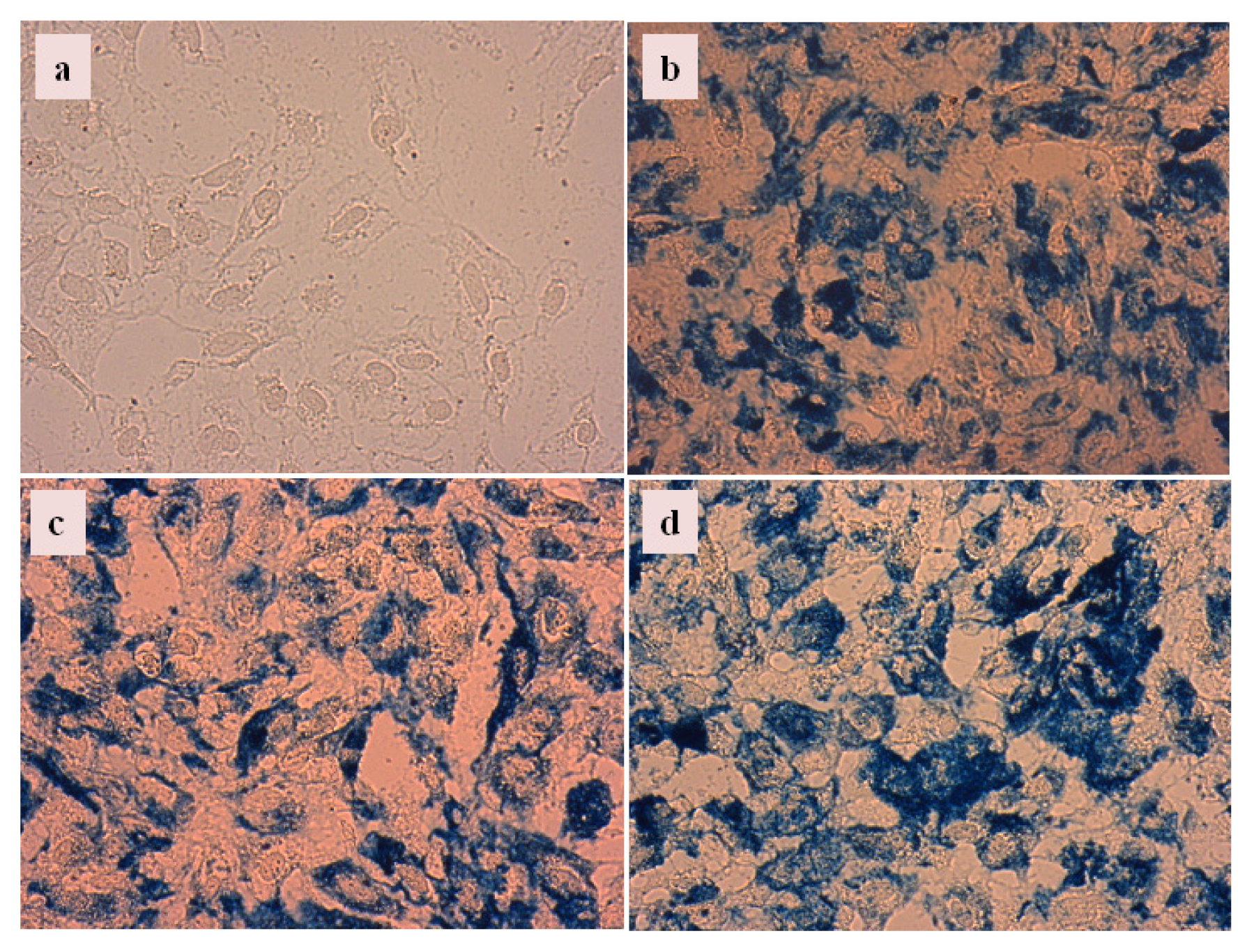
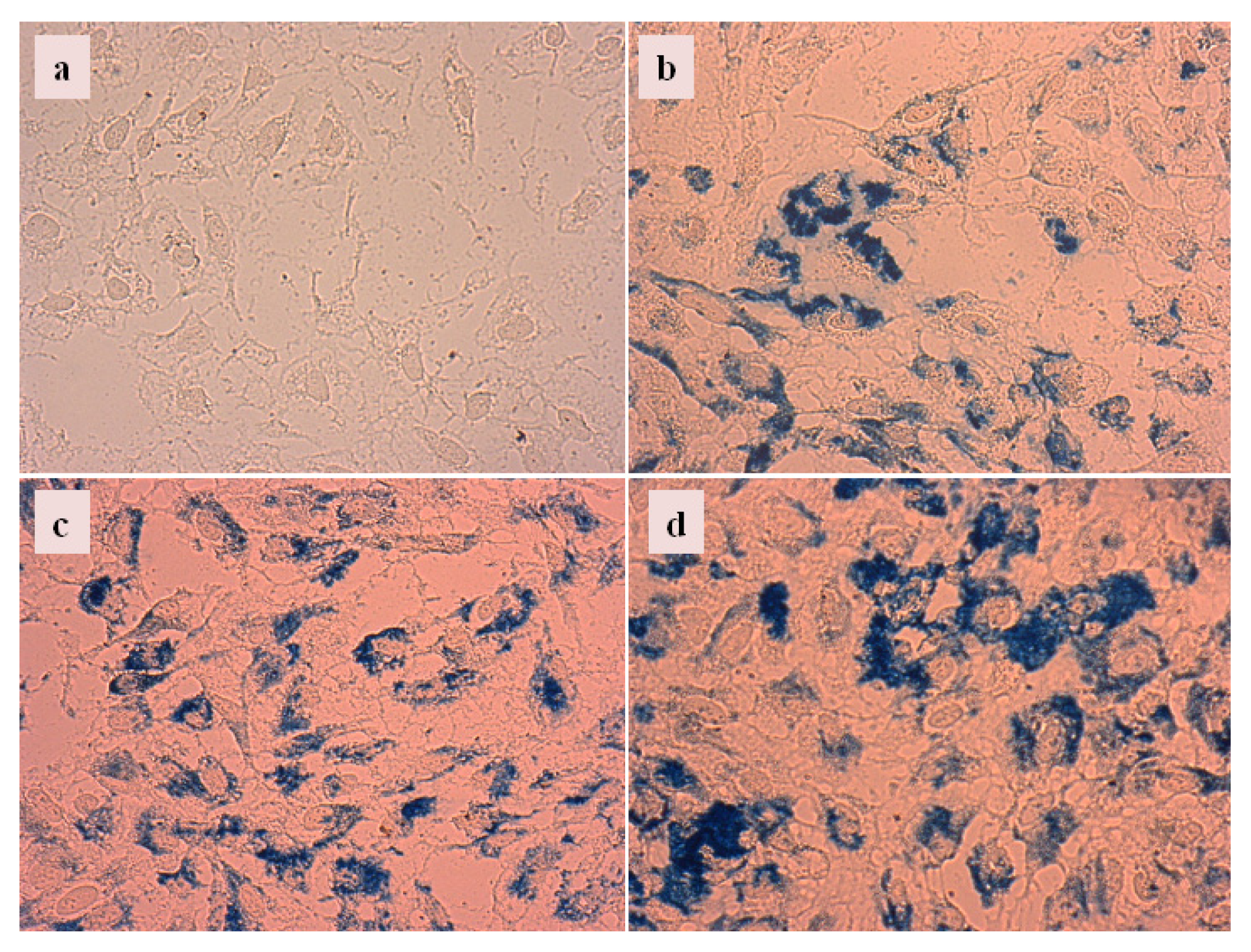
2.4.3. Cellular Uptake of Carboxylate@MNPs
3. Experimental Section
3.1. Materials
3.2. Methods
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Gupta, A.K.; Gupta, M. Synthesis and surface engineering of iron oxide nanoparticles for biomedical applications. Biomaterials 2005, 26, 3995–4021. [Google Scholar]
- Mura, S.; Couvreur, P. Nanotheranostics for personalized medicine. Adv. Drug Deliv. Rev 2012, 64, 1394–1416. [Google Scholar]
- Chia, X.; Huang, D.; Zhao, Z.; Zhou, Z.; Yin, Z.; Gao, J. Nanoprobes for in vitro diagnostics of cancer and infectious diseases. Biomaterials 2012, 33, 189–206. [Google Scholar]
- Ravindran, A.; Chandran, P.; Khan, S.S. Biofunctionalized silver nanoparticles: Advances and prospects. Colloids Surf. B 2013, 105, 342–352. [Google Scholar]
- Ordidge, K.L.; Duffy, B.A.; Wells, J.A.; Kalber, T.L.; Janes, S.M.; Lythgoe, M.F. Imaging the paediatric lung: What does nanotechnology have to offer? Paediatr. Respir. Rev 2012, 13, 84–88. [Google Scholar]
- Wang, M.; Thanou, M. Targeting nanoparticles to cancer. Pharmacol. Res 2010, 62, 90–99. [Google Scholar]
- Villa, C.; Erratico, S.; Razini, P.; Fiori, F.; Rustichelli, F.; Torrente, Y.; Belicchi, M. Stem Cell Tracking by Nanotechnologies. Int. J. Mol. Sci 2010, 11, 1070–1081. [Google Scholar]
- Sánchez-Moreno, P.; Ortega-Vinuesa, J.L.; Martín-Rodríguez, A.; Boulaiz, H.; Marchal-Corrales, J.A.; Peula-García, J.M. Characterization of different functionalized lipidic nanocapsules as potential drug carriers. Int. J. Mol. Sci 2012, 13, 2405–2424. [Google Scholar]
- Crommelina, D.J.A.; Florence, A.T. Towards more effective advanced drug delivery systems. Int. J. Pharm. 2013. [Google Scholar] [CrossRef]
- Etheridge, M.L.; Campbell, S.A.; Erdman, A.G.; Haynes, C.L.; Wolf, S.M.; McCullough, J. The big picture on nanomedicine: The state of investigational and approved nanomedicine products. Nanomedicine 2013, 9, 1–14. [Google Scholar]
- Prescription and Over-the-Counter Drug Product List, 32ND Edition ed. Available online: http://www.fda.gov/downloads/Drugs/InformationOnDrugs/UCM312738.pdf (on accessed 22 April 2013).
- U.S. Food and Drug Administration. Drugs to be Discontinued. Available online: http://www.fda.gov/Drugs/DrugSafety/DrugShortages/ucm050794.htm (on accessed 22 April 2013).
- European Medicines Agency. Questions and Answers on the Withdrawal of the Marketing Application for Sinerem, 2008. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Medicine_QA/2009/11/WC500015294.pdf (on accessed 22 April 2013).
- U.S. Food and Drug Administration. Available online: http://google2.fda.gov/search?q=Combidex&client=FDAgov&site=FDAgov&lr=&proxystylesheet=FDAgov&output=xml_no_dtd&getfields=*&filter=1&requiredfields=-archive%3AYes (on accessed 22 April 2013).
- U.S. food and drug administration. Feraheme (ferumoxytol) injection. Available online: http://www.fda.gov/Safety/MedWatch/SafetyInformation/ucm235636.htm (on accessed 22 April 2013).
- NCBI. Molecular Imaging and Contrast Agent Database (MICAD) [Internet]. Citrate-coated (184th variant) very small superparamagnetic iron oxide particles VSOP-C184. Available online: http://www.ncbi.nlm.nih.gov/books/NBK23512/ (on accessed 22 April 2013).
- Bayerpharma. Trial finder-clinical trial registry and results search. Available online: http://www.bayerpharma.com/en/research-and-development/clinical-trials/trial-finder/trialfinder_detail.php?trialid=90956&search=&product=Resovist%20%28Ferucarbotran,%20BAY86%E2%80%934884%29&overall_status=&country=&phase=&condition=&results=0&trials=1&btnSubmit=submit&num=10&show=1 (on accessed 22 April 2013).
- Dufort, S.; Sancey, L.; Coll, J.-L. Physicochemical parameters that govern nanoparticles fate also dictate rules for their molecular evolution. Adv. Drug Deliv. Rev 2012, 64, 179–189. [Google Scholar]
- Cornell, R.M.; Schwertmann, U. The Iron Oxides: Sturcutre, Properties, Reactions, Occurencies and Uses, 2nd ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2003; pp. 527–542. [Google Scholar]
- Creixell, M.; Herrera, A.P.; Latorre-Esteves, M.; Ayala, V.; Torres-Lugo, M.; Rinaldi, C. The effect of grafting method on the colloidal stability and in vitro cytotoxicity of carboxymethyl dextran coated magnetic nanoparticles. J. Mater. Chem. 2010, 20, 8539–8547. [Google Scholar]
- Musyanovych, A.; Dausend, J.; Dass, M.; Walther, P.; Mailänder, V.; Landfester, K. Criteria impacting the cellular uptake of nanoparticles: A study emphasizing polymer type and surfactant effects. Acta Biomat 2011, 7, 4160–4168. [Google Scholar]
- Hajdú, A.; Illés, E.; Tombácz, E.; Borbáth, I. Surface charging, polyanionic coating and colloid stability of magnetite nanoparticles. Colloids Surf. A 2009, 347, 104–108. [Google Scholar]
- Hajdú, A.; Szekeres, M.; Tóth, I.Y.; Bauer, R.A.; Mihály, J.; Zupkó, I.; Tombácz, E. Enhanced stability of polyacrylate-coated magnetite nanoparticles in biorelevant media. Colloids Surf. B 2012, 94, 242–249. [Google Scholar]
- Tóth, I.Y.; Illés, E.; Bauer, R.A.; Nesztor, D.; Szekeres, M.; Zupkó, I.; Tombácz, E. Designed polyelectrolyte shell on magnetite nanocore for dilution-resistant biocompatible magnetic fluids. Langmuir 2012, 28, 16638–16646. [Google Scholar]
- Tombácz, E.; Tóth, I.Y.; Nesztor, D.; Illés, E.; Hajdú, A.; Szekeres, M.; Vékás, L. Adsorption of organic acids on magnetite nanoparticles, pH-dependent colloidal stability and salt tolerance. Colloids Surf. A 2013, in press. [Google Scholar]
- Tombácz, E.; Illés, E.; Hajdú, A.; Tóth, I.Y.; Bauer, R.A.; Nesztor, D.; Szekeres, M.; Zupkó, I.; Vékás, L. Colloidal stability of carboxylated iron oxide nanomagnets for biomedical use. Period. Polytech. Chem. Eng. 2013, in press. [Google Scholar]
- Stephen, Z.R.; Kievit, F.M.; Zhang, M. Magnetite nanoparticles for medical MR imaging. Mater. Today 2011, 14, 330–338. [Google Scholar]
- Soenen, S.J.H.; Himmelreich, U.; Nuytten, N.; Pisanic, T.R., II; Ferrari, A.; de Cuyper, M. Intracellular nanoparticle coating stability determines nanoparticle diagnostics efficacy and cell functionality. Small 2010, 6, 2136–2145. [Google Scholar]
- Soenen, S.J.H.; Uwe Himmelreich, U.; Nuytten, E.; de Cuyper, M. Cytotoxic effects of iron oxide nanoparticles and implications for safety in cell labelling. Biomaterials 2011, 32, 195–205. [Google Scholar]
- Giles, C.H.; MacEwan, T.H.; Nakhwa, S.N.; Smith, D. Studies in adsorption. Part XI. A system of classification of solution adsorption isotherms. J. Chem. Soc 1960, 4, 3973–3993. [Google Scholar]
- Miles, W.C.; Huffstetler, P.P.; Goff, J.D.; Chen, A.Y.; Riffle, J.S.; Davis, R.M. Design of stable polyether-magnetite complexes in aqueous media: Effects of the anchor group, molecular weight, and chain density. Langmuir 2011, 27, 5456–5463. [Google Scholar]
- Santiago-Rodríguez, L.; Lafontaine, M.M.; Castro, C.; Méndez-Vega, J.; Latorre-Esteves, M.; Juan, E.J.; Mora, E.; Torres-Lugo, M.; Rinaldi, C. Synthesis, stability, cellular uptake, and blood circulation time of carboxymethyl-inulin coated magnetic nanoparticles. J. Mater. Chem. B 2013, 1, 2807–2817. [Google Scholar]
- Deng, Y.-F.; Zhou, Z.-H. Synthesis and crystal structure of a zinc citrate complex [Zn(H2cit)(H2O)]n. J. Coord. Chem 2009, 62, 1484–1491. [Google Scholar]
- Rodríguez, E.; Fernández, G.; Ledesma, B.; Álvarez, P.; Beltrán, F.J. Photocatalytic degradation of organics in water in the presence of iron oxides: Influence of carboxylic acids. Appl. Catal. B 2009, 92, 240–249. [Google Scholar]
- Cornell, R.M.; Schwertmann, U. The Iron Oxides: Sturcutre, Properties, Reactions, Occurencies and Uses, 2nd ed; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2003; pp. 211–213. [Google Scholar]
- Gutteridge, J.M.C.; Mumby, S.; Lamb, N.J. Iron binding and autoreduction by citrate: Are these involved in signalling by iron regulatory protein-I? Free Radic. Res 1998, 28, 319–322. [Google Scholar]
- Blesa, M.A.; Weisz, A.D.; Morando, P.J.; Salfity, J.A.; Magaz, G.E.; Regazzoni, A.E. The interaction of metal oxide surfaces with complexing agents dissolved in water. Coord. Chem. Rev 2000, 196, 31–63. [Google Scholar]
- Waite, T.D.; Morel, F.M.M. Photoreductive dissolution of colloidal iron oxide: Effect of citrate. J. Colloid Interface Sci. 1984, 102, 121–134. [Google Scholar]
- Behdadfar, B.; Kermanpur, A.; Sadeghi-Aliabadi, H.; Morales, M.P.; Mozaffari, M. Synthesis of high intrinsic loss power aqueous ferrofluids of iron oxide nanoparticles by citric acid-assisted hydrothermal-reduction route. J. Solid State Chem 2012, 187, 20–26. [Google Scholar]
- Colucci, J.; Montalvo, V.; Hernandez, R.; Poullet, C. Electrochemical oxidation potential of photocatalyst reducing agents. Electrochim. Acta 1999, 44, 2507–2514. [Google Scholar]
- Mahmoudi, M.; Simchi, A.; Milani, A.S.; Stroeve, P. Cell toxicity of superparamagnetic iron oxide nanoparticles. J. Colloid Interface Sci 2009, 336, 510–518. [Google Scholar]
- Meerod, S.; Tumcharern, G.; Wichai, U.; Rutnakornpituk, M. Magnetite nanoparticles stabilized with polymeric bilayer of poly(ethylene glycol) methyl ether-poly(3-caprolactone) copolymers. Polymer 2008, 49, 3950–3956. [Google Scholar]
- Haddad, P.S.; Martins, T.M.; D’Souza-Li, L.; Li, L.M.; Metze, K.; Adam, R.L.; Knobel, M.; Zanchet, D. Structural and morphological investigation of magnetic nanoparticles based on iron oxides for biomedical applications. Mater. Sci. Eng. C 2008, 28, 489–494. [Google Scholar]
- Sanchez, R.D.; Rivas, J.; Vaqueiro, P.; Lopez-Quintela, M.A.; Caeiro, D. Particle size effects on magnetic properties of yttrium iron garnets prepared by a sol-gel method. J. Magn. Magn. Mater 2002, 247, 92–98. [Google Scholar]
- Song, M.; Zhang, Y.; Hu, S.; Song, L.; Dong, J.; Chen, Z.; Gu, N. Influence of morphology and surface exchange reaction on magnetic properties of monodisperse magnetite nanoparticles. Colloids Surf. A 2012, 408, 114–121. [Google Scholar]
- Parvin, K.; Ma, J.; Ly, J.; Sun, X.C.; Nikles, D.E.; Sun, K.; Wang, L.M. Synthesis and magnetic properties of monodisperse Fe3O4 nanoparticles. J. Appl. Phys 2004, 95, 7121–7123. [Google Scholar]
- Shendruk, T.N.; Desautels, R.D.; Southern, B.W.; van Lierop, J. The effect of surface spin disorder on the magnetism of γ-Fe2O3nanoparticle dispersions. Nanotechnology 2007, 18. [Google Scholar] [CrossRef]
- Soenen, S.J.; Nuytten, N.; Himmelreich, U.; de Cuyper, M. Intracellular iron oxide nanoparticle coating stability determines nanoparticle usability and cell functionality. Drug Discov. Today 2010, 15, 1082. [Google Scholar]
- Safi, M.; Courtois, J.; Seigneuret, M.; Conjeaud, H.; Berret, J.-F. The effects of aggregation and protein corona on the cellular internalization of iron oxide nanoparticles. Biomaterials 2011, 32, 9353–9363. [Google Scholar]
- Lartigue, L.; Wilhelm, C.; Servais, J.; Factor, C.; Dencausse, A.; Bacri, J.-C.; Luciani, N.; Gazeau, F. Nanomagnetic sensing of blood plasma protein interactions with iron oxide nanoparticles: Impact on macrophage uptake. ACS Nano 2012, 6, 2665–2678. [Google Scholar]
- Jedlovszky-Hajdú, A.; Bombelli, F.B.; Monopoli, M.P.; Tombácz, E.; Dawson, K.A. Surface coatings shape the protein corona of SPIONs with relevance to their application in vivo. Langmuir 2012, 28, 14983–14991. [Google Scholar]
- Jackson, S.P. The growing complexity of platelet aggregation. Blood 2007, 109, 5087–5095. [Google Scholar]
- Jung, S.M.; Kinoshita, K.; Tanoue, K.; Isohisa, I.; Yamazaki, H. Role of surface negative charge in platelet function related to the hyperreactive state in estrogen-treated prostatic carcinoma. Thromb. Haemost 1982, 47, 203–209. [Google Scholar]
- Grøttum, K.A.; Jørgensen, L.; Jeremic, M. Decrease in platelet surface charge during phagocytosis of polystyrene latex particles or thorium dioxide. Scand. J. Haematol 1972, 9, 83–96. [Google Scholar]
- Grant, R.A.; Zucker, M.B. EDTA-induced increase in platelet surface charge associated with the loss of aggregability. Assessment by partition in aqueous two-phase polymer systems and electrophoretic mobility. Blood 1978, 52, 515–523. [Google Scholar]
- Smyth, S.S.; Whiteheart, S.; Italiano, J.E., Jr.; Coller, B.S. Platelet Morphology, Biochemistry & Function. In Williams Hematology, 8th ed; The McGraw-Hill Companies, Inc: New York, NY, USA, 2010; pp. 1735–1814. [Google Scholar]
- Bhattarai, S.R.; Kc, R.B.; Kim, S.Y.; Sharma, M.; Khil, M.S.; Hwang, P.H.; Chung, G.H.; Kim, H.Y. N-hexanoyl chitosan stabilized magnetic nanoparticles: Implication for cellular labeling and magnetic resonance imaging. J. Nanobiotechnol. 2008, 6. [Google Scholar] [CrossRef]
- Ruiz, A.; Salas, G.; Calero, M.; Hernández, Y.; Villanueva, A.; Herranz, F.; Veintemillas-Verdaguer, S.; Martínez, E.; Barber, D.F.; Morales, M.P. Short-chain PEG molecules strongly bound to magnetic nanoparticle for MRI long circulating agents. Acta Biomater 2013, 9, 6421–6430. [Google Scholar]
- Alexiou, C.; Jurgons, R.; Seliger, C.; Brunke, O.; Iro, H.; Odenbach, S. Delivery of superparamagnetic nanoparticles for local chemotherapy after intraarterial infusion and magnetic drug targeting. Anticancer Res 2007, 27, 2019–2022. [Google Scholar]
- Janic, B.; Rad, A.M.; Jordan, E.K.; Iskander, A.S.M.; Ali, M.M.; Varma, N.R.S.; Frank, J.A.; Arbab, A.S. Optimization and validation of FePro cell labeling method. PLoS One 2009, 4. [Google Scholar] [CrossRef]
- Misri, R.; Meier, D.; Yung, A.C.; Kozlowski, P.; Häfeli, U.O. Development and evaluation of a dual-modality (MRI/SPECT) molecular imaging bioprobe. Nanomedicine 2012, 8, 1007–1016. [Google Scholar]
- Illés, E.; Tombácz, E. The effect of humic acid adsorption on pH-dependent surface charging and aggregation of magnetite nanoparticles. J. Colloid Interface Sci 2006, 295, 115–123. [Google Scholar]
- Vékás, L.; Bica, D.; Marinica, O. Magnetic nanofluids stabilized with various chain length surfactants. Rom. Rep. Phys 2006, 58, 217–228. [Google Scholar]
- Bica, D.; Vékás, L.; Avdeev, M.V.; Marinica, O.; Socoliuc, V.; Balasoiu, M.; Garamus, V.M. Sterically stabilized water based magnetic fluids: Synthesis, structure and properties. J. Magn. Magn. Mater 2007, 311, 17–21. [Google Scholar]
- Tombácz, E.; Illés, E.; Majzik, A.; Hajdú, A.; Rideg, N.; Szekeres, M. Ageing in the inorganic nanoworld: Example of magnetite nanoparticles in aqueous medium. Croat. Chem. Acta 2007, 80, 503–515. [Google Scholar]
- Tombácz, E.; Hajdú, A.; Illés, E.; László, K.; Garberoglio, G.; Jedlovszky, P. Water in contact with magnetite nanoparticles, as seen from experiments and computer simulations. Langmuir 2009, 25, 13007–13014. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic points | CA | GA | PAA | PAM |
|---|---|---|---|---|
| mmol COOH/g MNP | ||||
| High-affinity adsorption limit | 0.33 | 0.17 | - | 0.32 |
| Isoelectric point (charge neutralization) | 0.45 | 0.05 | 0.13 | 0.17 |
| Complete overcharging | ~0.8 | 0.4 | 0.4 | 0.4 |
| Carboxylate coating | Averaged inhibition % | Dissolved iron mg/g MNP | CCC mM |
|---|---|---|---|
| CA | 18.3 ± 1.7 | 1.276 | 70 |
| GA | 1.5 ± 0.5 | 0 | 20 |
| PAA | 12.5 ± 1.4 | 0.038 | 500 |
| PAM | 1.5 ± 0.5 | 0 | 500 |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Szekeres, M.; Tóth, I.Y.; Illés, E.; Hajdú, A.; Zupkó, I.; Farkas, K.; Oszlánczi, G.; Tiszlavicz, L.; Tombácz, E. Chemical and Colloidal Stability of Carboxylated Core-Shell Magnetite Nanoparticles Designed for Biomedical Applications. Int. J. Mol. Sci. 2013, 14, 14550-14574. https://doi.org/10.3390/ijms140714550
Szekeres M, Tóth IY, Illés E, Hajdú A, Zupkó I, Farkas K, Oszlánczi G, Tiszlavicz L, Tombácz E. Chemical and Colloidal Stability of Carboxylated Core-Shell Magnetite Nanoparticles Designed for Biomedical Applications. International Journal of Molecular Sciences. 2013; 14(7):14550-14574. https://doi.org/10.3390/ijms140714550
Chicago/Turabian StyleSzekeres, Márta, Ildikó Y. Tóth, Erzsébet Illés, Angéla Hajdú, István Zupkó, Katalin Farkas, Gábor Oszlánczi, László Tiszlavicz, and Etelka Tombácz. 2013. "Chemical and Colloidal Stability of Carboxylated Core-Shell Magnetite Nanoparticles Designed for Biomedical Applications" International Journal of Molecular Sciences 14, no. 7: 14550-14574. https://doi.org/10.3390/ijms140714550